
Novel Molecular Subtyping Scheme Based on In Silico Analysis of Cuproptosis Regulator Gene Patterns Optimizes Survival Prediction and Treatment of Hepatocellular Carcinoma
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Identification of Cuproptosis Subtypes
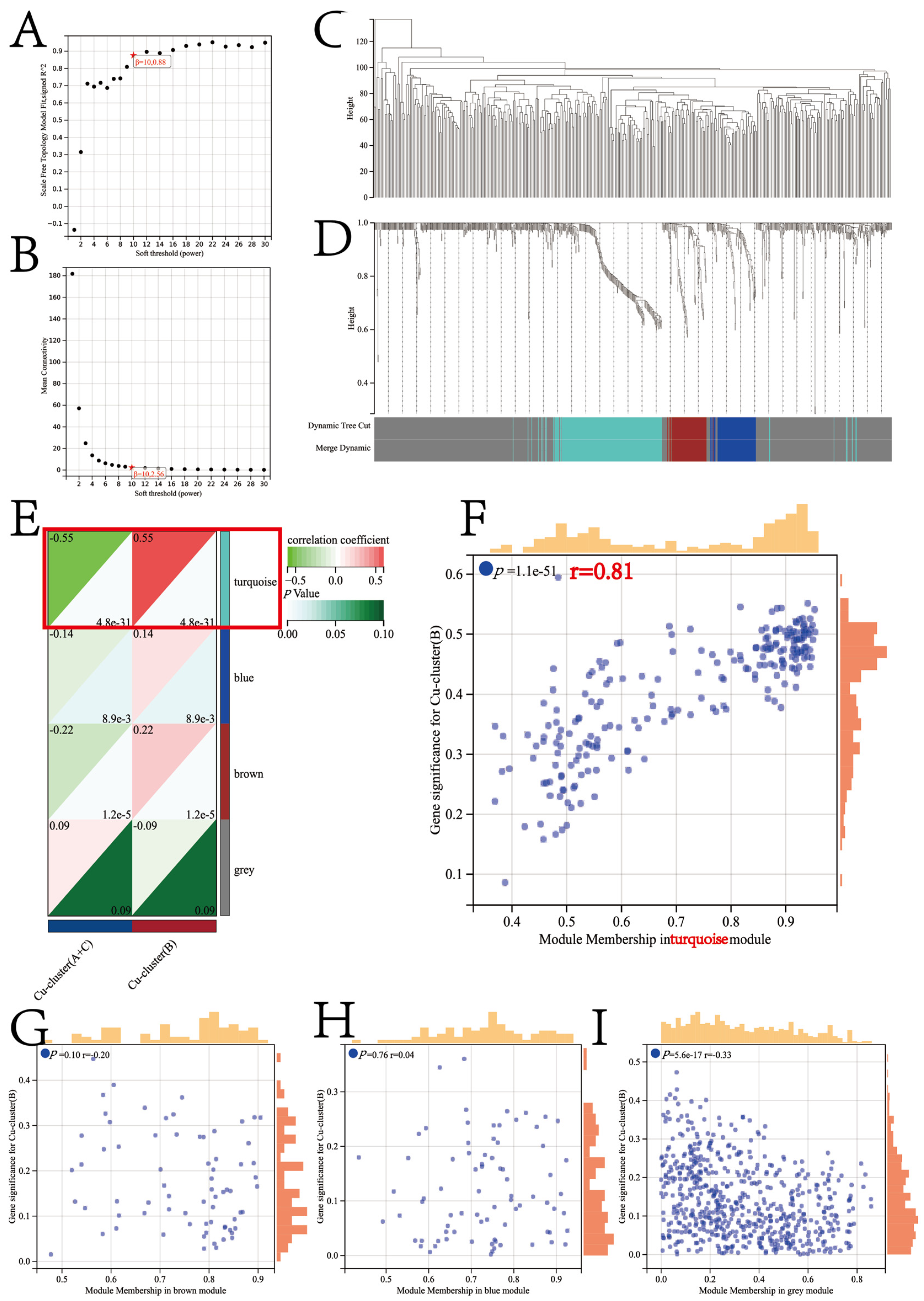
2.3. Co-Expression Network Construction
2.4. Pathway and Process Enrichment Analyses
2.5. Drug Sensitivity Prediction
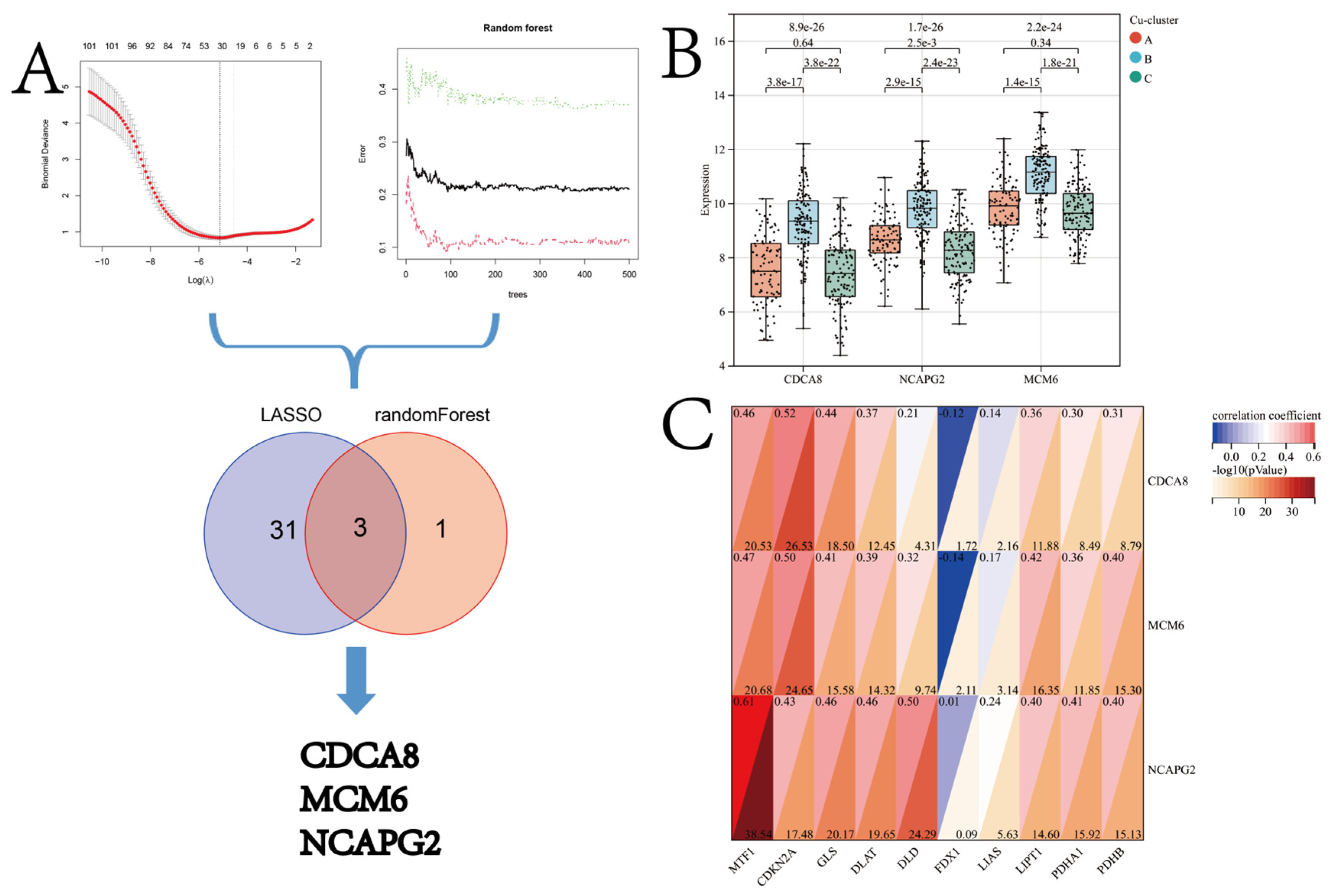
2.6. Machine Learning for the Candidate Cuproptosis Subtype-Specific Gene Signature (CSGS)
2.7. Protein Expression of CSGSs Using the Human Protein Atlas (HPA) Database
2.8. Statistical Analyses
3. Results
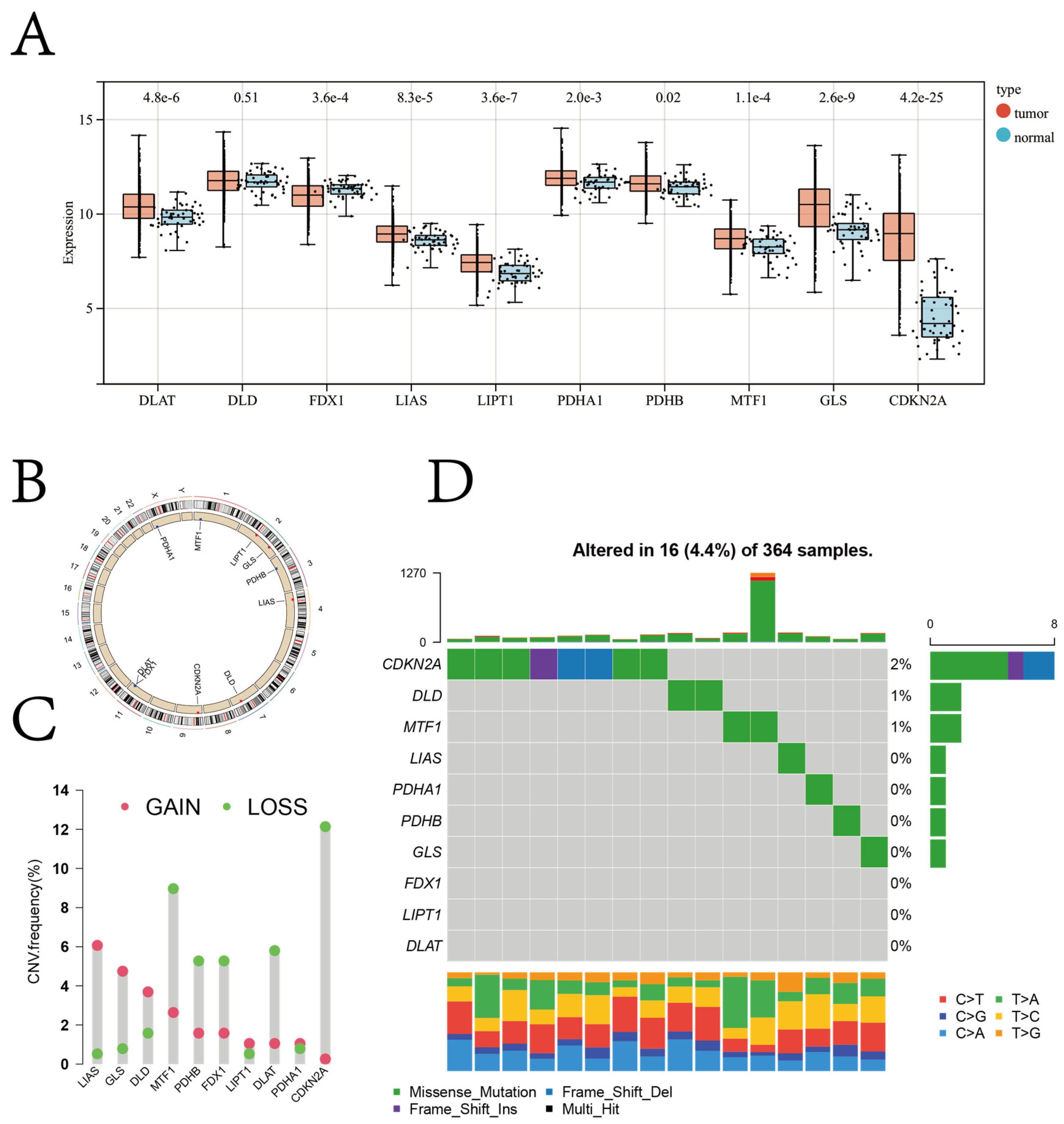
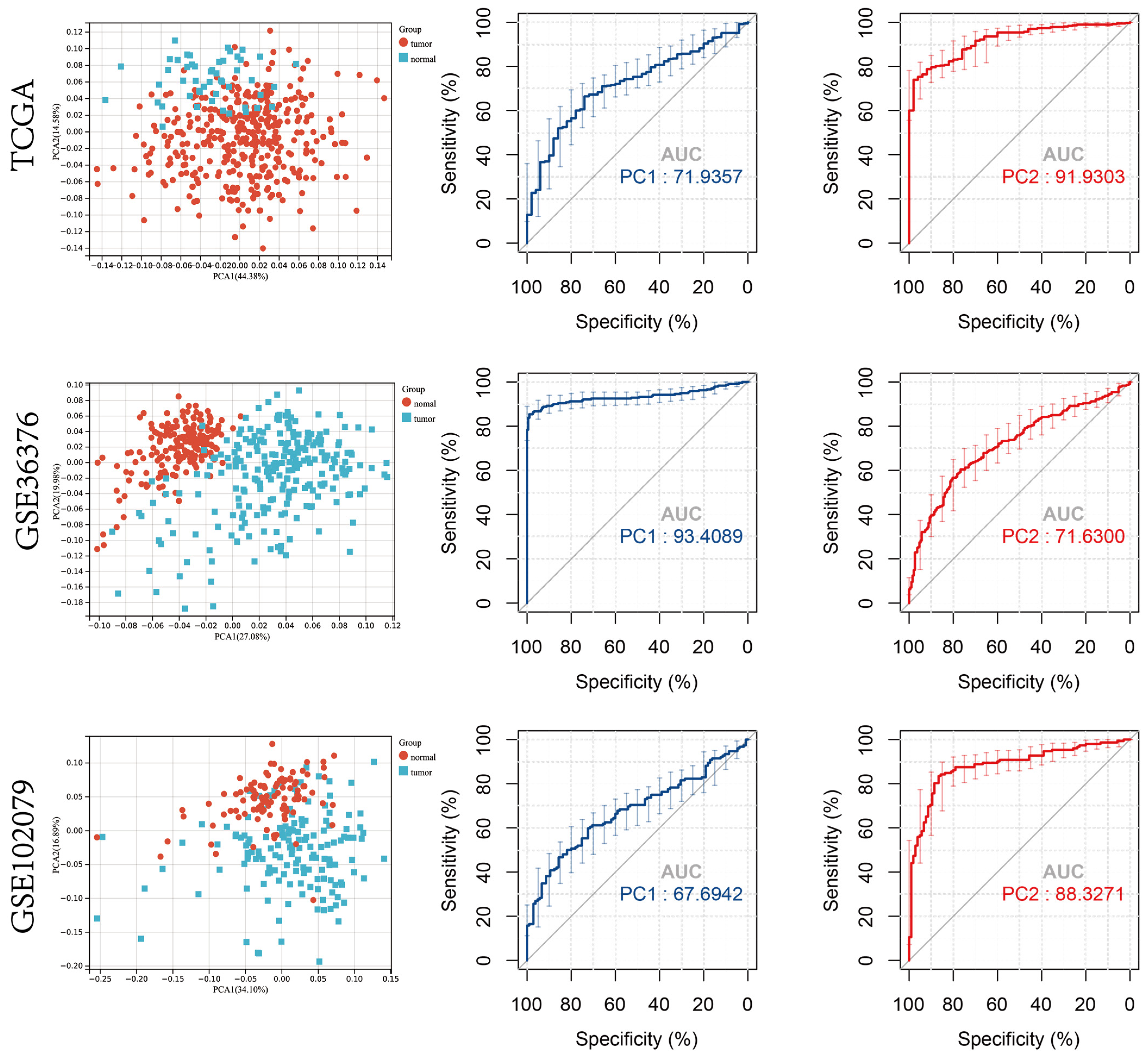
3.1. Diagnostic and Prognostic Role of Cuproptosis Regulator Genes in Patients with HCC
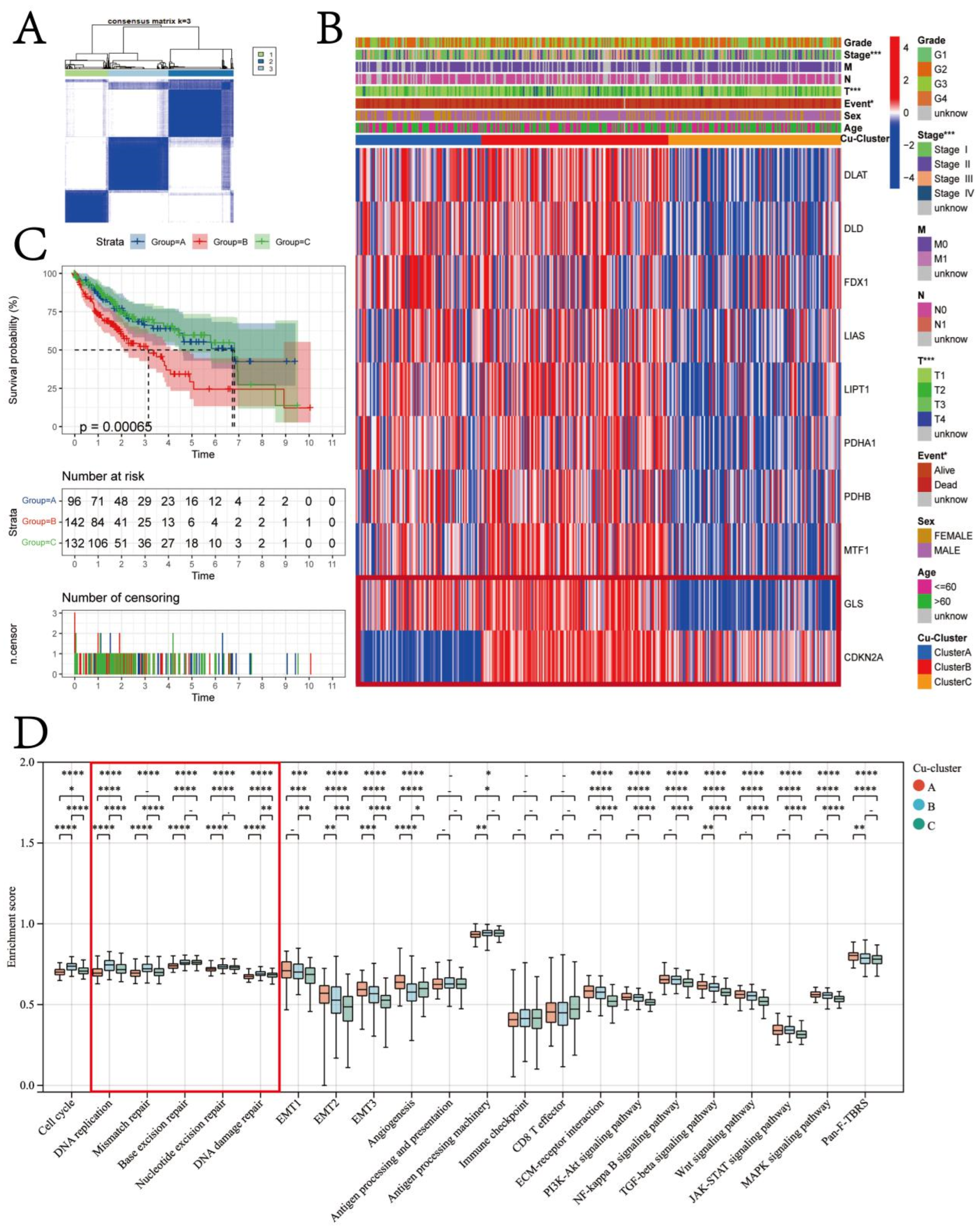
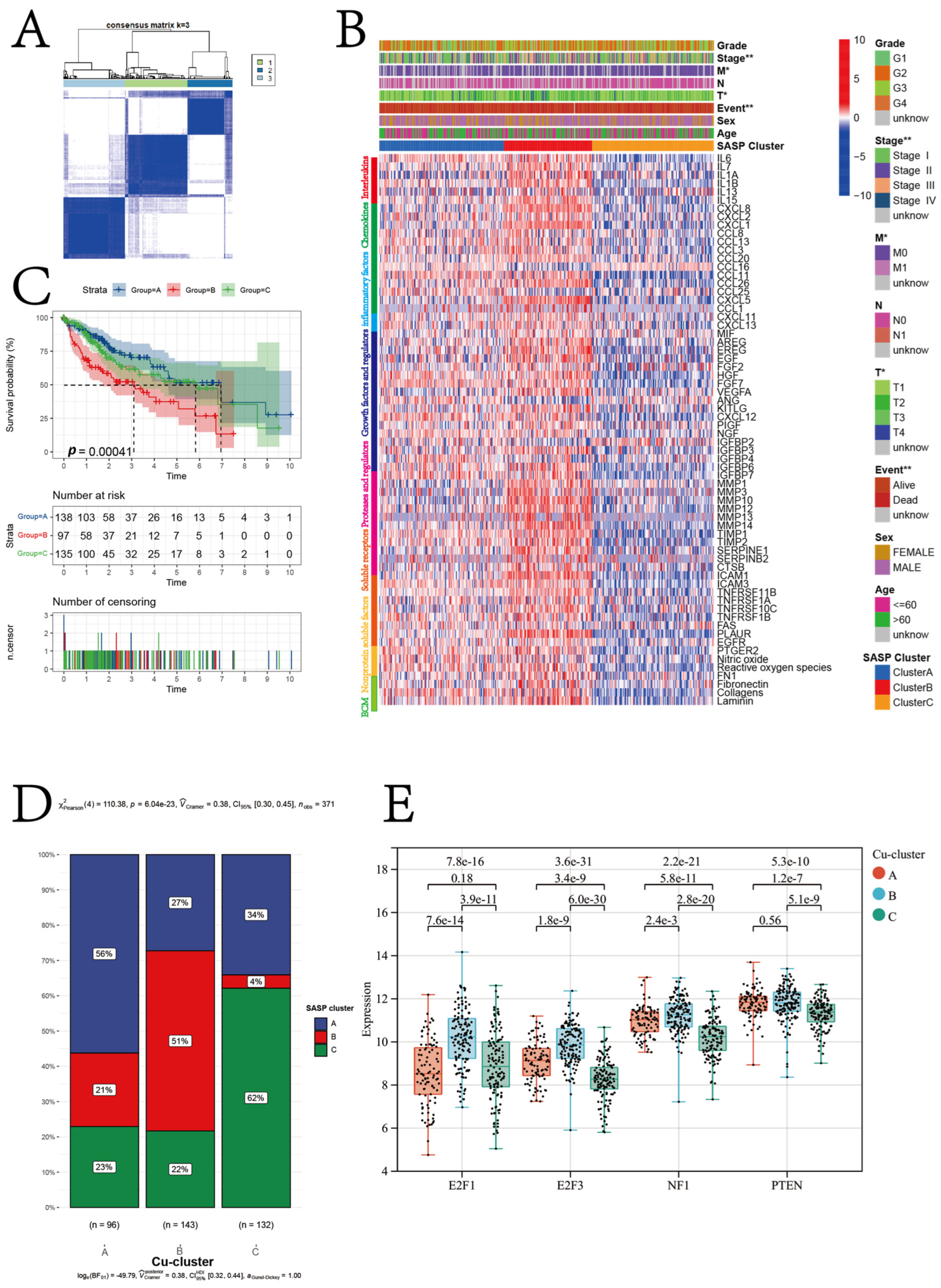
3.2. Cuproptosis Subtypes in HCC
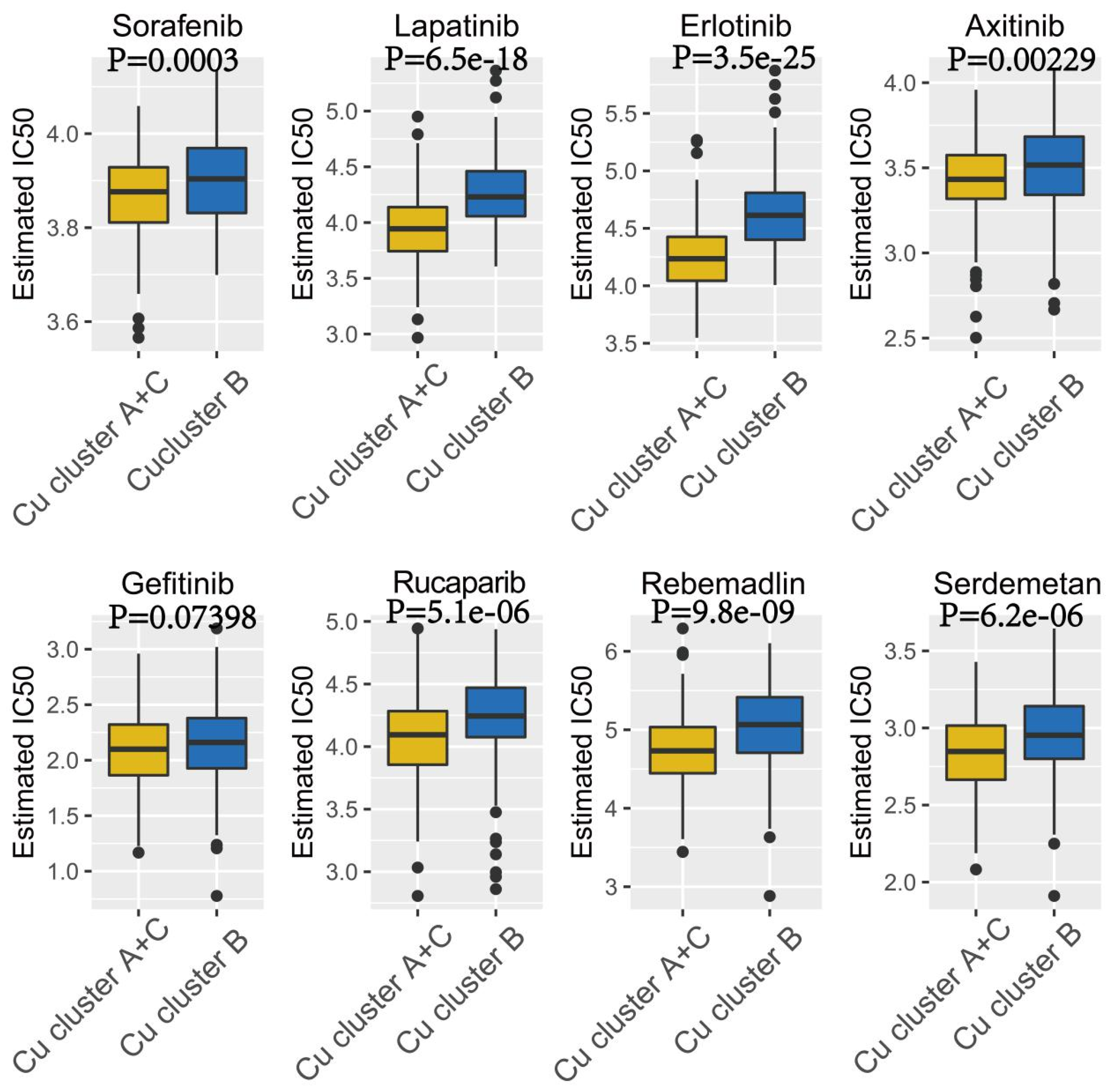
3.3. Therapeutic Potential of Cuproptosis Subtypes in HCC
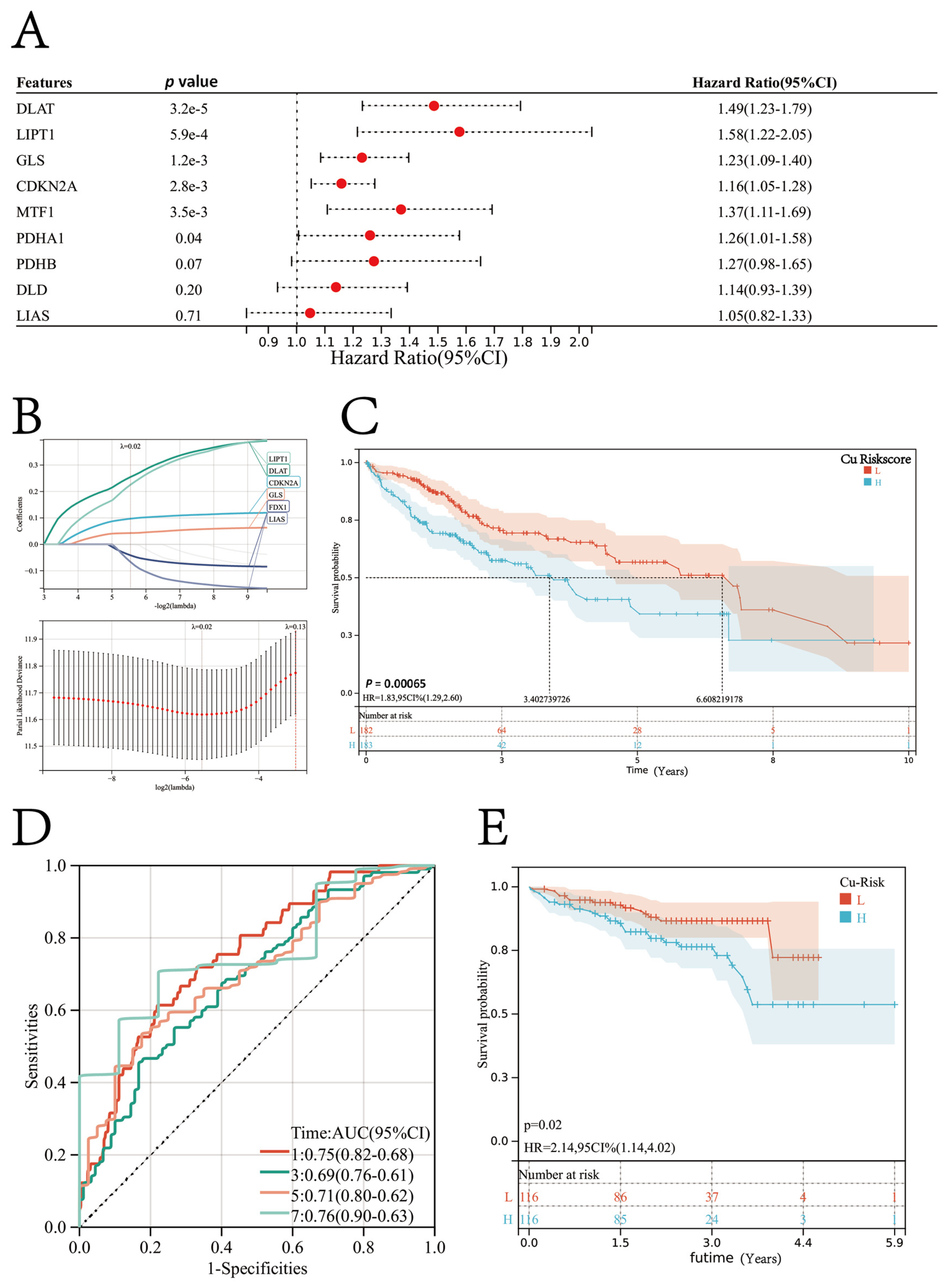
3.4. Identification of Candidate CSGSs using WGCNA and Machine Learning Techniques
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, Y.; Chen, H.; Gow, W.; Ma, L.; Jin, Y.; Hui, B.; Yang, Z.; Wang, Z. Potential biomarkers Ang II/AT1R and S1P/S1PR1 predict the prognosis of hepatocellular carcinoma. Oncol. Lett. 2020, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R. Deciphering Tumor Heterogeneity in Hepatocellular Carcinoma (HCC)—Multi-Omic and Singulomic Approaches. Semin. Liver Dis. 2021, 41, 9–18. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ho, Y.-J.; Salomao, M.A.; Dapito, D.H.; Bartolome, A.; Schwabe, R.F.; Lee, J.