The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies


Abstract
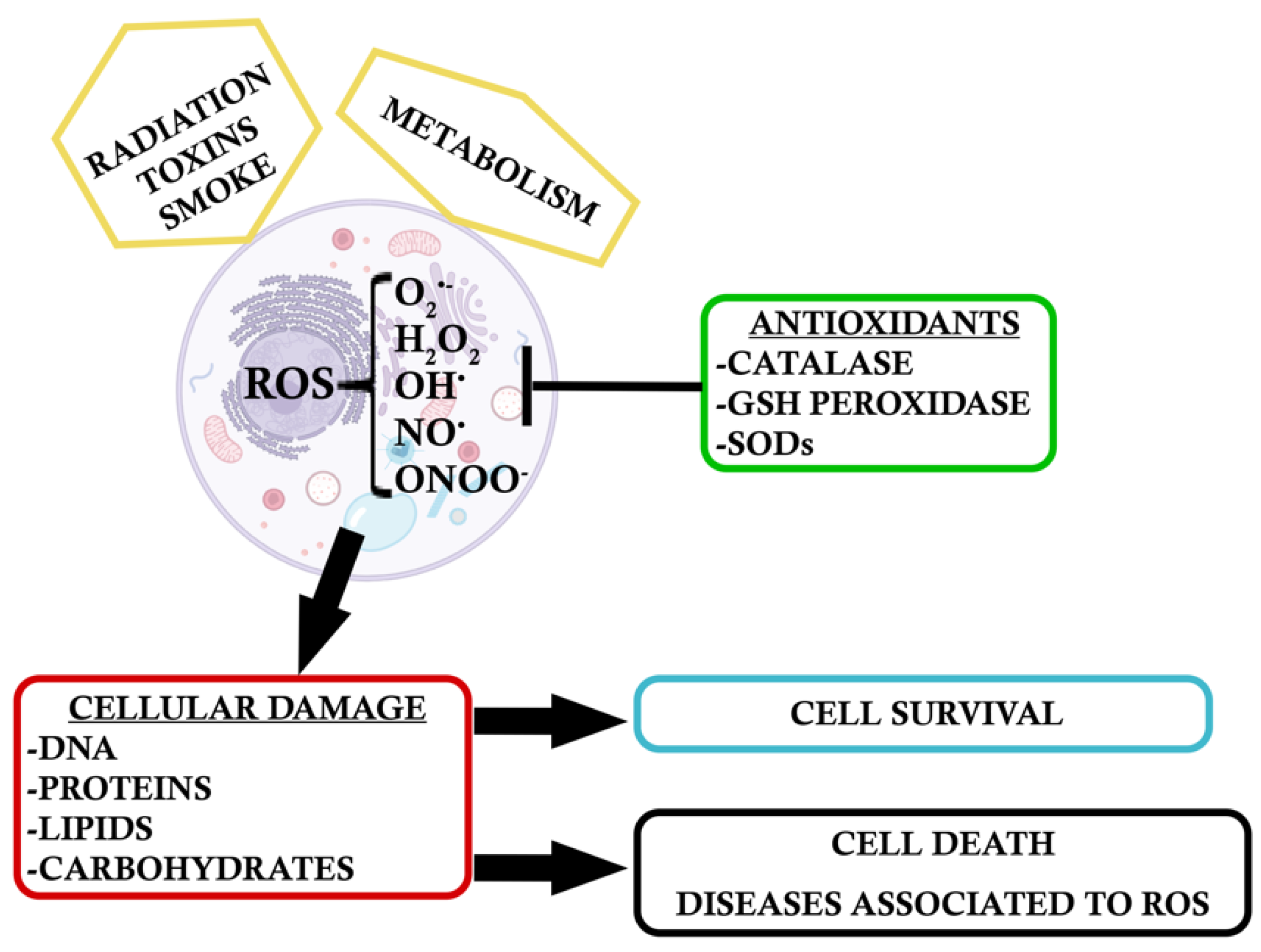
:1. Introduction
2. Environmental Oxidative Stress
2.1. Air Pollution and Cigarrete Smoke
2.2. Light-Induced Oxidative Stress
3. Intrinsic Response to Oxidative Stress
3.1. Inflammation and Gliosis
