Enzyme-Assisted Nucleic Acid Amplification in Molecular Diagnosis: A Review


 , and
, and
Abstract
:1. Introduction
2. Nucleic Acid Amplification Technologies
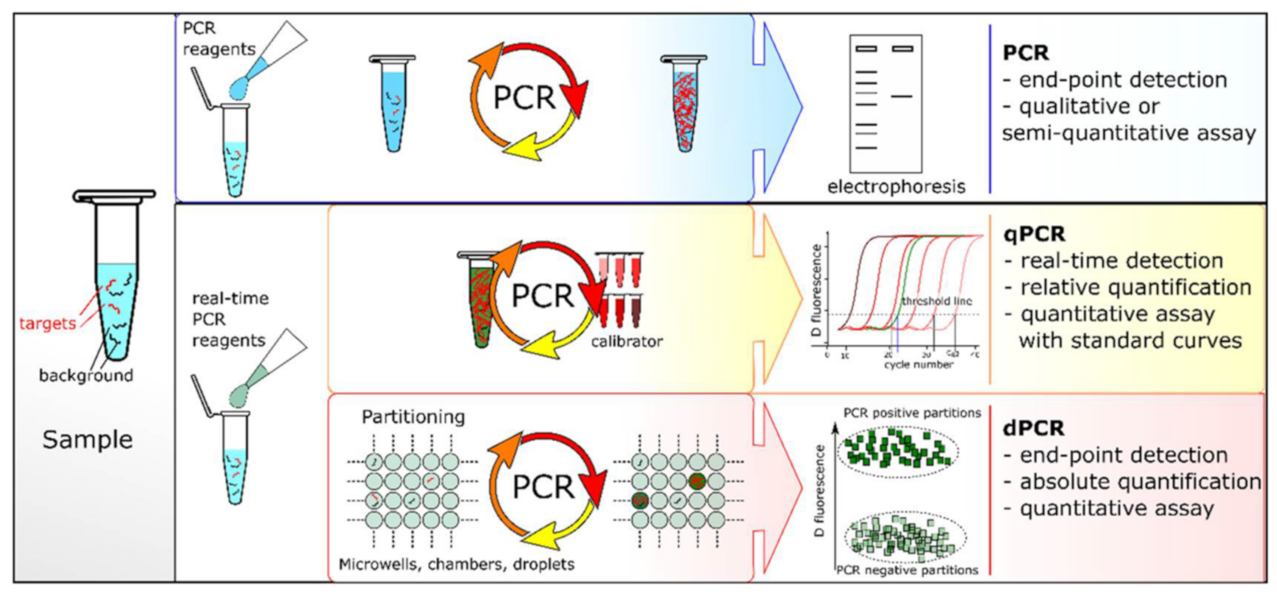
2.1. Polymerase Chain Reaction
2.1.1. Enzymes
2.1.2. Reaction Principle and Improved Methods
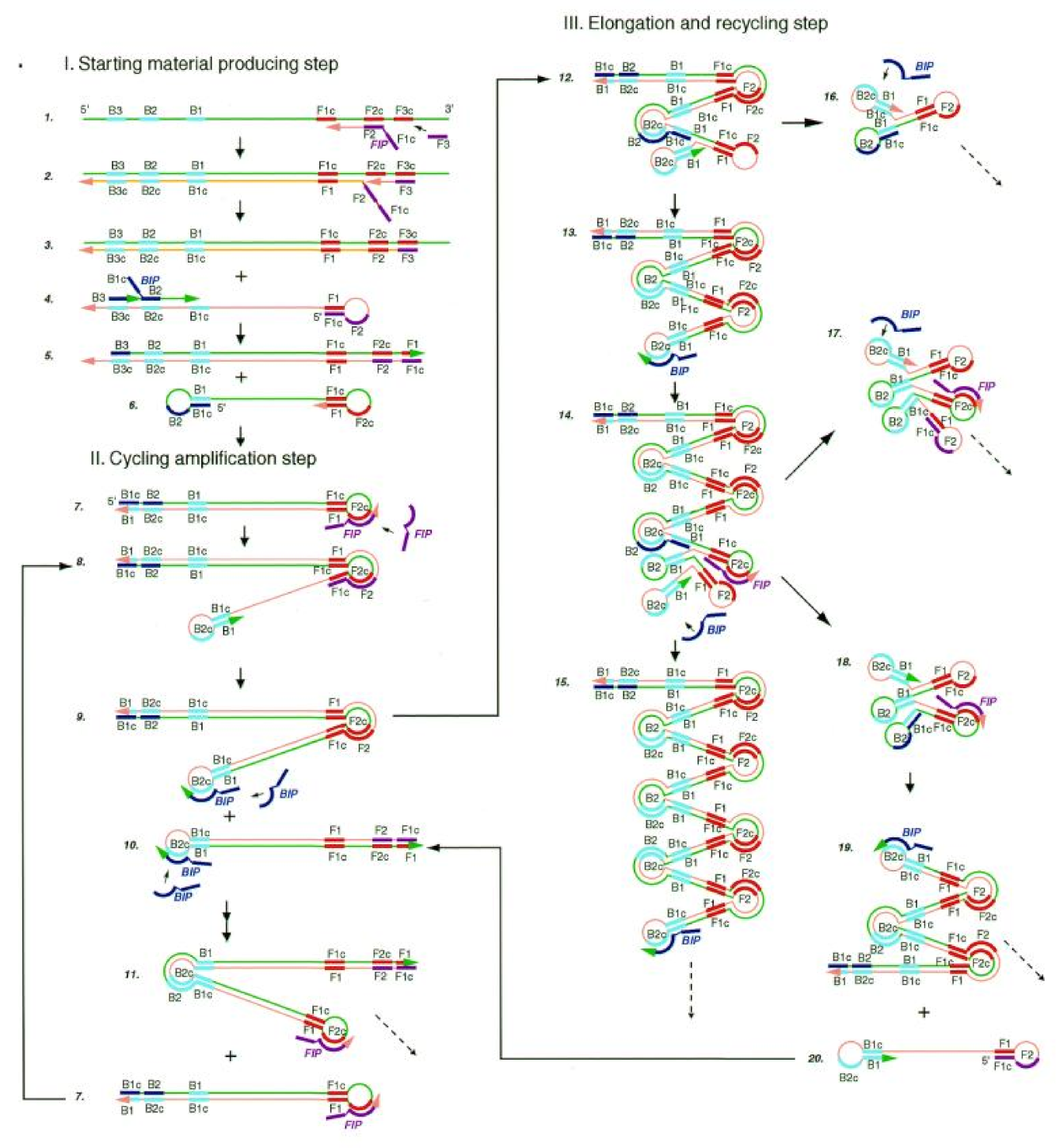
2.2. Loop-Mediated Isothermal Amplification
2.2.1. Enzymes
2.2.2. Reaction Principle and Improved Methods
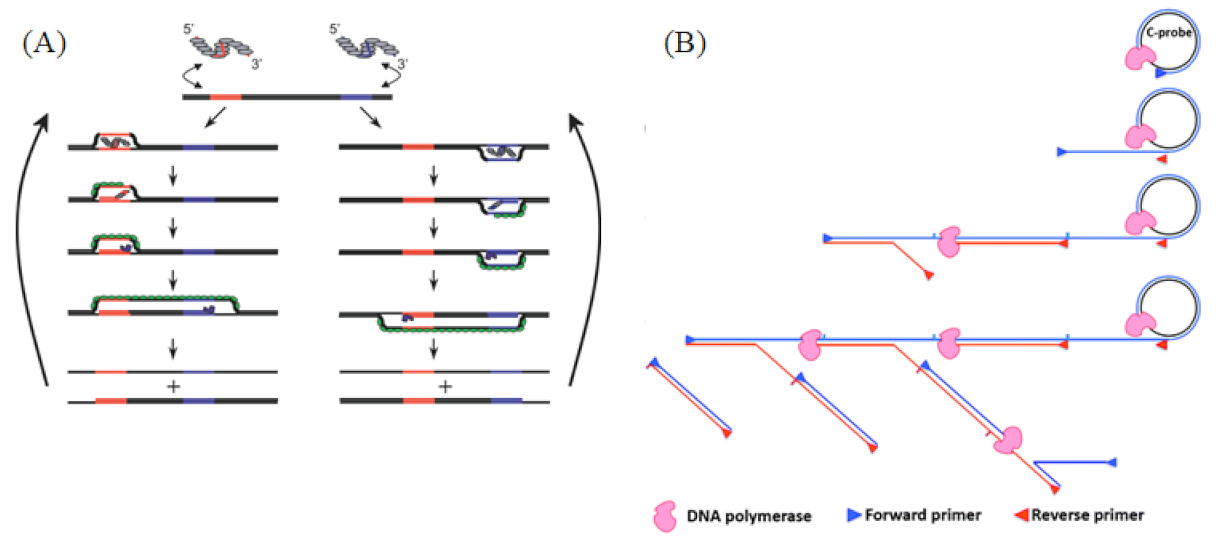
2.3. Recombinase Polymerase Amplification
2.3.1. Enzymes
2.3.2. Reaction Principle and Improved Methods

2.4. Rolling Circle Amplification
2.4.1. Enzymes
2.4.2. Reaction Principle and Improved Methods
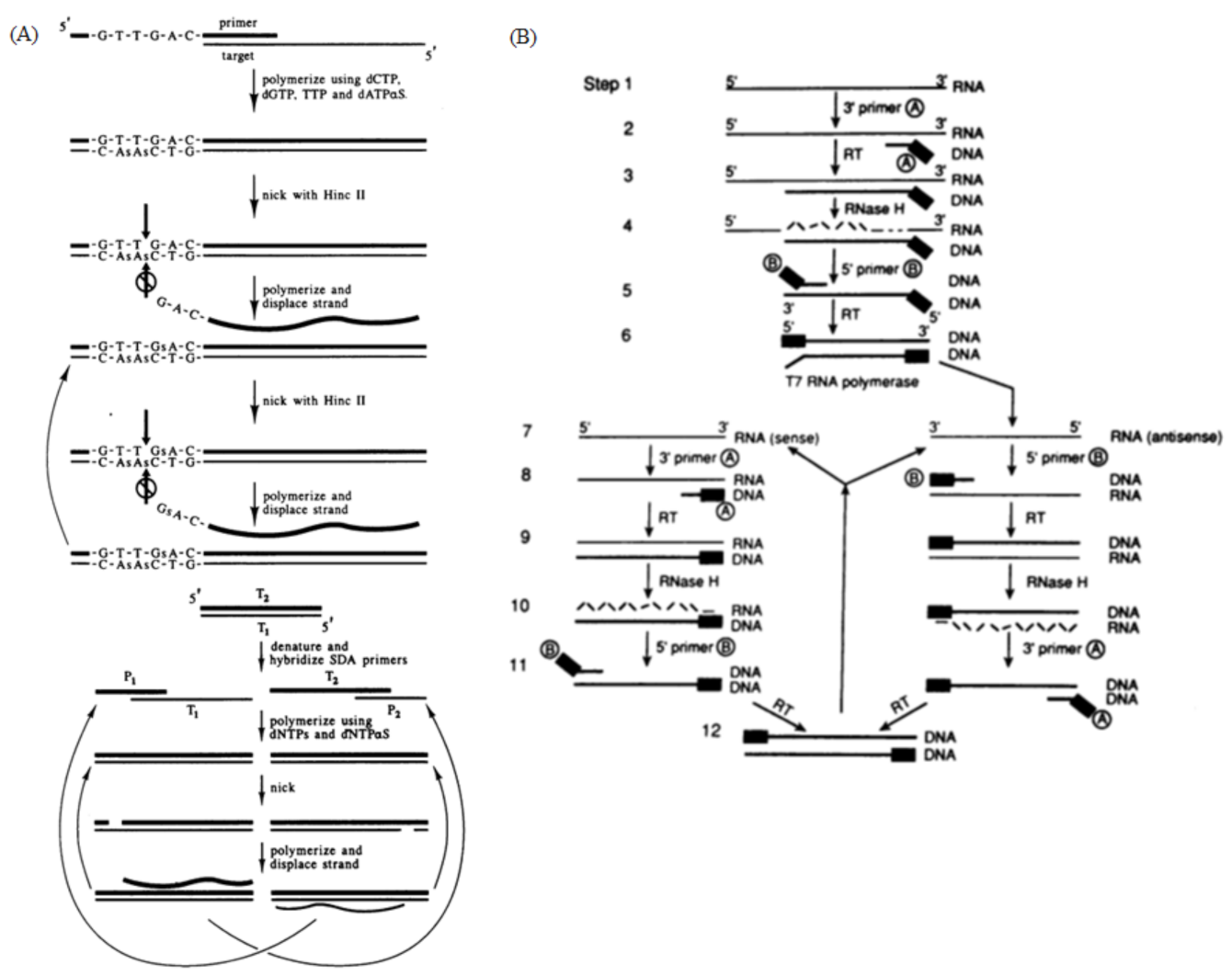
2.5. Strand Displacement Amplification
2.5.1. Enzymes
2.5.2. Reaction Principle and Improved Methods
2.6. Nucleic Acid Sequence-Based Amplification
2.6.1. Enzymes
2.6.2. Reaction Principle and Improved Methods
3. Enzymes-CRISPR
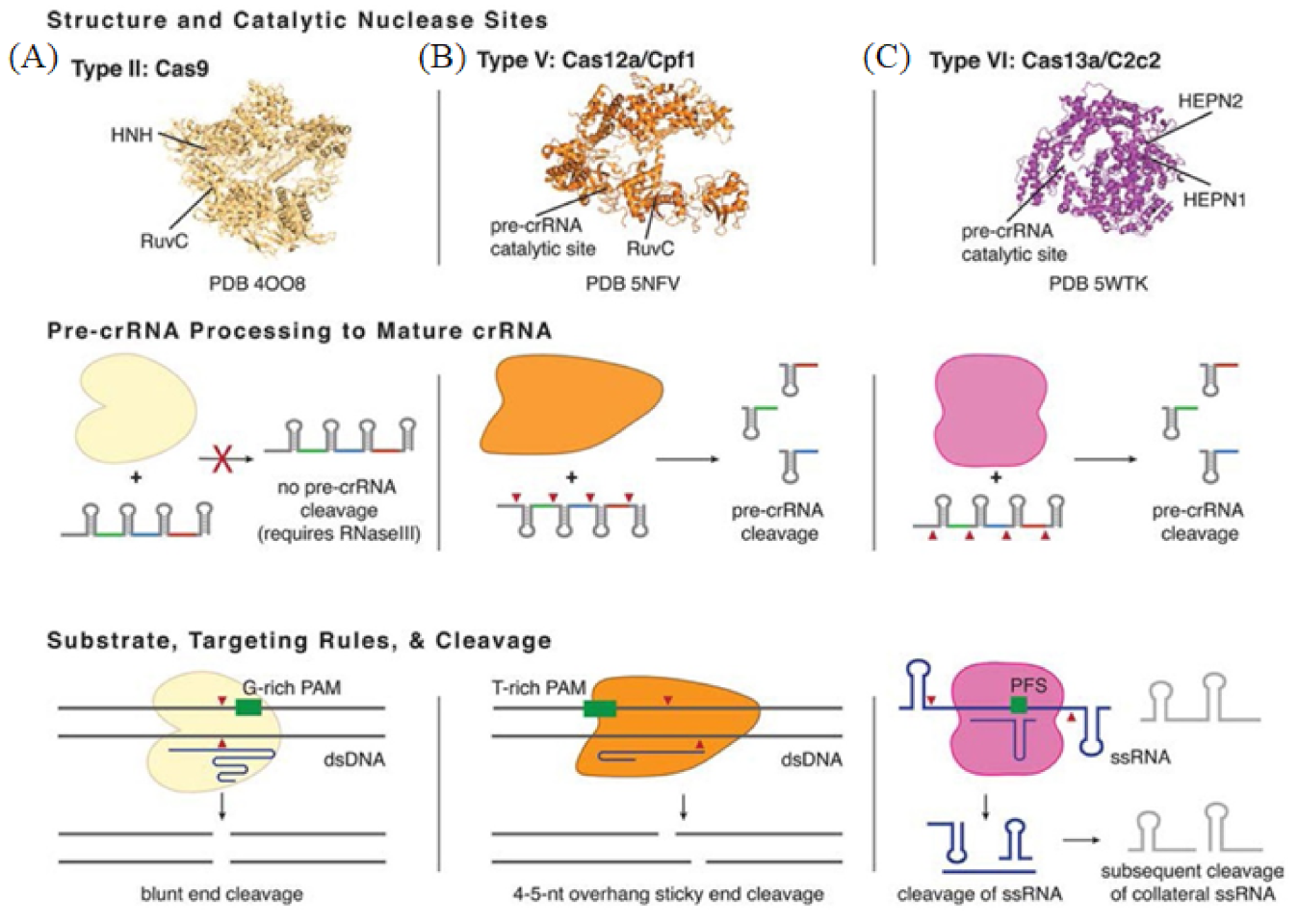
3.1. Cas9
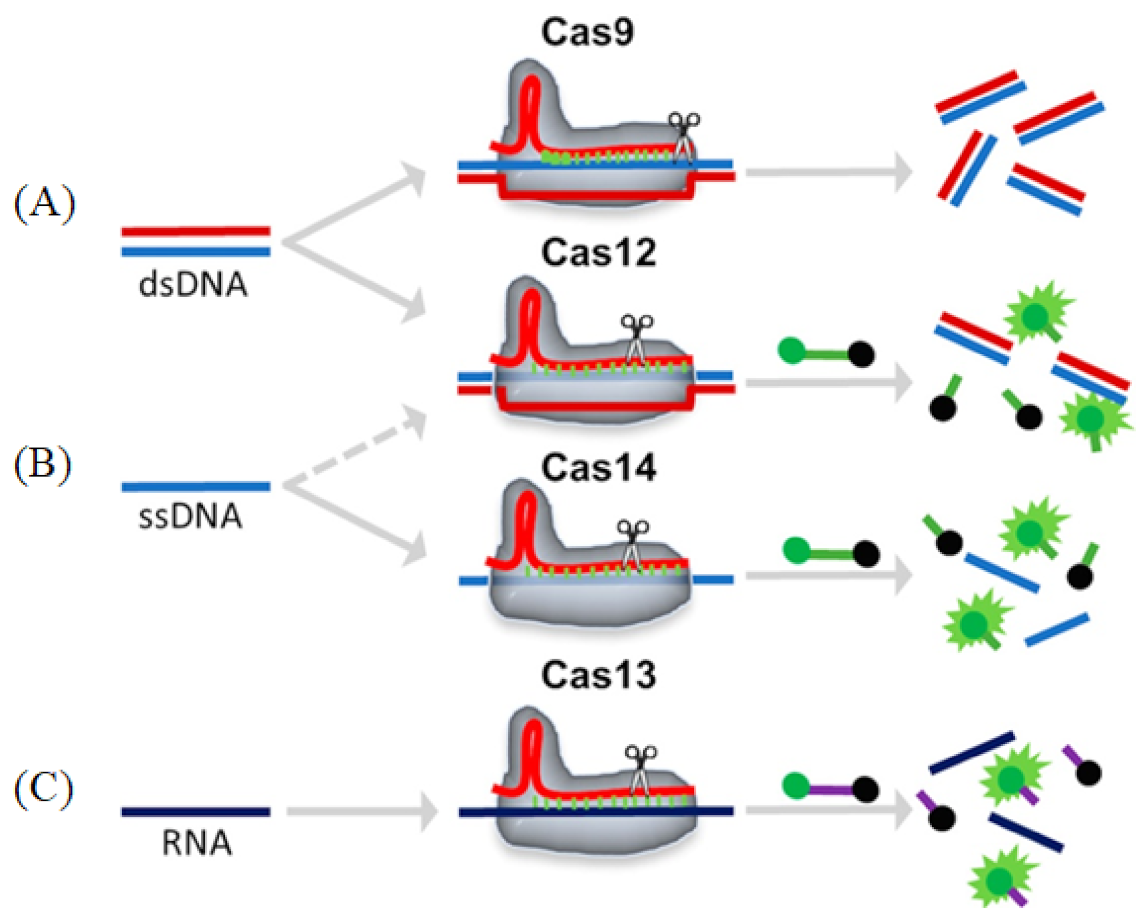
3.2. Cas12
3.3. Cas13
3.4. Cas14
4. Advances in Clinical Application
4.1. Infectious Disease
4.2. Tumor Diagnosis
4.3. Human Genetic Diseases
4.4. Chronic Disease
4.5. Personalized Medicine
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.; Chen, F.; Li, Q.; Wang, L.; Fan, C. Isothermal Amplification of Nucleic Acids. Chem. Rev. 2015, 115, 12491–12545. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.-M.; Kim, B.N.; Kwon, O.S.; Rho, W.-Y.; Jun, B.-H. Emerging ultrafast nucleic acid amplification technologies for next-generation molecular diagnostics. Biosens. Bioelectron. 2019, 141, 111448. [Google Scholar] [CrossRef]
- Craw, P.; Balachandran, W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: A critical review. Lab A Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef] [PubMed]
- Abramson, R.D.; Myers, T.W. Nucleic acid amplification technologies. Curr. Opin. Biotechnol. 1993, 4, 41–47. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Bosh, K.A.; Johnson, A.S.; Hernandez, A.L.; Prejean, J.; Taylor, J.; Wingard, R.; Valleroy, L.A.; Hall, H.I. Vital Signs: Deaths Among Persons with Diagnosed HIV Infection, United States, 2010–2018. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 1717–1724. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction, Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1986; pp. 263–273. [Google Scholar]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Xu, Y.; Laššáková, S.; Korabečná, M.; Neužil, P. PCR past, present and future. Biotechniques 2020, 69, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Pumford, E.A.; Lu, J.; Spaczai, I.; Prasetyo, M.E.; Zheng, E.M.; Zhang, H.; Kamei, D.T. Developments in integrating nucleic acid isothermal amplification and detection systems for point-of-care diagnostics. Biosens. Bioelectron. 2020, 170, 112674. [Google Scholar] [CrossRef]
- Obande, G.A.; Singh, K.K.B. Current and Future Perspectives on Isothermal Nucleic Acid Amplification Technologies for Diagnosing Infections. Infect. Drug Resist. 2020, 13, 455–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eboigbodin, K.E. Application of Loop-Mediated Isothermal Amplification Assay for the Detection of Chlamydia trachomatis and Neisseria gonorrhoeae. Methods Mol. Biol. 2019, 2042, 19–25. [Google Scholar] [CrossRef]
- White, T.J.; Arnheim, N.; Erlich, H.A. The polymerase chain reaction. Trends Genet. 1989, 5, 185–189. [Google Scholar] [CrossRef]
- Holland, P.M.; Abramson, R.D.; Watson, R.; Gelfand, D.H. Detection of specific polymerase chain reaction product by utilizing the 5′—3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 1991, 88, 7276–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.-P.; Othman, S.; Lau, Y.-L.; Radu, S.; Chee, H.-Y. Loop Mediated Isothermal Amplification (LAMP): A Versatile Technique for Detection of Microorganisms. J. Appl. Microbiol. 2018, 124, 626–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. TrAC Trends Anal. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Gao, F.; Jiang, J.-Z.; Wang, J.-Y.; Wei, H.-Y. Real-time quantitative isothermal detection of Ostreid herpesvirus-1 DNA in Scapharca subcrenata using recombinase polymerase amplification. J. Virol. Methods 2018, 255, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lizardi, P.M.; Huang, X.; Zhu, Z.; Bray-Ward, P.; Thomas, D.C.; Ward, D.C. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 1998, 19, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Boss, M.; Arenz, C. A Fast and Easy Method for Specific Detection of Circular RNA by Rolling-Circle Amplification. Chembiochem 2020, 21, 793–796. [Google Scholar] [CrossRef] [Green Version]
