

Multi-Mechanistic Approaches to the Treatment of Traumatic Brain Injury: A Review

Abstract
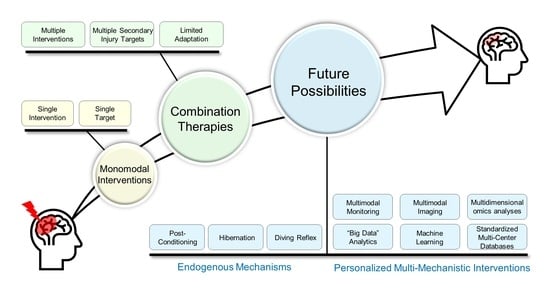
:
1. Introduction
2. Materials and Methods
2.1. Eligibility Criteria
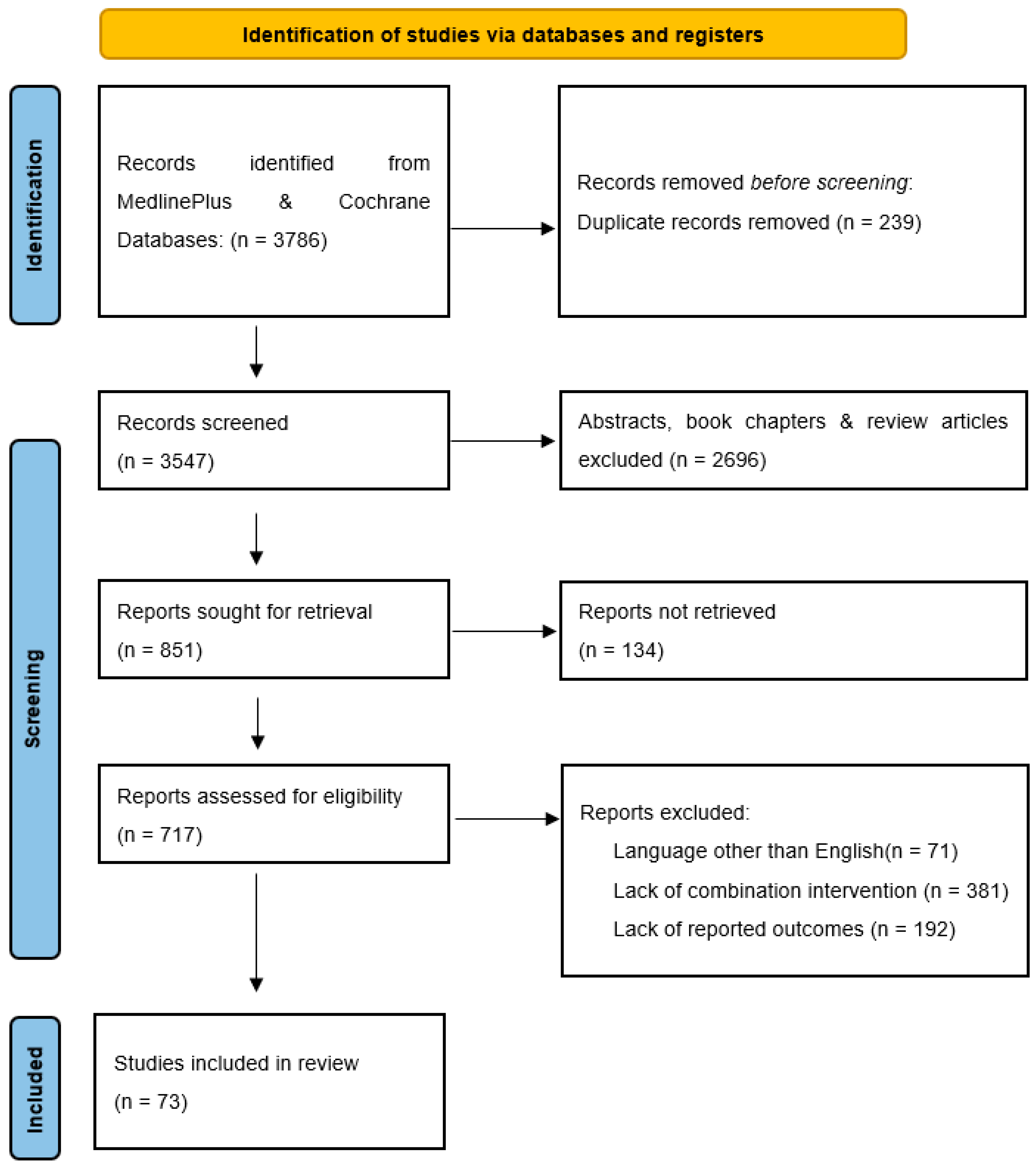
2.2. Literature Search
2.3. Study Selection
3. Results
3.1. Multi-Mechanistic Pharmacologic Interventions
3.1.1. Antioxidant Treatments
3.1.2. Anti-Excitatory Treatments
3.1.3. Anti-Inflammatory Treatments
3.1.4. Combined Multitarget Pharmacologic Therapies
3.2. Multimodal Nonpharmacologic Interventions
3.2.1. Clinical Trials
3.2.2. Preclinical Trials
3.3. Multimodal Combined Pharmacologic and Nonpharmacologic Interventions

{kind=link}
{kind=link}
| Model | Species | Sample Size | %Fem | Intervention | Outcome | Reference |
|---|---|---|---|---|---|---|
| Clinical Trials | ||||||
| Randomized Placebo Controlled Trial | Human Severe TBI | 107 | 15.9% | “Progesterone + Hypothermia” vs. progesterone or hypothermia alone | Worse long-term outcomes in combined group vs. individual therapies in acute TBI | Sinha, 2017 [100] |
| Preclinical Trials | ||||||
| rmCCI | Rat | 40 | 0% | “Amantadine + tDCS” vs. amantadine or tDCS alone | Improved neurobehavioral outcomes, decreased astrocyte activation | Han, 2022 [103] |
| CCI | Rat | 90 | 0% | “MSC + LITUS” vs. MSC or LITUS alone | Improved lesion volume and neurobehavioral outcomes, mediated through induction of BDNF and reduction of TNF-α and AQP4 | Yao, 2022 [102] |
| CCI | Rat | 68 | 0% | “Citalopram + EE” vs. citalopram or EE alone | Improved learning and cognitive flexibility | Minchew, 2021 [104] |
| FPI | Rat | 96 | 0% | “BMSC + Hypothermia” vs. BMSC or hypothermia alone | Decreased neuronal apoptosis and neurobehavioral defects | Song, 2020 [99] |
| CCI | Rat | 60 | 0% | “Amantadine + EE” vs. amantadine or EE alone | Improved lesion volume and neurobehavioral outcomes, no additional benefit versus monotherapy | Bleimeister, 2019 [94] |
| CCI | Rat | 72 | 0% | “Galantamine + EE” vs. galantamine or EE alone | Improved lesion volume and neurobehavioral outcomes, no additional benefit versus monotherapy | de la Tremblaye, 2017 [95] |
| CCI | Rat | 48 | 0% | “Methylphenidate + EE” vs. methylphenidate or EE alone | Improved neurobehavioral outcomes, no additional benefit versus monotherapy | Leary, 2017 [105] |
| CCI | Rat | 48 | 0% | “Citicoline + exercise” vs. citicoline or physical exercise alone | Improved lesion volume and neurobehavioral outcomes, no additional benefit versus monotherapy | Jacotte-Simancas, 2015 [101] |
| CCI | Rat | 78 | 0% | “Buspirone + EE” vs. EE or buspirone alone | Improved functional outcomes | Monaco, 2014 [96] |
| CCI | Rat | 60 | 0% | “Buspirone + EE” vs. EE or buspirone alone | Improved functional outcomes, no additional benefit versus monotherapy | Kline, 2012 [97] |
| CCI | Rat | 65 | 0% | “8-OH-DPAT + EE” vs. EE or 8-OH-DPAT alone | Decreased neuronal loss, no additional benefit versus monotherapy | Kline, 2010 [106] |
| CCI | Rat | 50 | 0% | “FGF-2 + Hypothermia” vs. Hypothermia or FGF-2 alone | Improved lesion volume and neurobehavioral outcomes, no additional benefit versus monotherapy | Yan, 2000 [98] |
3.4. Multimodal Neuromonitoring
Multimodal Neuromonitoring-Guided Treatment
4. Discussion
4.1. Current State of Multimodal TBI Treatment
4.2. Future Directions in Multimodal TBI Treatment
4.3. Imaging- and Neuromonitoring-Guided Treatment
4.4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [Green Version]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell. Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef]
- Stocchetti, N.; Carbonara, M.; Citerio, G.; Ercole, A.; Skrifvars, M.B.; Smielewski, P.; Zoerle, T.; Menon, D.K. Severe traumatic brain injury: Targeted management in the intensive care unit. Lancet Neurol. 2017, 16, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margulies, S.; Hicks, R. Combination Therapies for Traumatic Brain Injury: Prospective Considerations. J. Neurotrauma 2009, 26, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Margulies, S.; Anderson, G.; Atif, F.; Badaut, J.; Clark, R.; Empey, P.; Guseva, M.; Hoane, M.; Huh, J.; Pauly, J.; et al. Combination Therapies for Traumatic Brain Injury: Retrospective Considerations. J. Neurotrauma 2016, 33, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somayaji, M.R.; Przekwas, A.J.; Gupta, R.K. Combination Therapy for Multi-Target Manipulation of Secondary Brain Injury Mechanisms. Curr. Neuropharmacol. 2018, 16, 484–504. [Google Scholar] [CrossRef] [PubMed]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. [Google Scholar] [CrossRef]
- Cormio, M.; Robertson, C.S.; Narayan, R.K. Secondary insults to the injured brain. J. Clin. Neurosci. 1997, 4, 132–148. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Doppenberg, E.M.; Choi, S.C.; Bullock, R. Clinical trials in traumatic brain injury: Lessons for the future. J. Neurosurg. Anesthesiol. 2004, 16, 87–94. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Jackson, T.C.; Jha, R.M.; Clark, R.S.B.; Okonkwo, D.O.; Bayir, H.; Poloyac, S.M.; Wagner, A.K.; Empey, P.E.; Conley, Y.P.; et al. Paths to Successful Translation of New Therapies for Severe Traumatic Brain Injury in the Golden Age of Traumatic Brain Injury Research: A Pittsburgh Vision. J. Neurotrauma 2020, 37, 2353–2371. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef]
- Trimmel, H.; Herzer, G.; Derdak, C.; Kettenbach, J.; Grgac, I. A novel pharmacological treatment concept for neuroprotection in severe traumatic brain injury-Two case reports. Clin. Case Rep. 2022, 10, e6626. [Google Scholar] [CrossRef]
- Colton, K.; Yang, S.; Hu, P.F.; Chen, H.H.; Bonds, B.; Scalea, T.M.; Stein, D.M. Intracranial pressure response after pharmacologic treatment of intracranial hypertension. J. Trauma Acute Care Surg. 2014, 77, 47–53; discussion 53. [Google Scholar] [CrossRef]
- Clark, R.S.B.; Empey, P.E.; Bayir, H.; Rosario, B.L.; Poloyac, S.M.; Kochanek, P.M.; Nolin, T.D.; Au, A.K.; Horvat, C.M.; Wisniewski, S.R.; et al. Phase I randomized clinical trial of N-acetylcysteine in combination with an adjuvant probenecid for treatment of severe traumatic brain injury in children. PLoS ONE 2017, 12, e0180280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tian, H.; Niu, Y.; Yu, C.; Xie, L.; Jin, Z.; Niu, W.; Ren, J.; Fu, L.; Yao, Z. Combined cell grafting and VPA administration facilitates neural repair through axonal regeneration and synaptogenesis in traumatic brain injury. Acta Biochim. Biophys. Sin. 2022, 54, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.K.; Bathula, S.; Hsu, M.; Morris-Blanco, K.C.; Chokkalla, A.K.; Jeong, S.; Choi, J.; Subramanian, S.; Park, J.S.; Fabry, Z.; et al. An antioxidant and anti-ER stress combo therapy decreases inflammation, secondary brain damage and promotes neurological recovery following traumatic brain injury in mice. J. Neurosci. 2022, 42, 6810–6821. [Google Scholar] [CrossRef] [PubMed]
- Narouiepour, A.; Ebrahimzadeh-Bideskan, A.; Rajabzadeh, G.; Gorji, A.; Negah, S.S. Neural stem cell therapy in conjunction with curcumin loaded in niosomal nanoparticles enhanced recovery from traumatic brain injury. Sci. Rep. 2022, 12, 3572. [Google Scholar] [CrossRef]
- Whitney, K.; Nikulina, E.; Rahman, S.N.; Alexis, A.; Bergold, P.J. Delayed dosing of minocycline plus N-acetylcysteine reduces neurodegeneration in distal brain regions and restores spatial memory after experimental traumatic brain injury. Exp. Neurol. 2021, 345, 113816. [Google Scholar] [CrossRef] [PubMed]
- Chandran, R.; Mehta, S.L.; Vemuganti, R. Antioxidant Combo Therapy Protects White Matter After Traumatic Brain Injury. Neuromol. Med. 2021, 23, 344–347. [Google Scholar] [CrossRef]
- Alqahtani, F.; Assiri, M.A.; Mohany, M.; Imran, I.; Javaid, S.; Rasool, M.F.; Shakeel, W.; Sivandzade, F.; Alanazi, A.Z.; Al-Rejaie, S.S.; et al. Coadministration of Ketamine and Perampanel Improves Behavioral Function and Reduces Inflammation in Acute Traumatic Brain Injury Mouse Model. Biomed. Res. Int. 2020, 2020, 3193725. [Google Scholar] [CrossRef] [PubMed]
- Bayhan, I.; Turtay, M.G.; Ciftci, O.; Cetin, A.; Basak, N.; Namik Oztanir, M.; Oguzturk, H.; Gurbuz, S.; Guven, T. Comparison of immunological, histological and oxidative effects of felbamate and levetiracetam in traumatic brain injury. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7083–7091. [Google Scholar] [CrossRef]