-S.; Lowe, S.W.; Pajvani, U.B. Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J. Hepatol. 2021, 74, 613–626. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- Meury, M.; Knop, M.; Seebeck, F.P. Structural Basis for Copper-Oxygen Mediated C-H Bond Activation by the Formylglycine-Generating Enzyme. Angew. Chem. Int. Ed. 2017, 56, 8115–8119. [Google Scholar] [CrossRef]
- Pal, A. Copper toxicity induced hepatocerebral and neurodegenerative diseases: An urgent need for prognostic biomarkers. Neurotoxicology 2014, 40, 97–101. [Google Scholar] [CrossRef]
- Li, Y. Copper homeostasis: Emerging target for cancer treatment. IUBMB Life 2020, 72, 1900–1908. [Google Scholar] [CrossRef]
- Pavithra, V.; Sathisha, T.G.; Kasturi, K.; Mallika, D.S.; Amos, S.J.; Ragunatha, S. Serum Levels of Metal Ions in Female Patients with Breast Cancer. J. Clin. Diagn. Res. 2015, 9, BC25–BC27. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, M.; Zhao, Y. Association between serum copper levels and cervical cancer risk: A meta-analysis. Biosci. Rep. 2018, 38, BSR20180161. [Google Scholar] [CrossRef]
- Yaman, M.; Kaya, G.; Simsek, M. Comparison of trace element concentrations in cancerous and noncancerous human endometrial and ovary tissues. Int. J. Gynecol. Cancer 2007, 17, 220–228. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Q. Association between serum copper levels and lung cancer risk: A meta-analysis. J. Int. Med. Res. 2018, 46, 4863–4873. [Google Scholar] [CrossRef]
- Grubman, A.; White, A.R. Copper as a key regulator of cell signalling pathways. Expert Rev. Mol. Med. 2014, 16, e11. [Google Scholar] [CrossRef]