- Spargo, C.; Fraiser, M.; Van Cleve, M.; Wright, D.; Nycz, C.; Spears, P.; Walker, G. Detection ofM. tuberculosisDNA using Thermophilic Strand Displacement Amplification. Mol. Cell Probes 1996, 10, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.T.; Fraiser, M.S.; Schram, J.L.; Little, M.C.; Nadeau, J.G.; Malinowski, D.P. Strand displacement amplification—An isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992, 20, 1691–1696. [Google Scholar] [CrossRef]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Romano, J.W.; Williams, K.G.; Shurtliff, R.N.; Ginocchio, C.; Kaplan, M. Nasba Technology: Isothermal RNA Amplification in Qualitative and Quantitative Diagnostics. Immunol. Investig. 1997, 26, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, T.D.; Freeze, H. Thermus aquaticus gen. n. and sp. n., a Nonsporulating Extreme Thermophile. J. Bacteriol. 1969, 98, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.W.; Bender, M.L.; Williams, D.F.; Keigwin, L.D. Strontium-calcium ratios in Cenozoic planktonic foraminifera. Geochim. Cosmochim. Acta 1982, 46, 1281–1292. [Google Scholar] [CrossRef]
- Innis, M.A.; Myambo, K.B.; Gelfand, D.H.; Brow, M.A. DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 9436–9440. [Google Scholar] [CrossRef] [Green Version]
- Schochetman, G.; Ou, C.-Y.; Jones, W.K. Polymerase chain reaction. J. Infect. Dis. 1988, 158, 1154–1157. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, G.; Kapelner, S.; Sommer, S.S. Formamide can dramatically improve the specificity of PCR. Nucleic Acids Res. 1990, 18, 7465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. dPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, J.S.; Gibbs, R.A.; Rainer, J.E.; Nguyen, P.N.; Thomas, C. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988, 16, 11141–11156. [Google Scholar] [CrossRef] [Green Version]
- Ståhlberg, A.; Krzyzanowski, P.; Egyud, M.; Filges, S.; Stein, L.; Godfrey, T.E. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat. Protoc. 2017, 12, 664–682. [Google Scholar] [CrossRef] [PubMed]
- Aliotta, J.M.; Pelletier, J.J.; Ware, J.L.; Moran, L.S.; Benner, J.S.; Kong, H. Thermostable Bst DNA polymerase I lacks a 3′ → 5′ proofreading exonuclease activity. Genet. Anal. Biomol. Eng. 1996, 12, 185–195. [Google Scholar] [CrossRef]
- Riggs, M.G.; Tudor, S.; Sivaram, M.; McDonough, S.H. Construction of single amino acid substitution mutants of cloned Bacillus stearothermophilus DNA polymerase I which lack 5′ → 3′ exonuclease activity. Biochim. Et Biophys. Acta (BBA)-Gene Struct. Expr. 1996, 1307, 178–186. [Google Scholar] [CrossRef]
- McClary, J.; Ye, S.Y.; Hong, G.F.; Witney, F. Sequencing with the large fragment of DNA polymerase I from Bacillus stearothermophilus. DNA Seq. 1991, 1, 173–180. [Google Scholar] [CrossRef]
- Tanner, N.A.; Zhang, Y.; Evans, T.C., Jr. Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. BioTechniques 2012, 53, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Ghaith, D.M.; Abu Ghazaleh, R. Carboxamide and N-alkylcarboxamide additives can greatly reduce non specific amplification in Loop-Mediated Isothermal Amplification for Foot-and-Mouth disease Virus (FMDV) using Bst 3.0 polymerase. J. Virol. Methods 2021, 298, 114284. [Google Scholar] [CrossRef]
- Zhang, X.; Lowe, S.B.; Gooding, J.J. Brief review of monitoring methods for loop-mediated isothermal amplification (LAMP). Biosens. Bioelectron. 2014, 61, 491–499. [Google Scholar] [CrossRef]
- Kashir, J.; Yaqinuddin, A. Loop mediated isothermal amplification (LAMP) assays as a rapid diagnostic for COVID-19. Med. Hypotheses 2020, 141, 109786. [Google Scholar] [CrossRef]
- Teoh, B.-T.; Sam, S.-S.; Tan, K.-K.; Johari, J.; Danlami, M.B.; Hooi, P.-S.; Md-Esa, R.; AbuBakar, S. Detection of dengue viruses using reverse transcription-loop-mediated isothermal amplification. BMC Infect. Dis. 2013, 13, 387. [Google Scholar] [CrossRef] [Green Version]
- Gansen, A.; Herrick, A.M.; Dimov, I.K.; Lee, L.P.; Chiu, D.T. Digital LAMP in a sample self-digitization (SD) chip. Lab A Chip 2012, 12, 2247–2254. [Google Scholar] [CrossRef]
- Schuler, F.; Siber, C.; Hin, S.; Wadle, S.; Paust, N.; Zengerle, R.; von Stetten, F. Digital droplet LAMP as a microfluidic app on standard laboratory devices. Anal. Methods 2016, 8, 2750–2755. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Chao, Y.; Shum, H.C. Droplet and Microchamber-Based Digital Loop-Mediated Isothermal Amplification (dLAMP). Small 2020, 16, e1904469. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.C.; Spoto, G. Integration of isothermal amplification methods in microfluidic devices: Recent advances. Biosens. Bioelectron. 2017, 90, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Juma, K.M.; Akagi, S.; Hayashi, K.; Takita, T.; O’Sullivan, C.K.; Fujiwara, S.; Nakura, Y.; Yanagihara, I.; Yasukawa, K. Solvent engineering studies on recombinase polymerase amplification. J. Biosci. Bioeng. 2021, 131, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Kodadek, T.; Wong, M.L.; Alberts, B.M. The mechanism of homologous DNA strand exchange catalyzed by the bacteriophage T4 uvsX and gene 32 proteins. J. Biol. Chem. 1988, 263, 9427–9436. [Google Scholar] [CrossRef]