- Rana, A.; Singh, S.; Deshmukh, R.; Kumar, A. Pharmacological potential of tocopherol and doxycycline against traumatic brain injury-induced cognitive/motor impairment in rats. Brain Inj. 2020, 34, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, L.; Huang, X.; Wu, K.; Ding, S.; Wang, W.; Wang, B.; Smith, C.; Ren, C.; Ni, H.; et al. Calpain inhibitor MDL28170 improves the transplantation-mediated therapeutic effect of bone marrow-derived mesenchymal stem cells following traumatic brain injury. Stem Cell Res. Ther. 2019, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Black, A.M.B.; Milbourn, H.R.; Krakonja, S.; Nesbit, M.; Bartlett, C.A.; Fehily, B.; Takechi, R.; Yates, N.J.; Fitzgerald, M. The Effects of a Combination of Ion Channel Inhibitors in Female Rats Following Repeated Mild Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3408. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Shunmugavel, A.; Dhammu, T.S.; Khan, H.; Singh, I.; Singh, A.K. Combined treatment with GSNO and CAPE accelerates functional recovery via additive antioxidant activities in a mouse model of TBI. J. Neurosci. Res. 2018, 96, 1900–1913. [Google Scholar] [CrossRef]
- Sangobowale, M.; Nikulina, E.; Bergold, P.J. Minocycline plus N-acetylcysteine protect oligodendrocytes when first dosed 12 hours after closed head injury in mice. Neurosci. Lett. 2018, 682, 16–20. [Google Scholar] [CrossRef]
- Ameliorate, J.L.; Ghabriel, M.N.; Vink, R. Magnesium enhances the beneficial effects of NK1 antagonist administration on blood-brain barrier permeability and motor outcome after traumatic brain injury. Magnes. Res. 2017, 30, 88–97. [Google Scholar] [CrossRef]
- Ghazale, H.; Ramadan, N.; Mantash, S.; Zibara, K.; El-Sitt, S.; Darwish, H.; Chamaa, F.; Boustany, R.M.; Mondello, S.; Abou-Kheir, W.; et al. Docosahexaenoic acid (DHA) enhances the therapeutic potential of neonatal neural stem cell transplantation post-Traumatic brain injury. Behav. Brain Res. 2018, 340, 1–13. [Google Scholar] [CrossRef]
- Chandran, R.; Kim, T.; Mehta, S.L.; Udho, E.; Chanana, V.; Cengiz, P.; Kim, H.; Kim, C.; Vemuganti, R. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1818–1827. [Google Scholar] [CrossRef]
- Haber, M.; James, J.; Kim, J.; Sangobowale, M.; Irizarry, R.; Ho, J.; Nikulina, E.; Grin’kina, N.M.; Ramadani, A.; Hartman, I.; et al. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1312–1326. [Google Scholar] [CrossRef]
- Day, N.L.; Carle, M.S.; Floyd, C.L. Post-injury administration of a combination of memantine and 17beta-estradiol is protective in a rat model of traumatic brain injury. Neurochem. Int. 2017, 111, 57–68. [Google Scholar] [CrossRef]
- Busingye, D.S.; Turner, R.J.; Vink, R. Combined Magnesium/Polyethylene Glycol Facilitates the Neuroprotective Effects of Magnesium in Traumatic Brain Injury at a Reduced Magnesium Dose. CNS Neurosci. Ther. 2016, 22, 854–859. [Google Scholar] [CrossRef] [Green Version]
- Kota, D.J.; Prabhakara, K.S.; van Brummen, A.J.; Bedi, S.; Xue, H.; DiCarlo, B.; Cox, C.S., Jr.; Olson, S.D. Propranolol and Mesenchymal Stromal Cells Combine to Treat Traumatic Brain Injury. Stem Cells Transl. Med. 2016, 5, 33–44. [Google Scholar] [CrossRef]
- Baky, N.A.; Fadda, L.; Al-Rasheed, N.M.; Al-Rasheed, N.M.; Mohamed, A.; Yacoub, H. Neuroprotective effect of carnosine and cyclosporine-A against inflammation, apoptosis, and oxidative brain damage after closed head injury in immature rats. Toxicol. Mech. Methods 2016, 26, 1–10. [Google Scholar] [CrossRef]
- Lamprecht, M.R.; Morrison, B., 3rd. A Combination Therapy of 17beta-Estradiol and Memantine Is More Neuroprotective Than Monotherapies in an Organotypic Brain Slice Culture Model of Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1361–1368. [Google Scholar] [CrossRef]
- Paradells, S.; Zipancic, I.; Martinez-Losa, M.M.; Garcia Esparza, M.A.; Bosch-Morell, F.; Alvarez-Dolado, M.; Soria, J.M. Lipoic acid and bone marrow derived cells therapy induce angiogenesis and cell proliferation after focal brain injury. Brain Inj. 2015, 29, 380–395. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Hua, F.; Wang, J.; Yousuf, S.; Atif, F.; Sayeed, I.; Stein, D.G. Progesterone and vitamin D combination therapy modulates inflammatory response after traumatic brain injury. Brain Inj. 2015, 29, 1165–1174. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Sanberg, P.R.; Sanchez-Ramos, J.; Song, S.; Kaneko, Y.; Borlongan, C.V. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS ONE 2014, 9, e90953. [Google Scholar] [CrossRef]
- Ekici, M.A.; Uysal, O.; Cikriklar, H.I.; Ozbek, Z.; Turgut Cosan, D.; Baydemir, C.; Kazanci, B.; Hafizoglu, D. Effect of etanercept and lithium chloride on preventing secondary tissue damage in rats with experimental diffuse severe brain injury. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 10–27. [Google Scholar]
- Haber, M.; Abdel Baki, S.G.; Grin’kina, N.M.; Irizarry, R.; Ershova, A.; Orsi, S.; Grill, R.J.; Dash, P.; Bergold, P.J. Minocycline plus N-acetylcysteine synergize to modulate inflammation and prevent cognitive and memory deficits in a rat model of mild traumatic brain injury. Exp. Neurol. 2013, 249, 169–177. [Google Scholar] [CrossRef]
- Yu, F.; Wang, Z.; Tanaka, M.; Chiu, C.T.; Leeds, P.; Zhang, Y.; Chuang, D.M. Posttrauma cotreatment with lithium and valproate: Reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. J. Neurosurg. 2013, 119, 766–773. [Google Scholar] [CrossRef]
- Uysal, N.; Baykara, B.; Kiray, M.; Cetin, F.; Aksu, I.; Dayi, A.; Gurpinar, T.; Ozdemir, D.; Arda, M.N. Combined treatment with progesterone and magnesium sulfate positively affects traumatic brain injury in immature rats. Turk. Neurosurg. 2013, 23, 129–137. [Google Scholar] [CrossRef]
- Campolo, M.; Ahmad, A.; Crupi, R.; Impellizzeri, D.; Morabito, R.; Esposito, E.; Cuzzocrea, S. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J. Endocrinol. 2013, 217, 291–301. [Google Scholar] [CrossRef]
- Thal, S.C.; Schaible, E.V.; Neuhaus, W.; Scheffer, D.; Brandstetter, M.; Engelhard, K.; Wunder, C.; Forster, C.Y. Inhibition of proteasomal glucocorticoid receptor degradation restores dexamethasone-mediated stabilization of the blood-brain barrier after traumatic brain injury. Crit. Care Med. 2013, 41, 1305–1315. [Google Scholar] [CrossRef]
- Tang, H.; Hua, F.; Wang, J.; Sayeed, I.; Wang, X.; Chen, Z.; Yousuf, S.; Atif, F.; Stein, D.G. Progesterone and vitamin D: Improvement after traumatic brain injury in middle-aged rats. Horm. Behav. 2013, 64, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Ismailoglu, O.; Atilla, P.; Palaoglu, S.; Cakar, N.; Yasar, U.; Kilinc, K.; Kaptanoglu, E. The therapeutic effects of melatonin and nimodipine in rats after cerebral cortical injury. Turk. Neurosurg. 2012, 22, 740–746. [Google Scholar] [CrossRef]
- Hua, F.; Reiss, J.I.; Tang, H.; Wang, J.; Fowler, X.; Sayeed, I.; Stein, D.G. Progesterone and low-dose vitamin D hormone treatment enhances sparing of memory following traumatic brain injury. Horm. Behav. 2012, 61, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Thau-Zuchman, O.; Shohami, E.; Alexandrovich, A.G.; Leker, R.R. Combination of vascular endothelial and fibroblast growth factor 2 for induction of neurogenesis and angiogenesis after traumatic brain injury. J. Mol. Neurosci. 2012, 47, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Khaksari, M.; Soltani, Z.; Shahrokhi, N.; Moshtaghi, G.; Asadikaram, G. The role of estrogen and progesterone, administered alone and in combination, in modulating cytokine concentration following traumatic brain injury. Can. J. Physiol. Pharmacol. 2011, 89, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Kelso, M.L.; Scheff, N.N.; Scheff, S.W.; Pauly, J.R. Melatonin and minocycline for combinatorial therapy to improve functional and histopathological deficits following traumatic brain injury. Neurosci. Lett. 2011, 488, 60–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imer, M.; Omay, B.; Uzunkol, A.; Erdem, T.; Sabanci, P.A.; Karasu, A.; Albayrak, S.B.; Sencer, A.; Hepgul, K.; Kaya, M. Effect of magnesium, MK-801 and combination of magnesium and MK-801 on blood-brain barrier permeability and brain edema after experimental traumatic diffuse brain injury. Neurol. Res. 2009, 31, 977–981. [Google Scholar] [CrossRef]
- Cherian, L.; Robertson, C.S. L-arginine and free radical scavengers increase cerebral blood flow and brain tissue nitric oxide concentrations after controlled cortical impact injury in rats. J. Neurotrauma 2003, 20, 77–85. [Google Scholar] [CrossRef]