- Blockhuys, S.; Wittung-Stafshede, P. Roles of Copper-Binding Proteins in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 871. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.; Merajver, S.D.; Yoo, S.; Dick, R.D.; Brewer, G.J.; Lee, J.S.-J.; Teknos, T.N. Inhibition of the Growth of Squamous Cell Carcinoma by Tetrathiomolybdate-Induced Copper Suppression in a Murine Model. Arch. Otolaryngol. Neck Surg. 2003, 129, 781–785. [Google Scholar] [CrossRef]
- Cox, C.; Teknos, T.N.; Barrios, M.; Brewer, G.J.; Dick, R.D.; Merajver, S.D.; Bs, R.D.D. The Role of Copper Suppression as an Antiangiogenic Strategy in Head and Neck Squamous Cell Carcinoma. Laryngoscope 2001, 111, 696–701. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Yan, M.; Wang, H.; Zhang, C. Novel copper complexes as potential proteasome inhibitors for cancer treatment. Mol. Med. Rep. 2017, 15, 3–11. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics; Springer: New York, NY, USA, 2016; pp. 93–110. [Google Scholar]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium; Campbell, P.J.; Getz, G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wang, M.; Wang, L.; Pu, L.; Li, K.; Feng, T.; Zheng, P.; Li, S.; Sun, M.; Yao, Y.; Jin, L. LncRNAs related key pathways and genes in ischemic stroke by weighted gene co-expression network analysis (WGCNA). Genomics 2020, 112, 2302–2308. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R Package for Prediction of Clinical Chemotherapeutic Response from Tumor Gene Expression Levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Engebretsen, S.; Bohlin, J. Statistical predictions with glmnet. Clin. Epigenetics 2019, 11, 123. [Google Scholar] [CrossRef]