- Xu, H.; Beernink, H.T.H.; Morrical, S.W. DNA-binding properties of T4 UvsY recombination mediator protein: Polynucleotide wrapping promotes high-affinity binding to single-stranded DNA. Nucleic Acids Res. 2010, 38, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Chase, J.W.; Williams, K.R. Single-Stranded DNA Binding Proteins Required for DNA Replication. Annu. Rev. Biochem. 1986, 55, 103–136. [Google Scholar] [CrossRef]
- Formosa, T.; Alberts, B.M. Purification and characterization of the T4 bacteriophage uvsX protein. J. Biol. Chem. 1986, 261, 6107–6118. [Google Scholar] [CrossRef] [PubMed]
- Kodadek, T. The role of the bacteriophage T4 gene 32 protein in homologous pairing. J. Biol. Chem. 1990, 265, 20966–20969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tanner, N.A. Isothermal Amplification of Long, Discrete DNA Fragments Facilitated by Single-Stranded Binding Protein. Sci. Rep. 2017, 7, 8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Shahrajabian, M.H. Progress in recombinant polymerase nucleic acid amplification technology. J. Biol. Environ. Sci. 2019, 13, 173–183. [Google Scholar]
- Luo, G.-C.; Yi, T.-T.; Jiang, B.; Guo, X.-L.; Zhang, G.-Y. Betaine-assisted recombinase polymerase assay with enhanced specificity. Anal. Biochem. 2019, 575, 36–39. [Google Scholar] [CrossRef]
- Feng, W.; Peng, H.; Xu, J.; Liu, Y.; Pabbaraju, K.; Tipples, G.; Joyce, M.A.; Saffran, H.A.; Tyrrell, D.L.; Babiuk, S.; et al. Integrating Reverse Transcription Recombinase Polymerase Amplification with CRISPR Technology for the One-Tube Assay of RNA. Anal. Chem. 2021, 93, 12808–12816. [Google Scholar] [CrossRef] [PubMed]
- Xiong, E.; Jiang, L.; Tian, T.; Hu, M.; Yue, H.; Huang, M.; Lin, W.; Jiang, Y.; Zhu, D.; Zhou, X. Simultaneous Dual-Gene Diagnosis of SARS-CoV-2 Based on CRISPR/Cas9-Mediated Lateral Flow Assay. Angew. Chem. Int. Ed. 2021, 60, 5307–5315. [Google Scholar] [CrossRef]
- Yan, L.; Zhou, J.; Zheng, Y.; Gamson, A.S.; Roembke, B.T.; Nakayama, S.; Sintim, H.O. Isothermal amplified detection of DNA and RNA. Mol. BioSystems 2014, 10, 970–1003. [Google Scholar] [CrossRef] [Green Version]
- Blanco, L.; Bernad, A.; Lázaro, J.M.; Martín, G.; Garmendia, C.; Salas, M. Highly Efficient DNA Synthesis by the Phage ϕ 29 DNA Polymerase. J. Biol. Chem. 1989, 264, 8935–8940. [Google Scholar] [CrossRef]
- Li, X.-Y.; Du, Y.-C.; Zhang, Y.-P.; Kong, D.-M. Dual functional Phi29 DNA polymerase-triggered exponential rolling circle amplification for sequence-specific detection of target DNA embedded in long-stranded genomic DNA. Sci. Rep. 2017, 7, 6263. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Daubendiek, S.L.; Zillman, M.A.; Ryan, K.; Kool, E.T. Rolling Circle DNA Synthesis: Small Circular Oligonucleotides as Efficient Templates for DNA Polymerases. J. Am. Chem. Soc. 1996, 118, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Krzywkowski, T.; Kühnemund, M.; Di Wu, D.; Nilsson, M. Limited reverse transcriptase activity of phi29 DNA polymerase. Nucleic Acids Res. 2018, 46, 3625–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, L.L.; Pinheiro, V.B. Xenobiotic Nucleic Acid (XNA) Synthesis by Phi29 DNA Polymerase. Curr. Protoc. Chem. Biol. 2018, 10, e41. [Google Scholar] [CrossRef] [Green Version]
- De Vega, M.; Lazaro, J.M.; Salas, M.; Blanco, L. Primer-terminus stabilization at the 3′-5′ exonuclease active site of phi29 DNA polymerase. Involvement of two amino acid residues highly conserved in proofreading DNA polymerases. EMBO J. 1996, 15, 1182–1192. [Google Scholar] [CrossRef]
- Lagunavicius, A.; Merkiene, E.; Kiveryte, Z.; Savaneviciute, A.; Zimbaite-Ruskuliene, V.; Radzvilavicius, T.; Janulaitis, A. Novel application of Phi29 DNA polymerase: RNA detection and analysis in vitro and in situ by target RNA-primed RCA. RNA 2009, 15, 765–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallur, G.; Luo, C.; Fang, L.; Cooley, S.; Dave, V.; Lambert, J.; Kukanskis, K.; Kingsmore, S.; Lasken, R.; Schweitzer, B. Signal amplification by rolling circle amplification on DNA microarrays. Nucleic Acids Res. 2001, 29, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, Q.; Wu, J.; Chen, Y.; Wang, J. Detection of nucleic acids with a novel stem-loop primer rolling circle amplification technique. Biotechniques 2018, 64, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhang, L.; Tong, J.; Zhao, X.; Ren, J. CRISPR-Cas12a enhanced rolling circle amplification method for ultrasensitive miRNA detection. Microchem. J. 2020, 158, 105239. [Google Scholar] [CrossRef]