- Jenkins, L.W.; Lu, Y.C.; Johnston, W.E.; Lyeth, B.G.; Prough, D.S. Combined therapy affects outcomes differentially after mild traumatic brain injury and secondary forebrain ischemia in rats. Brain Res. 1999, 817, 132–144. [Google Scholar] [CrossRef]
- Lyeth, B.G.; Liu, S.; Hamm, R.J. Combined scopolamine and morphine treatment of traumatic brain injury in the rat. Brain Res. 1993, 617, 69–75. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, M.; Ko, D.G.; Choi, B.Y.; Suh, S.W. The Role of NADPH Oxidase in Neuronal Death and Neurogenesis after Acute Neurological Disorders. Antioxidants 2021, 10, 739. [Google Scholar] [CrossRef]
- Taran, S.; Pelosi, P.; Robba, C. Optimizing oxygen delivery to the injured brain. Curr. Opin. Crit. Care 2022, 28, 145–156. [Google Scholar] [CrossRef]
- Diringer, M.N.; Zazulia, A.R.; Powers, W.J. Does Ischemia Contribute to Energy Failure in Severe TBI? Transl. Stroke Res. 2011, 2, 517–523. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Taffe, K.M.; Abrahamson, E.E.; Dixon, C.E.; Kochanek, P.M.; Ikonomovic, M.D. Time course analysis of hippocampal nerve growth factor and antioxidant enzyme activity following lateral controlled cortical impact brain injury in the rat. J. Neurotrauma 2004, 21, 491–500. [Google Scholar] [CrossRef]
- Mader, M.M.; Czorlich, P. The role of L-arginine metabolism in neurocritical care patients. Neural Regen. Res. 2022, 17, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Sun, F.; Huang, R.; Sun, W.; Zhang, D.; Wang, Q. Inhibition of NADPH oxidase by apocynin prevents learning and memory deficits in a mouse Parkinson’s disease model. Redox Biol. 2019, 22, 101134. [Google Scholar] [CrossRef]
- Saykally, J.N.; Rachmany, L.; Hatic, H.; Shaer, A.; Rubovitch, V.; Pick, C.G.; Citron, B.A. The nuclear factor erythroid 2-like 2 activator, tert-butylhydroquinone, improves cognitive performance in mice after mild traumatic brain injury. Neuroscience 2012, 223, 305–314. [Google Scholar] [CrossRef]
- Sokka, A.L.; Putkonen, N.; Mudo, G.; Pryazhnikov, E.; Reijonen, S.; Khiroug, L.; Belluardo, N.; Lindholm, D.; Korhonen, L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J. Neurosci. 2007, 27, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, S.; Chi, Y.; Gao, K.; Kono, K.; Yao, J. eIF2alpha-Independent Inhibition of TNF-alpha-Triggered NF-kappaB Activation by Salubrinal. Biol. Pharm. Bull. 2015, 38, 1368–1374. [Google Scholar] [CrossRef] [Green Version]
- Hoffe, B.; Holahan, M.R. Hyperacute Excitotoxic Mechanisms and Synaptic Dysfunction Involved in Traumatic Brain Injury. Front. Mol. Neurosci. 2022, 15, 831825. [Google Scholar] [CrossRef] [PubMed]
- Jamjoom, A.A.B.; Rhodes, J.; Andrews, P.J.D.; Grant, S.G.N. The synapse in traumatic brain injury. Brain 2021, 144, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, L.S.; Sun, D.A.; Sombati, S.; Baranova, A.; Wilson, M.S.; Attkisson, E.; Hamm, R.J.; DeLorenzo, R.J. Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci. Lett. 2008, 441, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Neuberger, E.J.; Gupta, A.; Subramanian, D.; Korgaonkar, A.A.; Santhakumar, V. Converging early responses to brain injury pave the road to epileptogenesis. J. Neurosci. Res. 2019, 97, 1335–1344. [Google Scholar] [CrossRef]
- Sueiras, M.; Thonon, V.; Santamarina, E.; Sanchez-Guerrero, A.; Poca, M.A.; Quintana, M.; Riveiro, M.; Sahuquillo, J. Cortical Spreading Depression Phenomena Are Frequent in Ischemic and Traumatic Penumbra: A Prospective Study in Patients with Traumatic Brain Injury and Large Hemispheric Ischemic Stroke. J. Clin. Neurophysiol. 2021, 38, 47–55. [Google Scholar] [CrossRef]
- Lauritzen, M.; Dreier, J.P.; Fabricius, M.; Hartings, J.A.; Graf, R.; Strong, A.J. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab. 2011, 31, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Hanaya, R.; Arita, K. The New Antiepileptic Drugs: Their Neuropharmacology and Clinical Indications. Neurol. Med. Chir. 2016, 56, 205–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef] [Green Version]
- Kalra, S.; Malik, R.; Singh, G.; Bhatia, S.; Al-Harrasi, A.; Mohan, S.; Albratty, M.; Albarrati, A.; Tambuwala, M.M. Pathogenesis and management of traumatic brain injury (TBI): Role of neuroinflammation and anti-inflammatory drugs. Inflammopharmacology 2022, 30, 1153–1166. [Google Scholar] [CrossRef]
- Smith, C.; Gentleman, S.M.; Leclercq, P.D.; Murray, L.S.; Griffin, W.S.; Graham, D.I.; Nicoll, J.A. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol. Appl. Neurobiol. 2013, 39, 654–666. [Google Scholar] [CrossRef]
- Webster, G.; Del Rosso, J.Q. Anti-inflammatory activity of tetracyclines. Dermatol. Clin. 2007, 25, 133–135. [Google Scholar] [CrossRef]
- Li, J.; Jia, B.; Cheng, Y.; Song, Y.; Li, Q.; Luo, C. Targeting Molecular Mediators of Ferroptosis and Oxidative Stress for Neurological Disorders. Oxid. Med. Cell Longev. 2022, 2022, 3999083. [Google Scholar] [CrossRef]
- Mollica, A.; Greben, R.; Oriuwa, C.; Siddiqi, S.H.; Burke, M.J. Neuromodulation Treatments for Mild Traumatic Brain Injury and Post-concussive Symptoms. Curr. Neurol. Neurosci. Rep. 2022, 22, 171–181. [Google Scholar] [CrossRef]
- Surendrakumar, S.; Rabelo, T.K.; Campos, A.C.P.; Mollica, A.; Abrahao, A.; Lipsman, N.; Burke, M.J.; Hamani, C. Neuromodulation Therapies in Pre-Clinical Models of Traumatic Brain Injury: Systematic Review and Translational Applications. J. Neurotrauma 2022, 40, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Zhou, Y.; Greenwald, B.D. Update on Non-Pharmacological Interventions for Treatment of Post-Traumatic Headache. Brain Sci. 2022, 12, 1357. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, X.; Li, H.; Guo, R.; Wang, J.; Zhang, Y.; Lu, Z. Rehabilitation effect of rTMS combined with cognitive training on cognitive impairment after traumatic brain injury. Am. J. Transl. Res. 2021, 13, 11711–11717. [Google Scholar]
- Martino Cinnera, A.; Bonni, S.; Iosa, M.; Ponzo, V.; Fusco, A.; Caltagirone, C.; Koch, G. Clinical effects of non-invasive cerebellar magnetic stimulation treatment combined with neuromotor rehabilitation in traumatic brain injury. A single case study. Funct. Neurol. 2016, 31, 117–120. [Google Scholar]
- Shin, S.S.; Krishnan, V.; Stokes, W.; Robertson, C.; Celnik, P.; Chen, Y.; Song, X.; Lu, H.; Liu, P.; Pelled, G. Transcranial magnetic stimulation and environmental enrichment enhances cortical excitability and functional outcomes after traumatic brain injury. Brain Stimul. 2018, 11, 1306–1313. [Google Scholar] [CrossRef]
- Maegele, M.; Lippert-Gruener, M.; Ester-Bode, T.; Sauerland, S.; Schafer, U.; Molcanyi, M.; Lefering, R.; Bouillon, B.; Neiss, W.F.; Angelov, D.N.; et al. Reversal of neuromotor and cognitive dysfunction in an enriched environment combined with multimodal early onset stimulation after traumatic brain injury in rats. J. Neurotrauma 2005, 22, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegele, M.; Lippert-Gruener, M.; Ester-Bode, T.; Garbe, J.; Bouillon, B.; Neugebauer, E.; Klug, N.; Lefering, R.; Neiss, W.F.; Angelov, D.N. Multimodal early onset stimulation combined with enriched environment is associated with reduced CNS lesion volume and enhanced reversal of neuromotor dysfunction after traumatic brain injury in rats. Eur. J. Neurosci. 2005, 21, 2406–2418. [Google Scholar] [CrossRef]
- Cohen, S.L.; Bikson, M.; Badran, B.W.; George, M.S. A visual and narrative timeline of US FDA milestones for Transcranial Magnetic Stimulation (TMS) devices. Brain Stimul. 2022, 15, 73–75. [Google Scholar] [CrossRef]
- Pink, A.E.; Williams, C.; Alderman, N.; Stoffels, M. The use of repetitive transcranial magnetic stimulation (rTMS) following traumatic brain injury (TBI): A scoping review. Neuropsychol. Rehabil. 2021, 31, 479–505. [Google Scholar] [CrossRef]
- Luo, J.; Feng, Y.; Li, M.; Yin, M.; Qin, F.; Hu, X. Repetitive Transcranial Magnetic Stimulation Improves Neurological Function and Promotes the Anti-inflammatory Polarization of Microglia in Ischemic Rats. Front. Cell. Neurosci. 2022, 16, 878345. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Xu, C.; Hu, S.; Wen, G.; Lin, J.; Liu, T.; Xu, J. Repetitive Transcranial Magnetic Stimulation Improves Neuropathy and Oxidative Stress Levels in Rats with Experimental Cerebral Infarction through the Nrf2 Signaling Pathway. Evid. Based Complement. Altern. Med. 2021, 2021, 3908677. [Google Scholar] [CrossRef]