- Hanko, M.; Grendár, M.; Snopko, P.; Opšenák, R.; Šutovský, J.; Benčo, M.; Soršák, J.; Zeleňák, K.; Kolarovszki, B. Random Forest–Based Prediction of Outcome and Mortality in Patients with Traumatic Brain Injury Undergoing Primary Decompressive Craniectomy. World Neurosurg. 2021, 148, e450–e458. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Zhang, W.; Che, Y.; Bai, W.; Huang, G. LASSO-based Cox-PH model identifies an 11-lncRNA signature for prognosis prediction in gastric cancer. Mol. Med. Rep. 2018, 18, 5579–5593. [Google Scholar] [CrossRef] [PubMed]
- Gelbard, R.B.; Hensman, H.; Schobel, S.; Khatri, V.; Tracy, B.M.; Dente, C.J.; Buchman, T.; Kirk, A.; Elster, E. Random forest modeling can predict infectious complications following trauma laparotomy. J. Trauma Inj. Infect. Crit. Care 2019, 87, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Navani, S. Manual evaluation of tissue microarrays in a high-throughput research project: The contribution of Indian surgical pathology to the Human Protein Atlas (HPA) project. Proteomics 2016, 16, 1266–1270. [Google Scholar] [CrossRef]
- Chan, B.K.C. Data Analysis Using R Programming. Biostat. Hum. Genet. Epidemiol. 2018, 1082, 47–122. [Google Scholar] [CrossRef]
- Rich, J.T.; Neely, J.G.; Paniello, R.C.; Voelker, C.C.J.; Nussenbaum, B.; Wang, E.W. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Neck Surg. 2010, 143, 331–336. [Google Scholar] [CrossRef]
- Mandrekar, J.N. Receiver Operating Characteristic Curve in Diagnostic Test Assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Marsman, M.; Wagenmakers, E.-J. Analytic posteriors for Pearson’s correlation coefficient. Stat. Neerlandica 2017, 72, 4–13. [Google Scholar] [CrossRef]