- Lu, W.; Wang, Y.; Song, S.; Chen, C.; Yao, B.; Wang, M. A fishhook probe-based rolling circle amplification (FP-RCA) assay for efficient isolation and detection of microRNA without total RNA extraction. Analyst 2018, 143, 5046–5053. [Google Scholar] [CrossRef]
- Walker, G.T.; Little, M.C.; Nadeau, J.G.; Shank, D.D. Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc. Natl. Acad. Sci. 1992, 89, 392–396. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.T.; Linn, C.P.; Nadeau, J.G. DNA detection by strand displacement amplification and fluorescence polarization with signal enhancement using a DNA binding protein. Nucleic Acids Res. 1996, 24, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhu, M.; Liu, H.; Wei, J.; Zhou, X.; Xing, D. Invading stacking primer: A trigger for high-efficiency isothermal amplification reaction with superior selectivity for detecting microRNA variants. Biosens. Bioelectron. 2016, 81, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, B.; Wang, M.; Wang, J.; Yin, H.; Ai, S. Fluorometric determination of microRNA based on strand displacement amplification and rolling circle amplification. Microchim. Acta 2017, 184, 4359–4365. [Google Scholar] [CrossRef]
- DeStefano, J.J.; Buiser, R.G.; Mallaber, L.M.; Myers, T.W.; Bambara, R.A.; Fay, P.J. Polymerization and RNase H activities of the reverse transcriptases from avian myeloblastosis, human immunodeficiency, and Moloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem. 1991, 266, 7423–7431. [Google Scholar] [CrossRef]
- Yasukawa, K.; Nemoto, D.; Inouye, K. Comparison of the Thermal Stabilities of Reverse Transcriptases from Avian Myeloblastosis Virus and Moloney Murine Leukaemia Virus. J. Biochem. 2008, 143, 261–268. [Google Scholar] [CrossRef]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef]
- Oyama, F.; Kikuchi, R.; Omori, A.; Uchida, T. Avian myeloblastosis virus reverse transcriptase is easier to use than the Klenow fragment of DNA polymerase I for labeling the 3′-end of a DNA fragment. Anal. Biochem. 1988, 172, 444–450. [Google Scholar] [CrossRef]
- Champoux, J.J.; Schultz, S.J. Ribonuclease H: Properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 2009, 276, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Rittié, L.; Perbal, B. Enzymes used in molecular biology: A useful guide. J. Cell Commun. Signal. 2008, 2, 25–45. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Xu, J.; Lian, G.; Wang, X.; Gong, X.; Zhou, D.; Chang, J. A novel analytical principle using AP site-mediated T7 RNA polymerase transcription regulation for sensing uracil-DNA glycosylase activity. Analyst 2020, 145, 4321–4327. [Google Scholar] [CrossRef]
- Gholamalipour, Y.; Mudiyanselage, A.K.; Martin, C.T. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-Seq analyses. Nucleic Acids Res. 2018, 46, 9253–9263. [Google Scholar] [CrossRef]
- Borkotoky, S.; Murali, A. The highly efficient T7 RNA polymerase: A wonder macromolecule in biological realm. Int. J. Biol. Macromol. 2018, 118, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Chen, C.; Wu, C.N.; Wu, S.; Ou, B.; Lee, P. Rapid and sensitive detection of Taura syndrome virus using nucleic acid-based amplification. Dis. Aquat. Org. 2006, 73, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, J.H.; Ewert, M.S.; Belliot, G.; Glass, R.I.; Monroe, S.S. Development of a rapid method using nucleic acid sequence-based amplification for the detection of astrovirus. J. Virol. Methods 2003, 110, 119–127. [Google Scholar] [CrossRef]
- Abdolahzadeh, A.; Dolgosheina, E.V.; Unrau, P.J. RNA detection with high specificity and sensitivity using nested fluorogenic Mango NASBA. RNA 2019, 25, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, Z. Bioanalytical applications of isothermal nucleic acid amplification techniques. Anal. Chim. Acta 2015, 853, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Dimov, I.K.; Garcia-Cordero, J.L.; O’Grady, J.; Poulsen, C.R.; Viguier, C.; Kent, L.; Daly, P.; Lincoln, B.; Maher, M.; O’Kennedy, R.; et al. Integrated microfluidic tmRNA purification and real-time NASBA device for molecular diagnostics. Lab Chip 2008, 8, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; Wolters, L.; Schoone, G.; Schallig, H.; Sillekens, P.; Hermsen, R.; Sauerwein, R. Real-Time Nucleic Acid Sequence-Based Amplification Is More Convenient than Real-Time PCR for Quantification of Plasmodium falciparum. J. Clin. Microbiol. 