A CRISPR-Cas and Tat Peptide with Fluorescent RNA Aptamer System for Signal Amplification in RNA Imaging

{kind=link}
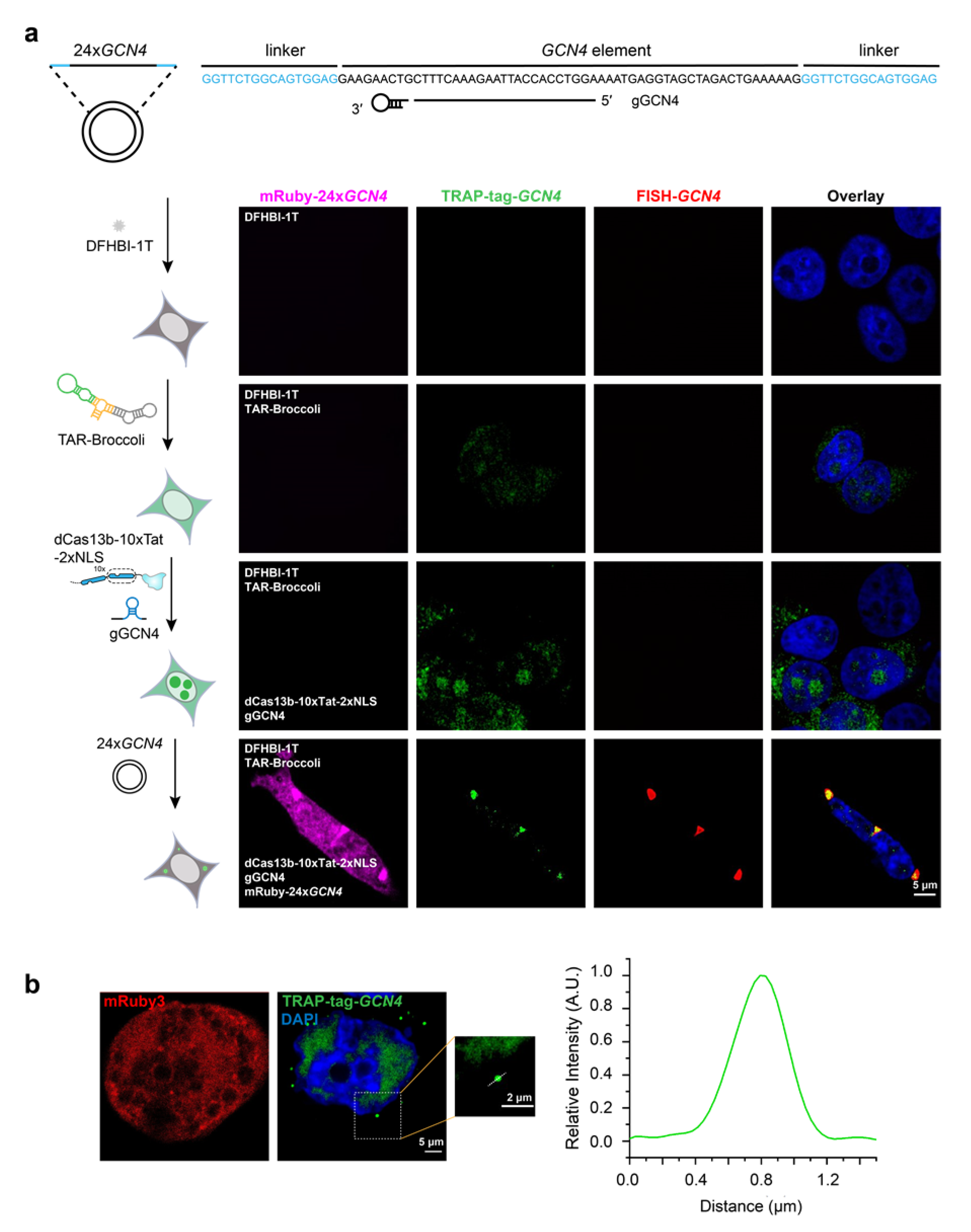{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design of the CRISPR-10 × Tat and TAR–Aptamer
2.2. Plasmid Construction
2.3. Cell Cultivation and Transfection
2.4. Single-Molecule Fluorescence In Situ Hybridization (smFISH)
2.5. Confocal Laser Scanning Microscopy
2.6. Fluorophore Synthesis
3. Results and Discussion
3.1. Verification of the RNA Imaging Feasibility of TRAP-Tag in Live Cells
3.2. Labeling Exogenous GCN4 mRNAs Using TRAP-Tag
3.3. Tracking Endogenous MUC4 mRNAs and Long Non-Coding RNA SatIII Using TRAP-Tag
3.4. TRAP-Tag Can Combine with other CRISPR Proteins in RNA Imaging
3.5. CRISPR-TRAP-Tag Systems Adaptive to another RNA Aptamer
3.6. CRISPR-TRAP-Tag Systems with Multiplex RNA Aptamers Improve the Signal-To-Noise Ratio (SNR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cai, Z.K.; Cao, C.C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.C.; Hu, N.J.; Yu, X.H.; et al. RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature 2020, 582, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Nelles, D.A.; Fang, M.Y.; O’Connell, M.R.; Xu, J.L.; Markmiller, S.J.; Doudna, J.A.; Yeo, G.W. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 2016, 165, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Deng, S.; Ge, T.; Ye, M.; Yu, J.; Lin, S.; Ma, W.; Songyang, Z. Live cell imaging and proteomic profiling of endogenous NEAT1 lncRNA by CRISPR/Cas9-mediated knock-in. Protein Cell 2020, 11, 641–660. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, S.; Zhang, L.; Zhao, Q.; Li, N.; Wu, Y. A sensitive and specific method for microRNA detection and in situ imaging based on a CRISPR–Cas9 modified catalytic hairpin assembly. RSC Adv. 2020, 10, 28037–28040. [Google Scholar] [CrossRef]
- Wang, M.; Chen, K.; Wu, Q.; Peng, R.; Zhang, R.; Li, J. RCasFISH: CRISPR/dCas9-Mediated in Situ Imaging of mRNA Transcripts in Fixed Cells and Tissues. Anal. Chem. 2020, 92, 2468–2475. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Li, S.Q.; Yao, R.W.; Luan, P.F.; Wu, H.; Carmichael, G.G.; Chen, L.L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997. [Google Scholar] [CrossRef]