- Frasca, D.; Tomaszczyk, J.; McFadyen, B.J.; Green, R.E. Traumatic brain injury and post-acute decline: What role does environmental enrichment play? A scoping review. Front. Hum. Neurosci. 2013, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleimeister, I.H.; Wolff, M.; Lam, T.R.; Brooks, D.M.; Patel, R.; Cheng, J.P.; Bondi, C.O.; Kline, A.E. Environmental enrichment and amantadine confer individual but nonadditive enhancements in motor and spatial learning after controlled cortical impact injury. Brain Res. 2019, 1714, 227–233. [Google Scholar] [CrossRef]
- de la Tremblaye, P.B.; Bondi, C.O.; Lajud, N.; Cheng, J.P.; Radabaugh, H.L.; Kline, A.E. Galantamine and Environmental Enrichment Enhance Cognitive Recovery after Experimental Traumatic Brain Injury but Do Not Confer Additional Benefits When Combined. J. Neurotrauma 2017, 34, 1610–1622. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.M.; Gebhardt, K.M.; Chlebowski, S.M.; Shaw, K.E.; Cheng, J.P.; Henchir, J.J.; Zupa, M.F.; Kline, A.E. A Combined Therapeutic Regimen of Buspirone and Environmental Enrichment Is More Efficacious than Either Alone in Enhancing Spatial Learning in Brain-Injured Pediatric Rats. J. Neurotrauma 2014, 31, 1934–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, A.E.; Olsen, A.S.; Sozda, C.N.; Hoffman, A.N.; Cheng, J.P. Evaluation of a combined treatment paradigm consisting of environmental enrichment and the 5-HT1A receptor agonist buspirone after experimental traumatic brain injury. J. Neurotrauma 2012, 29, 1960–1969. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.Q.; Yu, J.; Kline, A.E.; Letart, P.; Jenkins, L.W.; Marion, D.W.; Dixon, C.E. Evaluation of combined fibroblast growth factor-2 and moderate hypothermia therapy in traumatically brain injured rats. Brain Res. 2000, 887, 134–143. [Google Scholar] [CrossRef]
- Song, B.; Wang, X.X.; Yang, H.Y.; Kong, L.T.; Sun, H.Y. Temperature-sensitive bone mesenchymal stem cells combined with mild hypothermia reduces neurological deficit in rats of severe traumatic brain injury. Brain Inj. 2020, 34, 975–982. [Google Scholar] [CrossRef]
- Sinha, S.; Raheja, A.; Samson, N.; Goyal, K.; Bhoi, S.; Selvi, A.; Sharma, P.; Sharma, B.S. A randomized placebo-controlled trial of progesterone with or without hypothermia in patients with acute severe traumatic brain injury. Neurol. India 2017, 65, 1304–1311. [Google Scholar] [CrossRef]
- Jacotte-Simancas, A.; Costa-Miserachs, D.; Coll-Andreu, M.; Torras-Garcia, M.; Borlongan, C.V.; Portell-Cortes, I. Effects of voluntary physical exercise, citicoline, and combined treatment on object recognition memory, neurogenesis, and neuroprotection after traumatic brain injury in rats. J. Neurotrauma 2015, 32, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Wang, W.; Li, Y.; Cao, Z.; Wang, Y.; Yuan, Y.; Li, X.; Liang, X.; Yu, Y.; Liu, L. Study of the mechanism by which MSCs combined with LITUS treatment improve cognitive dysfunction caused by traumatic brain injury. Neurosci. Lett. 2022, 787, 136825. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Park, G.; Suh, J.H. Transcranial direct current stimulation combined with amantadine in repetitive mild traumatic brain injury in rats. BMC Neurosci. 2022, 23, 76. [Google Scholar] [CrossRef] [PubMed]
- Minchew, H.M.; Radabaugh, H.L.; LaPorte, M.L.; Free, K.E.; Cheng, J.P.; Bondi, C.O. A combined therapeutic regimen of citalopram and environmental enrichment ameliorates attentional set-shifting performance after brain trauma. Eur. J. Pharmacol. 2021, 904, 174174. [Google Scholar] [CrossRef]
- Leary, J.B.; Bondi, C.O.; LaPorte, M.J.; Carlson, L.J.; Radabaugh, H.L.; Cheng, J.P.; Kline, A.E. The Therapeutic Efficacy of Environmental Enrichment and Methylphenidate Alone and in Combination after Controlled Cortical Impact Injury. J. Neurotrauma 2017, 34, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Kline, A.E.; McAloon, R.L.; Henderson, K.A.; Bansal, U.K.; Ganti, B.M.; Ahmed, R.H.; Gibbs, R.B.; Sozda, C.N. Evaluation of a combined therapeutic regimen of 8-OH-DPAT and environmental enrichment after experimental traumatic brain injury. J. Neurotrauma 2010, 27, 2021–2032. [Google Scholar] [CrossRef] [Green Version]
- Lindblad, C.; Raj, R.; Zeiler, F.A.; Thelin, E.P. Current state of high-fidelity multimodal monitoring in traumatic brain injury. Acta Neurochir. 2022, 164, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Appavu, B.L.; Temkit, M.; Kensicki, J.F.; Kuwabara, M.; Burrows, B.T.; Adelson, P.D. Acute physiologic prediction of pediatric post-traumatic epilepsy. Epilepsy Res. 2022, 183, 106935. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.S.; Kumar, N.K.; Zhao, C.; Zhang, D.Y.; Tucker, A.M.; Storm, P.B.; Heuer, G.G.; Gajjar, A.A.; Kim, C.T.; Yuan, I.; et al. Invasive brain tissue oxygen and intracranial pressure (ICP) monitoring versus ICP-only monitoring in pediatric severe traumatic brain injury. J. Neurosurg. Pediatr. 2022, 30, 239–249. [Google Scholar] [CrossRef]
- Marini, C.P.; Stoller, C.; McNelis, J.; Del Deo, V.; Prabhakaran, K.; Petrone, P. Correlation of brain flow variables and metabolic crisis: A prospective study in patients with severe traumatic brain injury. Eur. J. Trauma Emerg. Surg. 2022, 48, 537–544. [Google Scholar] [CrossRef]
- Khellaf, A.; Garcia, N.M.; Tajsic, T.; Alam, A.; Stovell, M.G.; Killen, M.J.; Howe, D.J.; Guilfoyle, M.R.; Jalloh, I.; Timofeev, I.; et al. Focally administered succinate improves cerebral metabolism in traumatic brain injury patients with mitochondrial dysfunction. J. Cereb. Blood Flow Metab. 2022, 42, 39–55. [Google Scholar] [CrossRef]
- Robinson, M.B.; Shin, P.; Alunday, R.; Cole, C.; Torbey, M.T.; Carlson, A.P. Decision-making for decompressive craniectomy in traumatic brain injury aided by multimodality monitoring: Illustrative case. J. Neurosurg. Case Lessons 2021, 1, CASE2197. [Google Scholar] [CrossRef] [PubMed]
- Appavu, B.; Burrows, B.T.; Nickoles, T.; Boerwinkle, V.; Willyerd, A.; Gunnala, V.; Mangum, T.; Marku, I.; Adelson, P.D. Implementation of Multimodality Neurologic Monitoring Reporting in Pediatric Traumatic Brain Injury Management. Neurocrit. Care 2021, 35, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Petkus, V.; Preiksaitis, A.; Chaleckas, E.; Chomskis, R.; Zubaviciute, E.; Vosylius, S.; Rocka, S.; Rastenyte, D.; Aries, M.J.; Ragauskas, A.; et al. Optimal Cerebral Perfusion Pressure: Targeted Treatment for Severe Traumatic Brain Injury. J. Neurotrauma 2020, 37, 389–396. [Google Scholar] [CrossRef]
- Dellazizzo, L.; Demers, S.P.; Charbonney, E.; Williams, V.; Serri, K.; Albert, M.; Giguere, J.F.; Laroche, M.; Williamson, D.; Bernard, F. Minimal PaO2 threshold after traumatic brain injury and clinical utility of a novel brain oxygenation ratio. J. Neurosurg. 2018, 131, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, D.O.; Shutter, L.A.; Moore, C.; Temkin, N.R.; Puccio, A.M.; Madden, C.J.; Andaluz, N.; Chesnut, R.M.; Bullock, M.R.; Grant, G.A.; et al. Brain Oxygen Optimization in Severe Traumatic Brain Injury Phase-II: A Phase II Randomized Trial. Crit. Care Med. 2017, 45, 1907–1914. [Google Scholar] [CrossRef]
- Luca, L.; Rogobete, A.F.; Bedreag, O.H.; Sarandan, M.; Cradigati, C.A.; Papurica, M.; Gruneantu, A.; Patrut, R.; Vernic, C.; Dumbuleu, C.M.; et al. Intracranial Pressure Monitoring as a Part of Multimodal Monitoring Management of Patients with Critical Polytrauma: Correlation between Optimised Intensive Therapy According to Intracranial Pressure Parameters and Clinical Picture. Turk. J. Anaesthesiol. Reanim. 2015, 43, 412–417. [Google Scholar] [CrossRef]
- Dunham, C.M.; Ransom, K.J.; McAuley, C.E.; Gruber, B.S.; Mangalat, D.; Flowers, L.L. Severe brain injury ICU outcomes are associated with Cranial-Arterial Pressure Index and noninvasive Bispectral Index and transcranial oxygen saturation: A prospective, preliminary study. Crit. Care 2006, 10, R159. [Google Scholar] [CrossRef] [Green Version]
- Isa, R.; Wan Adnan, W.A.; Ghazali, G.; Idris, Z.; Ghani, A.R.; Sayuthi, S.; Awang, M.S.; Ghazali, M.M.; Naing, N.N.; Abdullah, J.M. Outcome of severe traumatic brain injury: Comparison of three monitoring approaches. Neurosurg. Focus 2003, 15, E1. [Google Scholar] [CrossRef]
- The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care. Indications for intracranial pressure monitoring. J. Neurotrauma 2000, 17, 479–491. [Google Scholar] [CrossRef]