2005, 43, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Genet. 2011, 9, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Pyzocha, N.K.; Chen, S. Diverse Class 2 CRISPR-Cas Effector Proteins for Genome Engineering Applications. ACS Chem. Biol. 2018, 13, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suea-Ngam, A.; Bezinge, L.; Mateescu, B.; Howes, P.D.; Demello, A.J.; Richards, D.A. Enzyme-Assisted Nucleic Acid Detection for Infectious Disease Diagnostics: Moving toward the Point-of-Care. ACS Sens. 2020, 5, 2701–2723. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; Bratovič, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Gao, P.; Rajashankar, K.R.; Patel, D.J. PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell 2016, 167, 1814–1828.e12. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R. Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR–Cas Systems. J. Mol. Biol. 2019, 431, 66–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, T.; Yin, J.; Yu, Y.; Xu, W.; Ding, J.; Patel, D.J.; Yang, H. Structural basis for self-cleavage prevention by tag:anti-tag pairing complementarity in type VI Cas13 CRISPR systems. Mol. Cell 2021, 81, 1100–1115.e5. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Aquino-Jarquin, G. CRISPR-Cas14 is now part of the artillery for gene editing and molecular diagnostic. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 428–431. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, W.; Chen, X.; Zhang, X.; Zhu, J.; Li, S.; Wu, C.; Tian, Z.; Sui, G. Fully Automated CRISPR-LAMP Platform for SARS-CoV-2 Delta and Omicron Variants. Anal. Chem. 2022, 94, 15472–15480. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Liu, J.; Zhu, W.; Zeng, M.; Xu, K.; Ding, J.; Zhou, H.; Zhu, J.; Ke, Y.; Li, L.Y.; et al. One-Pot Visual Detection of African Swine Fever Virus Using CRISPR-Cas12a. Front. Vet. Sci. 2022, 9, 962438. [Google Scholar] [CrossRef]
- Ackerman, C.M.; Myhrvold, C.; Thakku, S.G.; Freije, C.A.; Metsky, H.C.; Yang, D.K.; Ye, S.H.; Boehm, C.K.; Kosoko-Thoroddsen, T.-S.F.; Kehe, J.; et al. Massively multiplexed nucleic acid detection with Cas13. Nature 2020, 582, 277–282. [Google Scholar] [CrossRef]
- Strich, J.R.; Chertow, D.S. CRISPR-Cas Biology and Its Application to Infectious Diseases. J. Clin. Microbiol. 2019, 57, e01307-18. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Awadasseid, A.; Wu, Y.; Tanaka, Y.; Zhang, W. SARS-CoV-2 variants evolved during the early stage of the pandemic and effects of mutations on adaptation in Wuhan populations. Int. J. Biol. Sci. 2021, 17, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Zapor, M. Persistent Detection and Infectious Potential of SARS-CoV-2 Virus in Clinical Specimens from COVID-19 Patients. Viruses 2020, 12, 1384. [Google Scholar] [CrossRef]
- Liotti, F.M.; Menchinelli, G.; Lalle, E.; Palucci, I.; Marchetti, S.; Colavita, F.; La Sorda, M.; Sberna, G.; Bordi, L.; Sanguinetti, M.; et al. Performance of a novel diagnostic assay for rapid SARS-CoV-2 antigen detection in nasopharynx samples. Clin. Microbiol. Infect. 2021, 27, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.L.; Ismail, I.; Mustapa, N.I.; Lai, M.Y.; Soh, T.S.T.; Hassan, A.; Peariasamy, K.M.; Lee, Y.L.; Chong, Y.M.; Sam, I.-C.; et al. Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of SARS-CoV-2. Peer J. 2020, 8, e9278. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wu, X.; Wan, Z.; Li, Y.; Jin, X.; Zhang, C.; Lu, R.; Wu, X.; Wan, Z.; Li, Y.; et al. A Novel Reverse Transcription Loop-Mediated Isothermal Amplification Method for Rapid Detection of SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 2826. [Google Scholar] [CrossRef] [Green Version]
- Shelite, T.R.; Uscanga-Palomeque, A.C.; Castellanos-Gonzalez, A.; Melby, P.C.; Travi, B.L. Isothermal recombinase polymerase amplification-lateral flow detection of SARS-CoV-2, the etiological agent of COVID-19. J. Virol. Methods 2021, 296, 114227. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.; Ladha, A.; Saito, M.; Kim, N.-G.; Woolley, A.E.; Segel, M.; Barretto, R.P.; Ranu, A.; Macrae, R.K.; Faure, G.; et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. 2020, 383, 1492–1494. [Google Scholar] [CrossRef]
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149. [Google Scholar] [CrossRef]
- Magro, L.; Jacquelin, B.; Escadafal, C.; Garneret, P.; Kwasiborski, A.; Manuguerra, J.-C.; Monti, F.; Sakuntabhai, A.; Vanhomwegen, J.; Lafaye, P.; et al. Paper-based RNA detection and multiplexed analysis for Ebola virus diagnostics. Sci. Rep. 2017, 7, 1347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Wang, S.; Yang, K.; Liu, X.; Liu, W.; Guo, R.; Liang, W.; Yuan, F.; Liu, Z.; Gao, T.; et al. Rapid and simultaneous detection of Japanese encephalitis virus by real-time nucleic acid sequence-based amplification. Microb. Pathog. 2021, 150, 104724. [Google Scholar] [CrossRef]