- Groth, D.; Hartmann, S.; Klie, S.; Selbig, J. Principal Components Analysis. Comput. Toxicol. Vol. II 2013, 930, 527–547. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Y.; Guo, X.; Du, Y.; Zhu, Q.; Wang, X.; Duan, C. FDX1 can Impact the Prognosis and Mediate the Metabolism of Lung Adenocarcinoma. Front. Pharmacol. 2021, 12, 749134. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, H.; Yang, L.; Yi, P.; Wang, Q.; Huang, D. The role of FDX1 in granulosa cell of Polycystic ovary syndrome (PCOS). BMC Endocr. Disord. 2021, 21, 119. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Y.; Yang, L.; Sun, L.; Wen, Y.; Huang, Y.; Gao, K.; Yang, W.; Bai, F.; Ling, L.; et al. PM2.5 promotes NSCLC carcinogenesis through translationally and transcriptionally activating DLAT-mediated glycolysis reprograming. J. Exp. Clin. Cancer Res. 2022, 41, 229. [Google Scholar] [CrossRef] [PubMed]
- Sievers, P.; Hielscher, T.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Berghoff, A.S.; Neidert, M.C.; Wirsching, H.-G.; Mawrin, C.; Ketter, R.; et al. CDKN2A/B homozygous deletion is associated with early recurrence in meningiomas. Acta Neuropathol. 2020, 140, 409–413. [Google Scholar] [CrossRef]
- Ji, L.; Zhao, G.; Zhang, P.; Huo, W.; Dong, P.; Watari, H.; Jia, L.; Pfeffer, L.M.; Yue, J.; Zheng, J. Knockout of MTF1 Inhibits the Epithelial to Mesenchymal Transition in Ovarian Cancer Cells. J. Cancer 2018, 9, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.Q.J.; Ow, G.S.; Kuznetsov, V.A.; Chong, S.; Lim, Y.P. DLAT subunit of the pyruvate dehydrogenase complex is upregulated in gastric can-cer-implications in cancer therapy. Am. J. Transl. Res. 2015, 7, 1140–1151. [Google Scholar]