- Casault, C.; Couillard, P.; Kromm, J.; Rosenthal, E.; Kramer, A.; Brindley, P. Multimodal brain monitoring following traumatic brain injury: A primer for intensive care practitioners. J. Intensive Care Soc. 2022, 23, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Young, A.M.; Donnelly, J.; Czosnyka, M.; Jalloh, I.; Liu, X.; Aries, M.J.; Fernandes, H.M.; Garnett, M.R.; Smielewski, P.; Hutchinson, P.J.; et al. Continuous Multimodality Monitoring in Children after Traumatic Brain Injury-Preliminary Experience. PLoS ONE 2016, 11, e0148817. [Google Scholar] [CrossRef]
- Sykora, M.; Czosnyka, M.; Liu, X.; Donnelly, J.; Nasr, N.; Diedler, J.; Okoroafor, F.; Hutchinson, P.; Menon, D.; Smielewski, P. Autonomic Impairment in Severe Traumatic Brain Injury: A Multimodal Neuromonitoring Study. Crit. Care Med. 2016, 44, 1173–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturini, S.; Bhatti, F.; Timofeev, I.; Carpenter, K.L.H.; Hutchinson, P.J.; Guilfoyle, M.R.; Helmy, A. Microdialysis-Based Classifications of Abnormal Metabolic States after Traumatic Brain Injury: A Systematic Review of the Literature. J. Neurotrauma 2022, 40, 195–209. [Google Scholar] [CrossRef]
- Khellaf, A.; Khan, D.Z.; Helmy, A. Recent advances in traumatic brain injury. J. Neurol. 2019, 266, 2878–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef]
- Reddi, S.; Thakker-Varia, S.; Alder, J.; Giarratana, A.O. Status of precision medicine approaches to traumatic brain injury. Neural Regen. Res. 2022, 17, 2166–2171. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, N.; Ma, X.; Wang, P.; Dong, W.; Chen, Z.; Wu, M.; Wang, Z.; Wang, L.; Guan, D.; et al. Spatial-temporal changes of iron deposition and iron metabolism after traumatic brain injury in mice. Front. Mol. Neurosci. 2022, 15, 949573. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, Q.; Sun, Y.R.; Wu, X.; Du, Z.Y.; Li, Z.Q.; Wu, X.H.; Zhou, L.F.; Wu, G.; Hu, J. Effects of Deferoxamine Mesylate on Hematoma and Perihematoma Edema after Traumatic Intracerebral Hemorrhage. J. Neurotrauma 2017, 34, 2753–2759. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z.; Xi, G.; Keep, R.F.; Hua, Y. Deferoxamine attenuates acute hydrocephalus after traumatic brain injury in rats. Transl. Stroke Res. 2014, 5, 586–594. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, R.; Li, M.; Li, F.; Meng, H.; Zhu, G.; Lin, J.; Feng, H. Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurol. Sci. 2013, 34, 639–645. [Google Scholar] [CrossRef]
- Unterberg, A.W.; Kiening, K.L.; Hartl, R.; Bardt, T.; Sarrafzadeh, A.S.; Lanksch, W.R. Multimodal monitoring in patients with head injury: Evaluation of the effects of treatment on cerebral oxygenation. J. Trauma 1997, 42 (Suppl. 5), S32–S37. [Google Scholar] [CrossRef]
- Thapa, K.; Khan, H.; Singh, T.G.; Kaur, A. Traumatic Brain Injury: Mechanistic Insight on Pathophysiology and Potential Therapeutic Targets. J. Mol. Neurosci. 2021, 71, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 2019, 55, 100788. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.; Brooks, W.; Vukas, R.; Pierce, J.; Harris, J. Sex Differences in Traumatic Brain Injury: What We Know and What We Should Know. J. Neurotrauma 2019, 36, 3063–3091. [Google Scholar] [CrossRef]
- Inampudi, C.; Ciccotosto, G.D.; Cappai, R.; Crack, P.J. Genetic Modulators of Traumatic Brain Injury in Animal Models and the Impact of Sex-Dependent Effects. J. Neurotrauma 2020, 37, 706–723. [Google Scholar] [CrossRef]
- Doran, S.J.; Ritzel, R.M.; Glaser, E.P.; Henry, R.J.; Faden, A.I.; Loane, D.J. Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. J. Neurotrauma 2019, 36, 1040–1053. [Google Scholar] [CrossRef]
- Scott, M.C.; Prabhakara, K.S.; Walters, A.J.; Olson, S.D.; Cox, C.S., Jr. Determining Sex-Based Differences in Inflammatory Response in an Experimental Traumatic Brain Injury Model. Front. Immunol. 2022, 13, 753570. [Google Scholar] [CrossRef]
- Lazarus, R.C.; Buonora, J.E.; Jacobowitz, D.M.; Mueller, G.P. Protein carbonylation after traumatic brain injury: Cell specificity, regional susceptibility, and gender differences. Free Radic. Biol. Med. 2015, 78, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaksari, M.; Soltani, Z.; Shahrokhi, N. Effects of Female Sex Steroids Administration on Pathophysiologic Mechanisms in Traumatic Brain Injury. Transl. Stroke Res. 2018, 9, 393–416. [Google Scholar] [CrossRef]
- von Oettingen, G.; Bergholt, B.; Gyldensted, C.; Astrup, J. Blood flow and ischemia within traumatic cerebral contusions. Neurosurgery 2002, 50, 781–788; discussion 788–790. [Google Scholar] [CrossRef] [PubMed]
- Kawai, N.; Nakamura, T.; Tamiya, T.; Nagao, S. Metabolic disturbance without brain ischemia in traumatic brain injury: A positron emission tomography study. Acta Neurochir. Suppl. 2008, 102, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.K. Actions of glucocorticoids and related molecules after traumatic brain injury. Curr. Opin. Crit. Care 2003, 9, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.; Yates, D.; Sandercock, P.; Farrell, B.; Wasserberg, J.; Lomas, G.; Cottingham, R.; Svoboda, P.; Brayley, N.; Mazairac, G.; et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): Randomised placebo-controlled trial. Lancet 2004, 364, 1321–1328. [Google Scholar] [CrossRef]
- Chen, J.H.; Xu, Y.N.; Ji, M.; Li, P.P.; Yang, L.K.; Wang, Y.H. Multimodal monitoring combined with hypothermia for the management of severe traumatic brain injury: A case report. Exp. Ther. Med. 2018, 15, 4253–4258. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.T.; Zheng, M.; Wang, Y.; Diao, Y.; Zhao, W.; Wei, Z. Monitoring intracranial pressure utilizing a novel pattern of brain multiparameters in the treatment of severe traumatic brain injury. Neuropsychiatr. Dis. Treat. 2016, 12, 1517–1523. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Mao, J.; Yao, C.; Liu, Y.; Yan, H.; Jin, W. Neuroprotective effects of mild hypothermia against traumatic brain injury by the involvement of the Nrf2/ARE pathway. Brain Behav. 2022, 12, e2686. [Google Scholar] [CrossRef]
- Baillieul, S.; Chacaroun, S.; Doutreleau, S.; Detante, O.; Pepin, J.L.; Verges, S. Hypoxic conditioning and the central nervous system: A new therapeutic opportunity for brain and spinal cord injuries? Exp. Biol. Med. 2017, 242, 1198–1206. [Google Scholar] [CrossRef]
- Lu, M.; Wang, Y.; Yin, X.; Li, Y.; Li, H. Cerebral protection by remote ischemic post-conditioning in patients with ischemic stroke: A systematic review and meta-analysis of randomized controlled trials. Front. Neurol. 2022, 13, 905400. [Google Scholar] [CrossRef] [PubMed]
- Torres-Querol, C.; Quintana-Luque, M.; Arque, G.; Purroy, F. Preclinical evidence of remote ischemic conditioning in ischemic stroke, a metanalysis update. Sci. Rep. 2021, 11, 23706. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Azim, A.; Ibraheem, K.; Largent-Milnes, T.M.; Rhee, P.; Vanderah, T.W.; Joseph, B. Remote ischemic conditioning preserves cognition and motor coordination in a mouse model of traumatic brain injury. J. Trauma Acute Care Surg. 2017, 83, 1074–1081. [Google Scholar] [CrossRef]
- Pandit, V.; Khan, M.; Zakaria, E.R.; Largent-Milnes, T.M.; Hamidi, M.; O’Keeffe, T.; Vanderah, T.W.; Joseph, B. Continuous remote ischemic conditioning attenuates cognitive and motor deficits from moderate traumatic brain injury. J. Trauma Acute Care Surg. 2018, 85, 48–53. [Google Scholar] [CrossRef]
- Drew, K.L.; Rice, M.E.; Kuhn, T.B.; Smith, M.A. Neuroprotective adaptations in hibernation: Therapeutic implications for ischemia-reperfusion, traumatic brain injury and neurodegenerative diseases. Free Radic. Biol. Med. 2001, 31, 563–573. [Google Scholar] [CrossRef]
- Singhal, N.S.; Sun, C.H.; Lee, E.M.; Ma, D.K. Resilience to Injury: A New Approach to Neuroprotection? Neurotherapeutics 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Hirst, T.C.; Klasen, M.G.; Rhodes, J.K.; Macleod, M.R.; Andrews, P.J.D. A Systematic Review and Meta-Analysis of Hypothermia in Experimental Traumatic Brain Injury: Why Have Promising Animal Studies Not Been Replicated in Pragmatic Clinical Trials? J. Neurotrauma 2020, 37, 2057–2068. [Google Scholar] [CrossRef]