- Zhao, J.; Fang, S.; Liu, Y.; Zeng, L.; He, Z. A lateral flow biosensor based on gold nanoparticles detects four hemorrhagic fever viruses. Anal. Methods 2020, 12, 5613–5620. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-T.; Liu, Y.-J.; Wang, J.; Xu, Z.-G.; Yang, Y.; Shen, F.; Liu, X.-H.; Zhou, X.; Liu, S.-M. Next Generation Digital PCR Measurement of Hepatitis B Virus Copy Number in Formalin-Fixed Paraffin-Embedded Hepatocellular Carcinoma Tissue. Clin. Chem. 2015, 61, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Yan, L.; Tang, H.; Liao, Y. A sensitive and accurate quantification method for the detection of hepatitis B virus covalently closed circular DNA by the application of a droplet digital polymerase chain reaction amplification system. Biotechnol. Lett. 2015, 37, 2063–2073. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, Z.; Li, F.; Xie, X.; Ding, A. Rapid and sensitive detection of hepatitis B virus by lateral flow recombinase polymerase amplification assay. J. Virol. Methods 2021, 291, 114094. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Yan, W.; Liu, L.; Wang, S.; Zhang, X.; Lyu, M. Research Progress on Rolling Circle Amplification (RCA)-Based Biomedical Sensing. Pharmaceuticals 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yari, F.; Abiri, R.; Aryan, E.; Jouybari, T.A.; Navabi, J.; Alvandi, A. Loop-Mediated Isothermal Amplification as a Fast Noninvasive Method of Helicobacter pylori Diagnosis. J. Clin. Lab. Anal. 2016, 30, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Liu, S.; Li, X.; Yang, M. Electrochemical Detection of Circulating Tumor Cells Based on DNA Generated Electrochemical Current and Rolling Circle Amplification. Anal. Chem. 2019, 91, 11614–11619. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, Q.; Yang, M. Multivalent Duplexed-Aptamer Networks Regulated a CRISPR-Cas12a System for Circulating Tumor Cell Detection. Anal. Chem. 2021, 93, 12921–12929. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Paul, S.; Vázquez, L.A.B.; Uribe, S.P.; Reyes-Pérez, P.R.; Sharma, A. Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders. Cells 2020, 9, 1698. [Google Scholar] [CrossRef] [PubMed]
- Amini, S.; Abak, A.; Sakhinia, E.; Abhari, A. MicroRNA-221 and MicroRNA-222 in Common Human Cancers: Expression, Function, and Triggering of Tumor Progression as a Key Modulator. Lab. Med. 2019, 50, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Pak, T.R. miRNA degradation in the mammalian brain. Am. J. Physiol. Physiol. 2020, 319, C624–C629. [Google Scholar] [CrossRef]
- Kotyla, P.J.; Islam, A. MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS). Int. J. Mol. Sci. 2020, 21, 2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Li, P.; He, W.; Liu, C.; Li, Z. Rolling circle extension-actuated loop-mediated isothermal amplification (RCA-LAMP) for ultrasensitive detection of microRNAs. Biosens. Bioelectron. 2019, 128, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fang, X.; Bai, M.; Zhang, J.; Yu, H.; Chen, F.; Zhao, Y. A microfluidic surface-enhanced Raman scattering (SERS) sensor for microRNA in extracellular vesicles with nucleic acid-tyramine cascade amplification. Chin. Chem. Lett. 2022, 33, 2101–2104. [Google Scholar] [CrossRef]
- Mader, A.; Riehle, U.; Brandstetter, T.; Stickeler, E.; Ruehe, J. Universal nucleic acid sequence-based amplification for simultaneous amplification of messengerRNAs and microRNAs. Anal. Chim. Acta 2012, 754, 1–7. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, X.; Chen, X.; Qiu, X.; Qing, G.; Zhang, H.; Zhang, L.; Hu, X.; He, Z.; Zhong, D.; et al. Rolling Circular Amplification (RCA)-Assisted CRISPR/Cas9 Cleavage (RACE) for Highly Specific Detection of Multiple Extracellular Vesicle MicroRNAs. Anal. Chem. 2019, 92, 2176–2185. [Google Scholar] [CrossRef]
- Cui, Y.; Fan, S.; Yuan, Z.; Song, M.; Hu, J.; Qian, D.; Zhen, D.; Li, J.; Zhu, B. Ultrasensitive electrochemical assay for microRNA-21 based on CRISPR/Cas13a-assisted catalytic hairpin assembly. Talanta 2021, 224, 121878. [Google Scholar] [CrossRef]
- Chen, J.; Yan, J.; Feng, Q.; Miao, X.; Dou, B.; Wang, P. Label-free and enzyme-free fluorescence detection of microRNA based on sulfydryl-functionalized carbon dots via target-initiated hemin/G-quadruplex-catalyzed oxidation. Biosens. Bioelectron. 2021, 176, 112955. [Google Scholar] [CrossRef]