- Hsieh, Y.-H.; Hsu, J.-L.; Su, I.-J.; Huang, W. Genomic instability caused by hepatitis B virus: Into the hepatoma inferno. Front. Biosci. 2011, 16, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Scalise, J.R.; Poças, R.C.G.; Caneloi, T.P.; Lopes, C.O.; Kanno, D.T.; Marques, M.G.; Valdivia, J.C.M.; Maximo, F.R.; Pereira, J.A.; Ribeiro, M.L.; et al. DNA Damage Is a Potential Marker for TP53 Mutation in Colorectal Carcinogenesis. J. Gastrointest. Cancer 2016, 47, 409–416. [Google Scholar] [CrossRef]
- Lindemann, A.; Takahashi, H.; Patel, A.; Osman, A.; Myers, J. Targeting the DNA Damage Response in OSCC with TP53 Mutations. J. Dent. Res. 2018, 97, 635–644. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Stover, E.H.; Fuh, K.; Konstantinopoulos, P.A.; Matulonis, U.A.; Liu, J.F. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol. Oncol. 2020, 159, 887–898. [Google Scholar] [CrossRef]
- Wandt, V.K.; Winkelbeiner, N.; Bornhorst, J.; Witt, B.; Raschke, S.; Simon, L.; Ebert, F.; Kipp, A.P.; Schwerdtle, T. A matter of concern—Trace element dyshomeostasis and genomic stability in neurons. Redox Biol. 2021, 41, 101877. [Google Scholar] [CrossRef] [PubMed]
- Finke, H.; Winkelbeiner, N.; Lossow, K.; Hertel, B.; Wandt, V.K.; Schwarz, M.; Pohl, G.; Kopp, J.F.; Ebert, F.; Kipp, A.; et al. Effects of a Cumulative, Suboptimal Supply of Multiple Trace Elements in Mice: Trace Element Status, Genomic Stability, Inflammation, and Epigenetics. Mol. Nutr. Food Res. 2020, 64, e2000325. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Matsuno, Y. Genomic destabilization and its associated mutagenesis increase with senescence-associated phenotype expression. Cancer Sci. 2020, 112, 515–522. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Boilan, E.; Winant, V.; Dumortier, E.; Piret, J.-P.; Bonfitto, F.; Osiewacz, H.D.; Debacq-Chainiaux, F.; Toussaint, O. Role of p38MAPK and oxidative stress in copper-induced senescence. Age 2013, 35, 2255–2271. [Google Scholar] [CrossRef]