- Chen, H.; Wu, F.; Yang, P.; Shao, J.; Chen, Q.; Zheng, R. A meta-analysis of the effects of therapeutic hypothermia in adult patients with traumatic brain injury. Crit. Care 2019, 23, 396. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.Y.; Chen, S.B.; Wang, J.J.; Xu, C.; Zhao, M.L.; Dong, H.J.; Liang, H.Q.; Li, X.H.; Tu, Y.; Zhang, S.; et al. Establishment of an ideal time window model in hypothermic-targeted temperature management after traumatic brain injury in rats. Brain Res. 2017, 1669, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Z.; Wang, W.Y.; Zeng, J.; Zhou, Z.Y.; Peng, J.; Yang, H.; Deng, P.C.; Li, S.J.; Lu, C.D.; Jiang, H. Optimization of brain metabolism using metabolic-targeted therapeutic hypothermia can reduce mortality from traumatic brain injury. J. Trauma Acute Care Surg. 2017, 83, 296–304. [Google Scholar] [CrossRef]
- Tarahovsky, Y.S.; Fadeeva, I.S.; Komelina, N.P.; Khrenov, M.O.; Zakharova, N.M. Antipsychotic inductors of brain hypothermia and torpor-like states: Perspectives of application. Psychopharmacology 2017, 234, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Lapi, D.; Scuri, R.; Colantuoni, A. Trigeminal Cardiac Reflex and Cerebral Blood Flow Regulation. Front. Neurosci. 2016, 10, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureda, A.; Batle, J.M.; Tur, J.A.; Pons, A. Competitive apnea diving sessions induces an adaptative antioxidant response in mononucleated blood cells. J. Physiol. Biochem. 2015, 71, 373–380. [Google Scholar] [CrossRef]
- Bulmer, A.C.; Coombes, J.S.; Sharman, J.E.; Stewart, I.B. Effects of maximal static apnea on antioxidant defenses in trained free divers. Med. Sci. Sports Exerc. 2008, 40, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.; Gennser, M.; Harlow, P.S.; Lees, M.J. Physiology, pathophysiology and (mal)adaptations to chronic apnoeic training: A state-of-the-art review. Eur. J. Appl. Physiol. 2021, 121, 1543–1566. [Google Scholar] [CrossRef] [PubMed]
- Eftedal, I.; Flatberg, A.; Drvis, I.; Dujic, Z. Immune and inflammatory responses to freediving calculated from leukocyte gene expression profiles. Physiol. Genom. 2016, 48, 795–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, A.; Batten, A.J.; Levin, M.; Allen, K.N.; Fitzgerald, M.L.; Huckstadt, L.A.; Costa, D.P.; Buys, E.S.; Hindle, A.G. Intrinsic anti-inflammatory properties in the serum of two species of deep-diving seal. J. Exp. Biol. 2018, 221, jeb178491. [Google Scholar] [CrossRef] [Green Version]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef]
- Shah, K.A.; Sonti, A.N.; Wu, Y.C.; Powell, K.; Doobay, M.; Narayan, R.K.; Li, C. Electrical Stimulation of the Infraorbital Nerve Induces Diving Reflex in a Dose-Controlled Manner. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2020, 2020, 5208–5211. [Google Scholar] [CrossRef]
- Li, C.; Shah, K.A.; Powell, K.; Wu, Y.C.; Chaung, W.; Sonti, A.N.; White, T.G.; Doobay, M.; Yang, W.L.; Wang, P.; et al. CBF oscillations induced by trigeminal nerve stimulation protect the pericontusional penumbra in traumatic brain injury complicated by hemorrhagic shock. Sci. Rep. 2021, 11, 19652. [Google Scholar] [CrossRef]
- Li, C.; White, T.G.; Shah, K.A.; Chaung, W.; Powell, K.; Wang, P.; Woo, H.H.; Narayan, R.K. Percutaneous Trigeminal Nerve Stimulation Induces Cerebral Vasodilation in a Dose-Dependent Manner. Neurosurgery 2021, 88, E529–E536. [Google Scholar] [CrossRef]
- Shah, S.A.; Lowder, R.J.; Kuceyeski, A. Quantitative multimodal imaging in traumatic brain injuries producing impaired cognition. Curr. Opin. Neurol. 2020, 33, 691–698. [Google Scholar] [CrossRef]
- Shi, J.; Teng, J.; Du, X.; Li, N. Multi-Modal Analysis of Resting-State fMRI Data in mTBI Patients and Association with Neuropsychological Outcomes. Front. Neurol. 2021, 12, 639760. [Google Scholar] [CrossRef] [PubMed]
- Dean, P.J.; Sato, J.R.; Vieira, G.; McNamara, A.; Sterr, A. Multimodal imaging of mild traumatic brain injury and persistent postconcussion syndrome. Brain Behav. 2015, 5, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Lunkova, E.; Guberman, G.I.; Ptito, A.; Saluja, R.S. Noninvasive magnetic resonance imaging techniques in mild traumatic brain injury research and diagnosis. Hum. Brain Mapp. 2021, 42, 5477–5494. [Google Scholar] [CrossRef] [PubMed]
- Lippa, S.M.; Yeh, P.H.; Ollinger, J.; Brickell, T.A.; French, L.M.; Lange, R.T. White Matter Integrity Relates to Cognition in Service Members and Veterans after Complicated Mild, Moderate, and Severe Traumatic Brain Injury, but Not Uncomplicated Mild Traumatic Brain Injury. J. Neurotrauma 2022, 40, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Yan, F.; Zhang, H.; Zhang, E.; Wang, X.; Wei, M.; Pei, Y.; Yang, Z.; Li, Y.; et al. Investigating the mechanism and prognosis of patients with disorders of consciousness on the basis of brain networks between the thalamus and whole-brain. Front. Neurol. 2022, 13, 990686. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, L.; Li, F.; Chen, Y.C. Neuropathological Mechanisms of Mild Traumatic Brain Injury: A Perspective from Multimodal Magnetic Resonance Imaging. Front. Neurosci. 2022, 16, 923662. [Google Scholar] [CrossRef]
- Harris, N.G.; Paydar, A.; Smith, G.S.; Lepore, S. Diffusion MR imaging acquisition and analytics for microstructural delineation in pre-clinical models of TBI. J. Neurosci. Res. 2022, 100, 1128–1139. [Google Scholar] [CrossRef]
- Minchew, H.M.; Ferren, S.L.; Christian, S.K.; Hu, J.; Keselman, P.; Brooks, W.M.; Andrews, B.T.; Harris, J.L. Comparing imaging biomarkers of cerebral edema after TBI in young adult male and female rats. Brain Res. 2022, 1789, 147945. [Google Scholar] [CrossRef]
- Mayer, A.R.; Bellgowan, P.S.; Hanlon, F.M. Functional magnetic resonance imaging of mild traumatic brain injury. Neurosci. Biobehav. Rev. 2015, 49, 8–18. [Google Scholar] [CrossRef]
- Storti, S.F.; Formaggio, E.; Franchini, E.; Bongiovanni, L.G.; Cerini, R.; Fiaschi, A.; Michel, C.M.; Manganotti, P. A multimodal imaging approach to the evaluation of post-traumatic epilepsy. MAGMA 2012, 25, 345–360. [Google Scholar] [CrossRef] [PubMed]
- French, J.A.; Bebin, M.; Dichter, M.A.; Engel, J., Jr.; Hartman, A.L.; Jozwiak, S.; Klein, P.; McNamara, J., Sr.; Twyman, R.; Vespa, P. Antiepileptogenesis and disease modification: Clinical and regulatory issues. Epilepsia Open 2021, 6, 483–492. [Google Scholar] [CrossRef]
- Stovell, M.G.; Yan, J.L.; Sleigh, A.; Mada, M.O.; Carpenter, T.A.; Hutchinson, P.J.A.; Carpenter, K.L.H. Assessing Metabolism and Injury in Acute Human Traumatic Brain Injury with Magnetic Resonance Spectroscopy: Current and Future Applications. Front. Neurol. 2017, 8, 426. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, P.; Morrison, T.R.; Cai, X.; Iriah, S.; Simon, N.; Sabrick, J.; Neuroth, L.; Ferris, C.F. Neuroradiological Changes Following Single or Repetitive Mild TBI. Front. Syst. Neurosci. 2019, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.L.; Yeh, H.W.; Choi, I.Y.; Lee, P.; Berman, N.E.; Swerdlow, R.H.; Craciunas, S.C.; Brooks, W.M. Altered neurochemical profile after traumatic brain injury: (1)H-MRS biomarkers of pathological mechanisms. J. Cereb. Blood Flow Metab. 2012, 32, 2122–2134. [Google Scholar] [CrossRef] [Green Version]
- Yasmin, A.; Pitkanen, A.; Jokivarsi, K.; Poutiainen, P.; Grohn, O.; Immonen, R. MRS Reveals Chronic Inflammation in T2w MRI-Negative Perilesional Cortex—A 6-Months Multimodal Imaging Follow-Up Study. Front. Neurosci. 2019, 13, 863. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.W.; Vivash, L.; Mudududdla, R.; Nguyen, N.; Hermans, S.J.; Shackleford, D.M.; Field, J.; Xue, L.; Aprico, A.; Hancock, N.C.; et al. Development of [(18)F]MIPS15692, a radiotracer with in vitro proof-of-concept for the imaging of MER tyrosine kinase (MERTK) in neuroinflammatory disease. Eur. J. Med. Chem. 2021, 226, 113822. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, F.A.; Iturria-Medina, Y.; Thelin, E.P.; Gomez, A.; Shankar, J.J.; Ko, J.H.; Figley, C.R.; Wright, G.E.B.; Anderson, C.M. Integrative Neuroinformatics for Precision Prognostication and Personalized Therapeutics in Moderate and Severe Traumatic Brain Injury. Front. Neurol. 2021, 12, 729184. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.; Hukkelhoven, C.W.; Marshall, L.F.; Steyerberg, E.W. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: A comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery 2005, 57, 1173–1182; discussion 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, R.; Siironen, J.; Skrifvars, M.B.; Hernesniemi, J.; Kivisaari, R. Predicting outcome in traumatic brain injury: Development of a novel computerized tomography classification system (Helsinki computerized tomography score). Neurosurgery 2014, 75, 632–646; discussion 637–646. [Google Scholar] [CrossRef]