- Wang, H.F.; Nakamura, M.; Abbott, T.R.; Zhao, D.H.; Luo, K.W.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Rauch, S.; He, E.; Srienc, M.; Zhou, H.; Zhang, Z.; Dickinson, B.C. Programmable RNA-Guided RNA Effector Proteins Built from Human Parts. Cell 2019, 178, 122–134. [Google Scholar] [CrossRef]
- Rauch, S.; Jones, K.A.; Dickinson, B.C. Small Molecule-Inducible RNA-Targeting Systems for Temporal Control of RNA Regulation. ACS Cent. Sci. 2020, 6, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Paige, J.S.; Wu, K.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Peng, J.; Peng, S.; Wang, Q.; Jiang, X.; Xue, X.; Tao, Y.; Xiang, L.; Ji, Q.; Liu, S.-M.; et al. Live-cell RNA imaging using the CRISPR-dCas13 system with modified sgRNAs appended with fluorescent RNA aptamers. Chem. Sci. 2022, 13, 14032–14040. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016, 214, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Tu, L.C.; Naseri, A.; Chung, Y.C.; Grunwald, D.; Zhang, S.; Pederson, T. CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging. Nat. Methods 2018, 15, 928–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braselmann, E.; Wierzba, A.J.; Polaski, J.T.; Chrominski, M.; Holmes, Z.E.; Hung, S.T.; Batan, D.; Wheeler, J.R.; Parker, R.; Jimenez, R.; et al. A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells. Nat. Chem. Biol. 2018, 14, 964–971. [Google Scholar] [CrossRef]
- Filonov, G.S.; Moon, J.D.; Svensen, N.; Jaffrey, S.R. Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014, 136, 16299–16308. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Strack, R.L.; Svensen, N.; Jaffrey, S.R. Plug-and-play fluorophores extend the spectral properties of Spinach. J. Am. Chem. Soc. 2014, 136, 1198–1201. [Google Scholar] [CrossRef]
- Cawte, A.D.; Unrau, P.J.; Rueda, D.S. Live cell imaging of single RNA molecules with fluorogenic Mango II arrays. Nat. Commun. 2020, 11, 1283. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, D.; Su, N.; Bao, B.; Xie, X.; Zuo, F.; Yang, L.; Wang, H.; Jiang, L.; Lin, Q.; et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 2019, 37, 1287–1293. [Google Scholar] [CrossRef]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Larson, D.R.; Zenklusen, D.; Wu, B.; Chao, J.A.; Singer, R.H. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 2011, 332, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Mo, J.; Chen, Z.; Qin, S.; Li, S.; Liu, C.; Zhang, L.; Ran, R.; Kong, Y.; Wang, F.; Liu, S.; et al. TRADES: Targeted RNA Demethylation by SunTag System. Adv. Sci. 2020, 7, 2001402. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, J.D.; Chen, L.; Blanchard, S.; Frankel, A.D. Solution Structure of a Bovine Immunodeficiency Virus Tat-TAR Peptide-RNA Complex. Science 1995, 270, 1200–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zaccara, S.; Khuperkar, D.; Kim, H.; Tanenbaum, M.E.; Jaffrey, S.R. Live imaging of mRNA using RNA-stabilized fluorogenic proteins. Nat. Methods. 2019, 16, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Litke, J.L.; Jaffrey, S.R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 2019, 37, 667–675. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Raj, A.; Tyagi, S. Detection of Individual Endogenous RNA Transcripts In Situ Using Multiple Singly Labeled Probes. In Methods in Enzymology; Walter, N.G., Ed.; Academic Press: London, UK, 2010. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Nollet, S.; Moniaux, N.; Maury, J.; Petitprez, D.; Degand, P.; LAine, A.; Porchet, N.; Aubert, J.P. Human mucin gene MUC4: Organization of its 5’-region and polymorphism of its central tandem repeat array. Biochem. J. 1998, 332, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.H.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Valgardsdottir, R.; Chiodi, I.; Giordano, M.; Rossi, A.; Bazzini, S.; Ghigna, C.; Riva, S.; Biamonti, G. Transcription of Satellite III non-coding RNAs is a general stress response in human cells. Nucleic Acids Res. 2008, 36, 423–434. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Peng, J.; Jiang, X.; Peng, S.; Wang, F.; Weng, X.; Zhou, X. A CRISPR-Cas and Tat Peptide with Fluorescent RNA Aptamer System for Signal Amplification in RNA Imaging. Biosensors 2023, 13, 293. https://doi.org/10.3390/bios13020293
Tang H, Peng J, Jiang X, Peng S, Wang F, Weng X, Zhou X. A CRISPR-Cas and Tat Peptide with Fluorescent RNA Aptamer System for Signal Amplification in RNA Imaging. Biosensors. 2023; 13(2):293. https://doi.org/10.3390/bios13020293
Chicago/Turabian StyleTang, Heng, Junran Peng, Xin Jiang, Shuang Peng, Fang Wang, Xiaocheng Weng, and Xiang Zhou. 2023. "A CRISPR-Cas and Tat Peptide with Fluorescent RNA Aptamer System for Signal Amplification in RNA Imaging" Biosensors 13, no. 2: 293. https://doi.org/10.3390/bios13020293