- Wang, G.; Guo, Y.; Liu, Y.; Zhou, W.; Wang, G. Algorithm-Assisted Detection and Imaging of microRNAs in Living Cancer Cells via the Disassembly of Plasmonic Core-Satellite Probes Coupled with Strand Displacement Amplification. ACS Sens. 2021, 6, 958–966. [Google Scholar] [CrossRef]
- Chang, J.; Lv, W.; Wu, J.; Li, H.; Li, F. Simultaneous photoelectrochemical detection of dual microRNAs by capturing CdS quantum dots and methylene blue based on target-initiated strand displaced amplification. Chin. Chem. Lett. 2021, 32, 775–778. [Google Scholar] [CrossRef]
- Wang, R.; Lan, L.; Liu, L.; Cheng, L. Asymmetric polymerase chain reaction and loop-mediated isothermal amplification (AP-LAMP) for ultrasensitive detection of microRNAs. Chin. Chem. Lett. 2020, 31, 159–162. [Google Scholar] [CrossRef]
- Guerriero, C.; Matera, C.; Del Bufalo, D.; De Amici, M.; Conti, L.; Dallanoce, C.; Tata, A. The Combined Treatment with Chemotherapeutic Agents and the Dualsteric Muscarinic Agonist Iper-8-Naphthalimide Affects Drug Resistance in Glioblastoma Stem Cells. Cells 2021, 10, 1877. [Google Scholar] [CrossRef] [PubMed]
- MacLean, E.; Kohli, M.; Weber, S.F.; Suresh, A.; Schumacher, S.G.; Denkinger, C.M.; Pai, M. Advances in Molecular Diagnosis of Tuberculosis. J. Clin. Microbiol. 2020, 58, e01582-19. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.; Ford, N.; Padgen, P.; Cox, H. Time to treatment for rifampicin-resistant tuberculosis: Systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2017, 21, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Theron, G.; Zijenah, L.; Chanda, D.; Clowes, P.; Rachow, A.; Lesosky, M.; Bara, W.; Mungofa, S.; Pai, M.; Hoelscher, M.; et al. Feasibility, accuracy, and clinical effect of point-of-care Xpert MTB/RIF testing for tuberculosis in primary-care settings in Africa: A multicentre, randomised, controlled trial. Lancet 2014, 383, 424–435. [Google Scholar] [CrossRef]
- Gliddon, H.D.; Frampton, D.; Munsamy, V.; Heaney, J.; Pataillot-Meakin, T.; Nastouli, E.; Pym, A.S.; Steyn, A.J.C.; Pillay, D.; McKendry, R.A. A Rapid Drug Resistance Genotyping Workflow for Mycobacterium tuberculosis, Using Targeted Isothermal Amplification and Nanopore Sequencing. Microbiol. Spectr. 2021, 9, e00610-21. [Google Scholar] [CrossRef]
- Galbiati, S.; Brisci, A.; Lalatta, F.; Seia, M.; Makrigiorgos, G.M.; Ferrari, M.; Cremonesi, L. Full COLD-PCR Protocol for Noninvasive Prenatal Diagnosis of Genetic Diseases. Clin. Chem. 2011, 57, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, P.; Grifa, A.; Origone, P.; Coviello, D.; Antonacci, R.; Rocchi, M. Detection of a neurofibromatosis type I (NF1) homologous sequence by PCR: Implications for the diagnosis and screening of genetic diseases. Mol. Cell Probes 1993, 7, 415–418. [Google Scholar] [CrossRef]
- Tan, C.; Chen, X.; Wang, F.; Wang, D.; Cao, Z.; Zhu, X.; Lu, C.; Yang, W.; Gao, N.; Gao, H.; et al. A multiplex droplet digital PCR assay for non-invasive prenatal testing of fetal aneuploidies. Analyst 2019, 144, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Almasi, M.A.; Almasi, G. Loop mediated isothermal amplification (LAMP) for embryo sex determination in pregnant women at eight weeks of pregnancy. J. Reprod. Infertil. 2017, 18, 197. [Google Scholar] [PubMed]
- Shastry, B.S. SNPs: Impact on Gene Function and Phenotype. Methods Mol. Biol. 2009, 578, 3–22. [Google Scholar] [CrossRef]
- Ramírez-Bello, J.; Jiménez-Morales, M. Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases. Gac. Med. Mex. 2017, 153, 238–250. [Google Scholar] [PubMed]
- Liu, X.; Zhang, C.; Zhao, M.; Liu, K.; Li, H.; Li, N.; Gao, L.; Yang, X.; Ma, T.; Zhu, J.; et al. A direct isothermal amplification system adapted for rapid SNP genotyping of multifarious sample types. Biosens. Bioelectron. 2018, 115, 70–76. [Google Scholar] [CrossRef]
- Dhar, B.C.; Steimberg, N.; Mazzoleni, G. Point-of-Care Pathogen Detection with CRISPR-based Programmable Nucleic Acid Binding Proteins. Chemmedchem 2020, 16, 1566–1575. [Google Scholar] [CrossRef]
- Chen, Y.; Mei, Y.; Jiang, X. Universal and high-fidelity DNA single nucleotide polymorphism detection based on a CRISPR/Cas12a biochip. Chem. Sci. 2021, 12, 4455–4462. [Google Scholar] [CrossRef]
- Ding, S.; Chen, R.; Chen, G.; Li, M.; Wang, J.; Zou, J.; Du, F.; Dong, J.; Cui, X.; Huang, X.; et al. One-step colorimetric genotyping of single nucleotide polymorphism using probe-enhanced loop-mediated isothermal amplification (PE-LAMP). Theranostics 2019, 9, 3723–3731. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Li, Y.; Wang, X.; Li, R.; Zeng, X.; Yang, M.; Xu, X.; Ye, T.; Bao, L.; Huang, Y. TaqMan-MGB probe quantitative PCR assays to genotype and quantify three mtDNA mutations of Leber hereditary optic neuropathy. Sci. Rep. 2020, 10, 12264. [Google Scholar] [CrossRef]