- Sheth, K.N.; Mazurek, M.H.; Yuen, M.M.; Cahn, B.A.; Shah, J.T.; Ward, A.; Kim, J.A.; Gilmore, E.J.; Falcone, G.J.; Petersen, N.; et al. Assessment of Brain Injury Using Portable, Low-Field Magnetic Resonance Imaging at the Bedside of Critically Ill Patients. JAMA Neurol. 2020, 78, 41–47. [Google Scholar] [CrossRef]
- Turpin, J.; Unadkat, P.; Thomas, J.; Kleiner, N.; Khazanehdari, S.; Wanchoo, S.; Samuel, K.; Moclair, B.O.; Black, K.; Dehdashti, A.R.; et al. Portable Magnetic Resonance Imaging for ICU Patients. Crit. Care Explor. 2020, 2, e0306. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.M.; Yuh, E.L.; Mac Donald, C.L.; Bourla, I.; Wren-Jarvis, J.; Sun, X.; Vassar, M.J.; Diaz-Arrastia, R.; Giacino, J.T.; Okonkwo, D.O.; et al. Diffusion Tensor Imaging Reveals Elevated Diffusivity of White Matter Microstructure that Is Independently Associated with Long-Term Outcome after Mild Traumatic Brain Injury: A TRACK-TBI Study. J. Neurotrauma 2022, 39, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.; Alamri, A.; Mohamed, M.; Khalid, N.; O’Halloran, P.; Staartjes, V.; Uff, C. Prognosticating outcome using magnetic resonance imaging in patients with moderate to severe traumatic brain injury: A machine learning approach. Brain Inj. 2022, 36, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Vergara, V.M.; Mayer, A.R.; Damaraju, E.; Kiehl, K.A.; Calhoun, V. Detection of Mild Traumatic Brain Injury by Machine Learning Classification Using Resting State Functional Network Connectivity and Fractional Anisotropy. J. Neurotrauma 2017, 34, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Daley, M.; Cameron, S.; Ganesan, S.L.; Patel, M.A.; Stewart, T.C.; Miller, M.R.; Alharfi, I.; Fraser, D.D. Pediatric severe traumatic brain injury mortality prediction determined with machine learning-based modeling. Injury 2022, 53, 992–998. [Google Scholar] [CrossRef]
- Hoffman, H.; Abi-Aad, K.; Bunch, K.M.; Beutler, T.; Otite, F.O.; Chin, L.S. Outcomes associated with brain tissue oxygen monitoring in patients with severe traumatic brain injury undergoing intracranial pressure monitoring. J. Neurosurg. 2021, 135, 1799–1806. [Google Scholar] [CrossRef]
- Andrew, R.D.; Hartings, J.A.; Ayata, C.; Brennan, K.C.; Dawson-Scully, K.D.; Farkas, E.; Herreras, O.; Kirov, S.A.; Muller, M.; Ollen-Bittle, N.; et al. The Critical Role of Spreading Depolarizations in Early Brain Injury: Consensus and Contention. Neurocrit. Care 2022, 37 (Suppl. 1), 83–101. [Google Scholar] [CrossRef]
- Hinzman, J.M.; Andaluz, N.; Shutter, L.A.; Okonkwo, D.O.; Pahl, C.; Strong, A.J.; Dreier, J.P.; Hartings, J.A. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 2014, 137, 2960–2972. [Google Scholar] [CrossRef] [Green Version]
- Sueiras, M.; Thonon, V.; Santamarina, E.; Sanchez-Guerrero, A.; Riveiro, M.; Poca, M.A.; Quintana, M.; Gandara, D.; Sahuquillo, J. Is Spreading Depolarization a Risk Factor for Late Epilepsy? A Prospective Study in Patients with Traumatic Brain Injury and Malignant Ischemic Stroke Undergoing Decompressive Craniectomy. Neurocrit. Care 2021, 34, 876–888. [Google Scholar] [CrossRef]
- Andrew, R.D.; Farkas, E.; Hartings, J.A.; Brennan, K.C.; Herreras, O.; Muller, M.; Kirov, S.A.; Ayata, C.; Ollen-Bittle, N.; Reiffurth, C.; et al. Questioning Glutamate Excitotoxicity in Acute Brain Damage: The Importance of Spreading Depolarization. Neurocrit. Care 2022, 37 (Suppl. 1), 11–30. [Google Scholar] [CrossRef]
- Rueda Carrillo, L.; Garcia, K.A.; Yalcin, N.; Shah, M. Ketamine and Its Emergence in the Field of Neurology. Cureus 2022, 14, e27389. [Google Scholar] [CrossRef]
- Sanchez-Porras, R.; Kentar, M.; Zerelles, R.; Geyer, M.; Trenado, C.; Hartings, J.A.; Woitzik, J.; Dreier, J.P.; Santos, E. Eighteen-hour inhibitory effect of s-ketamine on potassium- and ischemia-induced spreading depolarizations in the gyrencephalic swine brain. Neuropharmacology 2022, 216, 109176. [Google Scholar] [CrossRef]
- Podell, J.; Pergakis, M.; Yang, S.; Felix, R.; Parikh, G.; Chen, H.; Chen, L.; Miller, C.; Hu, P.; Badjatia, N. Leveraging Continuous Vital Sign Measurements for Real-Time Assessment of Autonomic Nervous System Dysfunction After Brain Injury: A Narrative Review of Current and Future Applications. Neurocrit. Care 2022, 37 (Suppl. 2), 206–219. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Baker, W.; Mahanna-Gabrielli, E.; Kofke, A.W.; Balu, R. Hierarchical Cluster Analysis Identifies Distinct Physiological States after Acute Brain Injury. Neurocrit. Care 2022, 36, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, C.; Ajith, A.; Mansour, A.; Okonkwo, D.O.; Diaz-Arrastia, R.; Mayampurath, A. Prediction of Intracranial Hypertension and Brain Tissue Hypoxia Utilizing High-Resolution Data from the BOOST-II Clinical Trial. Neurotrauma Rep. 2022, 3, 473–478. [Google Scholar] [CrossRef]
- Wilde, E.A.; Wanner, I.B.; Kenney, K.; Gill, J.; Stone, J.R.; Disner, S.; Schnakers, C.; Meyer, R.; Prager, E.M.; Haas, M.; et al. A Framework to Advance Biomarker Development in the Diagnosis, Outcome Prediction, and Treatment of Traumatic Brain Injury. J. Neurotrauma 2022, 39, 436–457. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Schwartzbauer, G.; Jia, X. Brain Monitoring in Critically Neurologically Impaired Patients. Int. J. Mol. Sci. 2016, 18, 43. [Google Scholar] [CrossRef] [Green Version]
- Seule, M.; Sikorski, C.; Sakowitz, O.; von Campe, G.; Santos, E.; Orakcioglu, B.; Unterberg, A.; Keller, E. Evaluation of a New Brain Tissue Probe for Intracranial Pressure, Temperature, and Cerebral Blood Flow Monitoring in Patients with Aneurysmal Subarachnoid Hemorrhage. Neurocrit. Care 2016, 25, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Pease, M.; Nwachuku, E.; Goldschmidt, E.; Elmer, J.; Okonkwo, D.O. Complications from Multimodal Monitoring Do not Affect Long-Term Outcomes in Severe Traumatic Brain Injury. World Neurosurg. 2022, 161, e109–e117. [Google Scholar] [CrossRef] [PubMed]
- Robba, C.; Pozzebon, S.; Moro, B.; Vincent, J.L.; Creteur, J.; Taccone, F.S. Multimodal non-invasive assessment of intracranial hypertension: An observational study. Crit. Care 2020, 24, 379. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, F.A.; Ziesmann, M.T.; Goeres, P.; Unger, B.; Park, J.; Karakitsos, D.; Blaivas, M.; Vergis, A.; Gillman, L.M. A unique method for estimating the reliability learning curve of optic nerve sheath diameter ultrasound measurement. Crit. Ultrasound J. 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butts, C.; Wilson, J.; Lasseigne, L.; Oral, E.; Kaban, N. Ultrasound of the Optic Nerve Does Not Appear to Be a Consistently Reliable or Generalizable Method to Monitor Changes in Intracranial Pressure. J. Intensive Care Med. 2022, 37, 663–670. [Google Scholar] [CrossRef] [PubMed]

| Model | Population | Sample Size | %Fem | Intervention | Outcome of Combination Therapy | Reference |
|---|---|---|---|---|---|---|
| Clinical Trials | ||||||
| Case Series | Severe Human TBI | 2 | 0% | Cerebrolysin, citicoline | Improved long-term neurologic recovery with combined therapy in acute TBI | Trimmel, 2022 [15] |
| Retrospective Observational | Severe Human TBI | 117 | 20.6% | HTS, mannitol, barbiturates, propofol, fentanyl | Propofol and fentanyl reduced ICP, but less than HTS in patients with acute TBI | Colton, 2014 [16] |
| Randomized Placebo Controlled | Severe Human TBI | 14 | 21.4% | Probenecid, NAC | No harmful effects from combo in Phase I trial in acute pediatric TBI | Clark, 2017 [17] |
| Preclinical Trials | ||||||
| CCI | Mouse | 36 | 0% | NSC, olfactory ensheathing cells, valproic acid | Improved behavioral function and NSC neuronal differentiation | Liu, 2022 [18] |
| CCI | Mouse | 292 | 45.9% | Apocynin, salubrinal, TBHQ | Improved functional outcomes, brain lesion development, and reduced inflammation | Davis, 2022 [19] |
| Biopsy Punch | Rat | 45 | 0% | NSC, curcumin nanoparticles | Reduced glial activation and edema, improved recovery | Narouiepour, 2022 [20] |
| CHI | Mouse | 26 | 0% | Minocycline, NAC | Improved memory function and reduced neuronal loss | Whitney, 2021 [21] |
| CCI | Mouse | 10 | 0% | Apocynin, TBHQ | Reduced white matter disruption | Chandran, 2021 [22] |