- Matos, L.; Gouveia, A.M.; Almeida, H. Resveratrol Attenuates Copper-Induced Senescence by Improving Cellular Proteostasis. Oxidative Med. Cell. Longev. 2017, 2017, 3793817. [Google Scholar] [CrossRef]
- Munk, R.; Panda, A.C.; Grammatikakis, I.; Gorospe, M.; Abdelmohsen, K. Senescence-Associated MicroRNAs. Int. Rev. Cell Mol. Biol. 2017, 334, 177–205. [Google Scholar] [CrossRef]
- Mayer, R.L.; Schwarzmeier, J.D.; Gerner, M.C.; Bileck, A.; Mader, J.C.; Meier-Menches, S.M.; Gerner, S.M.; Schmetterer, K.G.; Pukrop, T.; Reichle, A.; et al. Proteomics and metabolomics identify molecular mechanisms of aging potentially predisposing for chronic lymphocytic leukemia. Mol. Cell. Proteom. 2018, 17, 290–303. [Google Scholar] [CrossRef]
- Tong, Y.; Guo, D.; Lin, S.-H.; Liang, J.; Yang, D.; Ma, C.; Shao, F.; Li, M.; Yu, Q.; Jiang, Y.; et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol. Cell 2021, 81, 2303–2316.e8. [Google Scholar] [CrossRef]
- Crane, E.K.; Kwan, S.-Y.; Izaguirre, D.I.; Tsang, Y.T.M.; Mullany, L.K.; Zu, Z.; Richards, J.S.; Gershenson, D.M.; Wong, K.-K. Nutlin-3a: A Potential Therapeutic Opportunity for TP53 Wild-Type Ovarian Carcinomas. PLoS ONE 2015, 10, e0135101. [Google Scholar] [CrossRef]
- Lai, Z.-Y.; Tsai, K.-Y.; Chang, S.-J.; Chuang, Y.-J. Gain-of-Function Mutant TP53 R248Q Overexpressed in Epithelial Ovarian Carcinoma Alters AKT-Dependent Regulation of Intercellular Trafficking in Responses to EGFR/MDM2 Inhibitor. Int. J. Mol. Sci. 2021, 22, 8784. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Morizane, C.; Ueno, M.; Okusaka, T.; Ishii, H.; Furuse, J. Chemotherapy for hepatocellular carcinoma: Current status and future perspectives. Jpn. J. Clin. Oncol. 2018, 48, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Xi, G.-M.; Zhang, B.; Wu, Y.; Liu, H.-B.; Liu, Y.-F.; Xu, W.-J.; Zhu, Q.; Cai, F.; Zhou, Z.-J.; et al. NCAPG2 promotes tumour proliferation by regulating G2/M phase and associates with poor prognosis in lung adenocarcinoma. J. Cell. Mol. Med. 2017, 21, 665–676. [Google Scholar] [CrossRef]
- Lee, J.; Choi, A.; Cho, S.; Jun, Y.; Na, D.; Lee, A.; Jang, G.; Kwon, J.Y.; Kim, J.; Lee, S.; et al. Genome-scale CRISPR screening identifies cell cycle and protein ubiquitination processes as druggable targets for erlotinib-resistant lung cancer. Mol. Oncol. 2021, 15, 487–502. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Chen, H.; Wang, Y.; Qian, Y. Novel Molecular Subtyping Scheme Based on In Silico Analysis of Cuproptosis Regulator Gene Patterns Optimizes Survival Prediction and Treatment of Hepatocellular Carcinoma. J. Clin. Med. 2023, 12, 5767. https://doi.org/10.3390/jcm12185767
Jiang H, Chen H, Wang Y, Qian Y. Novel Molecular Subtyping Scheme Based on In Silico Analysis of Cuproptosis Regulator Gene Patterns Optimizes Survival Prediction and Treatment of Hepatocellular Carcinoma. Journal of Clinical Medicine. 2023; 12(18):5767. https://doi.org/10.3390/jcm12185767
Chicago/Turabian StyleJiang, Heng, Hao Chen, Yao Wang, and Yeben Qian. 2023. "Novel Molecular Subtyping Scheme Based on In Silico Analysis of Cuproptosis Regulator Gene Patterns Optimizes Survival Prediction and Treatment of Hepatocellular Carcinoma" Journal of Clinical Medicine 12, no. 18: 5767. https://doi.org/10.3390/jcm12185767