- Kohli, S.; Saxena, R.; Thomas, E.; Singh, K.; Mahay, S.B.; Puri, R.D.; Verma, I.C. Mutation Spectrum of Dystrophinopathies in India: Implications for Therapy. Indian J. Pediatr. 2020, 87, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Corrigendum: Diagnosis of the accurate genotype of HKaa carriers in patients with thalassemia using multiplex ligation-dependent probe amplification combined with nested polymerase chain reaction. Chin. Med. J. 2020, 133, 1631. [CrossRef]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, C.I.; Limone, B.; Sobieraj, D.M.; Lee, S.; Roberts, M.S.; Kaur, R.; Alam, T. Dosing Frequency and Medication Adherence in Chronic Disease. J. Manag. Care Pharm. 2012, 18, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Hori, M.; Harada-Shiba, M.; Kataoka, Y.; Onozuka, D.; Nishimura, K.; Nishikawa, R.; Tsuda, K.; Ogura, M.; Son, C.; et al. Patients With LDLR and PCSK9 Gene Variants Experienced Higher Incidence of Cardiovascular Outcomes in Heterozygous Familial Hypercholesterolemia. J. Am. Heart Assoc. 2021, 10, e018263. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Wang, D.; Dai, Y.; Wang, X.; Yu, P.; Qu, S.; Liu, Z.; Cao, Y.; Zhang, L.; Ping, Y.; Liu, W.; et al. Determination of plasma β-amyloids by rolling circle amplification chemiluminescent immunoassay for noninvasive diagnosis of Alzheimer’s disease. Microchim. Acta 2021, 188, 24. [Google Scholar] [CrossRef]
- Coombes, B.; Mahony, B.K.C.A.J.B. Nucleic Acid Sequence Based Amplification (NASBA) of Chlamydia pneumoniae Major Outer Membrane Protein (ompA) mRNA with Bioluminescent Detection. Comb. Chem. High Throughput Screen. 2000, 3, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Secq, V.; Villeret, J.; Fina, F.; Carmassi, M.; Carcopino, X.; Garcia, S.; Metellus, I.; Boubli, L.; Iovanna, J.; Charpin, C. Triple negative breast carcinoma EGFR amplification is not associated with EGFR, Kras or ALK mutations. Br. J. Cancer 2014, 110, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.; Mao, X.; Wang, Z.; Sun, K.; Ma, R.; Wu, J.; Cao, H. Next-generation sequencing-based detection of EGFR, KRAS, BRAF, NRAS, PIK3CA, Her-2 and TP53 mutations in patients with non-small cell lung cancer. Mol. Med. Rep. 2018, 18, 2191–2197. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; You, M.; Peng, P.; Tong, H.; He, W.; Li, A.; Mao, P.; Xu, T.; Xu, F.; Yao, C. Taqman-MGB nanoPCR for Highly Specific Detection of Single-Base Mutations. Int. J. Nanomed. 2021, 16, 3695–3705. [Google Scholar] [CrossRef]
- Jiang, W.; Xiang, L.; Pei, X.; He, T.; Shen, X.; Wu, X.; Yang, H. Mutational analysis of KRAS and its clinical implications in cervical cancer patients. J. Gynecol. Oncol. 2018, 29, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex Picodroplet Digital PCR to Detect KRAS Mutations in Circulating DNA from the Plasma of Colorectal Cancer Patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, L.; Guo, T.; Wu, X.; Cai, H. Rapid diagnosis of cisplatin-sensitive and resistant cervical squamous cell carcinomas by reverse transcription loop-mediated isothermal amplification. Int. J. Clin. Exp. Pathol. 2018, 11, 882. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleic Acid Amplification | Enzymes | Reaction Temperature (°C) | Reaction Time (min) | Target | Primers | Amplification Capacity | Refs. |
|---|---|---|---|---|---|---|---|
| PCR | Taq polymerase | 95,55,72 | 45–120 | DNA | 2 | 107–1010 | [7,14] |
| LAMP | Bst DNA Polymerase | 60–65 | 60 | DNA | 4 | 109 | [15,16] |
| RPA | Bsu DNA polymerase recombinase and ssDNA binding protein | 37–42 | 20 | DNA/RNA | 2 | 10 | [17,18] |
| RCA | Phi29 DNA Polymerase | 30–65 | 60–180 | DNA/RNA | 2 | 109 | [19,20] |
| SDA | Restriction endonuclease and DNA polymerase | 37–40 | 120 | DNA | 4 | 107 | [21,22] |
| NASBA | Reverse transcriptase and RNA polymerase (RNaseH) | 41 | 60–120 | RNA | 2 | 106 | [23,24] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Liu, H.; Ren, J.; Huang, Y.; Deng, Y.; Liu, Y.; Chen, Z.; Chow, F.W.-N.; Leung, P.H.-M.; Li, S. Enzyme-Assisted Nucleic Acid Amplification in Molecular Diagnosis: A Review. Biosensors 2023, 13, 160. https://doi.org/10.3390/bios13020160
Wang M, Liu H, Ren J, Huang Y, Deng Y, Liu Y, Chen Z, Chow FW-N, Leung PH-M, Li S. Enzyme-Assisted Nucleic Acid Amplification in Molecular Diagnosis: A Review. Biosensors. 2023; 13(2):160. https://doi.org/10.3390/bios13020160
Chicago/Turabian StyleWang, Meiling, Hongna Liu, Jie Ren, Yunqi Huang, Yan Deng, Yuan Liu, Zhu Chen, Franklin Wang-Ngai Chow, Polly Hang-Mei Leung, and Song Li. 2023. "Enzyme-Assisted Nucleic Acid Amplification in Molecular Diagnosis: A Review" Biosensors 13, no. 2: 160. https://doi.org/10.3390/bios13020160