| Weight Drop | Mouse | 44 | 0% | Ketamine, perampanel | Reduced neurobehavioral dysfunction and NF-kB/iNOS expression | Alqahtani, 2020 [23] |
| Weight Drop | Rat | 32 | 0% | Felbamate, levetiracetam | Reduced pro-inflammatory cytokines and histologic damage | Bayhan, 2020 [24] |
| Weight Drop | Rat | 42 | 0% | Doxycycline, tocopherol | Reduced neurobehavioral deficits, ROS, and pro-inflammatory cytokines | Rana, 2020 [25] |
| Weight Drop | Rat | 30 | 0% | MDL28170, BMSC | Reduced inflammation and improved survival of stem cells, with reduced neurobehavioral impairment | Hu, 2019 [26] |
| Weight Drop | Rat | 24 | 100% | Lomerizine, YM872, Brilliant Blue G | Decreased microglial activation and myelin disruption without affecting neurobehavioral impairment | Mao, 2018 [27] |
| CCI | Mouse | 48 | 0% | GSNO, CAPE | Reduced oxidative stress and mitochondrial dysfunction, improved neurobehavioral function | Khan, 2018 [28] |
| CCI | Mouse | NR | 0% | Minocycline, NAC | Prevented loss of oligodendrocytes following CCI | Sangobowale, 2018 [29] |
| CCI | Rat | 24 | 0% | Magnesium, NAT | Reduced BBB disruption and improved functional outcomes | Ameliorate, 2017 [30] |
| CCI | Mouse | 31 | 0% | DHA, NSC | Improved neurogenesis and functional outcomes, with increased astrocyte and microglia activation | Ghazale, 2018 [31] |
| CCI | Mouse | NR | 0% | Apocynin, TBHQ | Improved function and lesion volume | Chandran, 2018 [32] |
| mCCI | Rat | NR | 0% | Minocycline, NAC | Protected oligodendrocytes and increased M1/M2 microglial activation | Haber, 2018 [33] |
| FPI | Rat | 70 | 0% | Memantine, estradiol | Improved functional deficits and reduced neuronal degeneration | Day, 2017 [34] |
| CCI | Rat | 96 | 0% | Magnesium, PEG | Improved CNS penetration of magnesium, increased neuroprotection | Busingye, 2016 [35] |
| CCI | Rat | 207 | 0% | BMSC, propranolol | Decreased long-term neurobehavioral deficits | Kota, 2016 [36] |
| CHI | Rat | 35 | 0% | Carnosine, Cyclosporine | Decreased pro-inflammatory cytokines and neuronal apoptosis | Baky, 2016 [37] |
| In-Vitro TBI | Rat Brain Slices | NR | 0% | Memantine, estradiol | Reduced neuronal death | Lamprecht, 2015 [38] |
| Cryo-Injury | Mouse | 70 | 0% | BMDC, lipoic acid | Increased cell growth in perilesional penumbra, decreased astrocyte infiltration, increased microglial activation | Paradells, 2015 [39] |
| CCI | Rat | 40 | 0% | Progesterone, vitamin D | Reduced neuronal loss and astrocyte activation, mediated through downregulations in TLR4/NF-kB | Tang, 2015 [40] |
| CCI | Rat | 31 | 0% | G-CSF, hUCB | Reduced activation of microglia and improved neurogenesis and functional recovery | Acosta, 2014 [41] |
| CCI | Rat | 40 | 0% | Etanercept, lithium | Reduced edema and neuronal/glial apoptosis | Ekici, 2014 [42] |
| mCCI | Rat | NR | NR | Minocycline, NAC | Reduced neuroinflammation and neurobehavioral deficits | Haber, 2013 [43] |
| CCI | Mouse | 126 | 0% | Lithium, valproic acid | Reduced BBB disruption, lesion volume, neuronal degeneration, and functional deficits | Yu, 2013 [44] |
| CCI | Rat | 74 | NR | Progesterone, magnesium | Reduced neuronal apoptosis and neurobehavioral deficits | Uysal, 2013 [45] |
| CCI | Mouse | 50 | 0% | Melatonin, dexamethasone | Reduced lesion volume, oxidative stress, and functional deficits | Campolo, 2013 [46] |
| CCI | Mouse | 44 | 0% | Dexamethasone, bortezomib | Reduced edema and BBB disruption | Thal, 2013 [47] |
| CCI | Rat | 128 | 0% | Progesterone, vitamin D | Reduced neuronal loss and astrocyte activation | Tang, 2013 [48] |
| CCI | Rat | 38 | 0% | Nimodipine, melatonin | Worsened edema and neuronal necrosis compared to melatonin alone | Ismailoglu, 2012 [49] |
| CCI | Rat | 46 | 0% | Progesterone, vitamin D | Improved neurobehavioral function and increased astrocyte activation | Hua, 2012 [50] |
| CHI | Mouse | 39 | 0% | VEGF, FGF2 | Improved functional outcomes, no additional benefit versus monotherapy | Thau-Zuchman, 2012 [51] |
| CHI | Rat | 35 | 100% | Estrogen, progesterone | Less reduction of edema and anti-inflammatory cytokines versus estrogen alone | Khaksari, 2011 [52] |
| CCI | Rat | 50 | 0% | Minocycline, melatonin | No significant effect | Kelso, 2012 [53] |
| CHI | Rat | 32 | 0% | Magnesium, MK801 | Reduced edema and BBB disruption, but no greater effect than monotherapy | Imer, 2009 [54] |
| CCI | Rat | NR | NR | L-arginine, D-arginine, SOD, catalase | Increased nitric oxide and cerebral blood flow after TBI | Cherian, 2003 [55] |
| mCCI | Rat | 30 | 0% | MK801, scopolamine | Improved hippocampal neuronal death and associated memory deficits | Jenkins, 1999 [56] |
| FPI | Rat | 42 | NR | Morphine, scopolamine | Improved functional outcomes | Lyeth, 1993 [57] |
| Model | Population | Sample Size | %Fem | Intervention | Outcome of Combination Therapy | Reference |
|---|---|---|---|---|---|---|
| Clinical Trials | ||||||
| Randomized Prospective | Mild- Moderate Human TBI | 166 | 44% | Cognitive training + rTMS vs. Cognitive training alone | Improved neurologic and functional outcomes in chronic TBI rehabilitation | Zhou, 2021 [84] |
| Case Report | Severe Human TBI | 1 | 0% | rTMS + Neuromotor training | Improved motor function in chronic TBI rehabilitation | Martino Cinnera, 2016 [85] |
| Preclinical Trials | ||||||
| CCI | Rat | 97 | 0% | TMS + EE vs. TMS alone | Improved motor and sensory function | Shin, 2018 [86] |
| FPI | Rat | 46 | 0% | EE + MEOS vs. EE alone | Improved neurocognitive dysfunction | Maegele, 2005 [87] |
| FPI | Rat | 24 | 0% | EE + MEOS vs. EE alone | Improved neurocognitive dysfunction, reduced neuronal apoptosis and astrocyte activation | Maegele, 2005 [88] |
| TBI Severity | Study Design | Sample Size | %Fem | Neuromonitoring Parameters | Outcome of MMM-Guided Treatment | Reference |
|---|---|---|---|---|---|---|
| Clinical Trials | ||||||
| Moderate- Severe | Retrospective Observational | 61 | 29.5% | ICP, CPP, PRx | May predict need for long-term treatment of seizures after TBI | Appavu, 2022 [108] |
| Severe | Retrospective Cohort | 49 | 20.4% | ICP, PbtO2, CPP | Improved treatment of cerebral hypoxia and hypertension without improvement in long-term outcomes | Lang, 2022 [109] |
| Severe | Retrospective Observational | 20 | 15% | ICP, CPP, PbtO2, LPR | Enabled diagnosis and treatment of cerebral metabolic crisis | Marini, 2022 [110] |
| Moderate- Severe | Prospective Interventional | 5 | 100% | ICP, PbtO2, LPR | Improved cerebral metabolic dysfunction | Khellaf, 2022 [111] |
| Severe | Case Report | 1 | 0% | ICP, PbtO2, CPP, PRx, | Guided need for surgical intervention | Robinson, 2021 [112] |
| Moderate- Severe | Retrospective Observational | 85 | 31.8% | ICP, CPP, PRx | Helped guide clinical treatment in pediatric TBI, reduced length of time on mechanical ventilation | Appavu, 2021 [113] |
| Severe | Retrospective Observational | 81 | 19.8% | ICP, CPP, PRx | Improved clinical outcomes | Petkus, 2020 [114] |
| Moderate- Severe | Retrospective Observational | 38 | 32% | ICP, CPP, PbtO2, PaO2 | Characterized etiology of cerebral hypoxemia and guided treatment | Dellazizzo, 2018 [115] |
| Severe | Prospective Randomized Cohort | 119 | 21% | ICP, PbtO2 | Reduced hypoxia with trend toward better outcomes than ICP alone | Okonkwo, 2017 [116] |
| Moderate- Severe | Retrospective Cohort | 30 | 10% | ICP, CPP | Reduced mortality and length of ICU stay | Luca, 2015 [117] |
| Severe | Prospective Observational | 18 | 38.9% | ICP, CPP | Reduced mortality and improved long-term neurologic outcomes | Dunham, 2006 [118] |
| Severe | Prospective Randomized Cohort | 82 | 15.9% | ICP, CPP, PbtO2, CBF, pH, SvjO2, PRx | Treatment guided by MMM associated with improved outcomes | Isa, 2003 [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynch, D.G.; Narayan, R.K.; Li, C. Multi-Mechanistic Approaches to the Treatment of Traumatic Brain Injury: A Review. J. Clin. Med. 2023, 12, 2179. https://doi.org/10.3390/jcm12062179
Lynch DG, Narayan RK, Li C. Multi-Mechanistic Approaches to the Treatment of Traumatic Brain Injury: A Review. Journal of Clinical Medicine. 2023; 12(6):2179. https://doi.org/10.3390/jcm12062179
Chicago/Turabian StyleLynch, Daniel G., Raj K. Narayan, and Chunyan Li. 2023. "Multi-Mechanistic Approaches to the Treatment of Traumatic Brain Injury: A Review" Journal of Clinical Medicine 12, no. 6: 2179. https://doi.org/10.3390/jcm12062179