3.2. Endoplasmic Reticulum Stress
3.3. High Metabolic Rate of the Retina
4. Endogenous Antioxidants: Dual Role of NF-κB and the Antioxidant NRF2
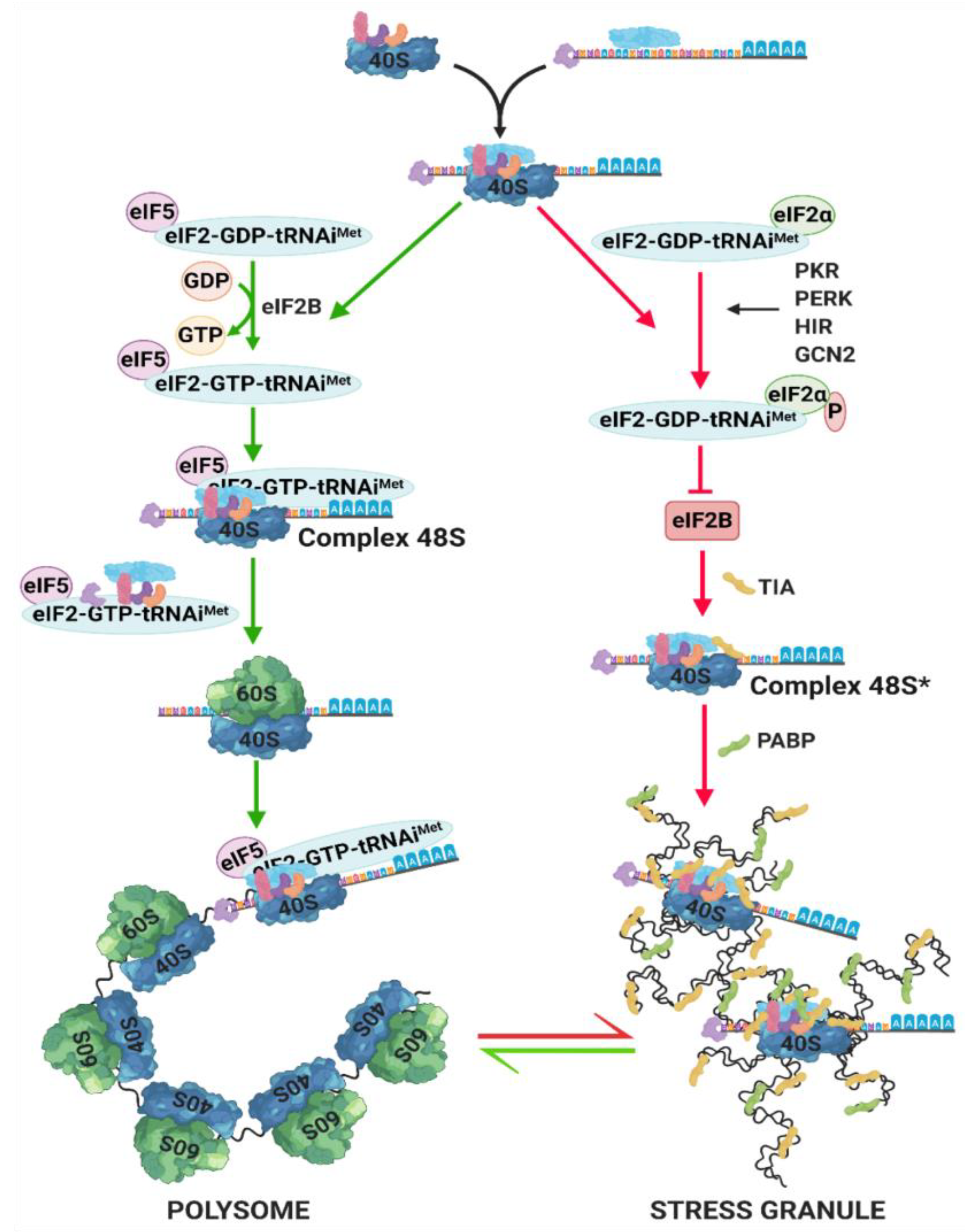
5. Stress Granules (SG) Formation
6. Oxidative Stress Impact on Autophagy and Mitophagy
6.1. Autophagy and Oxidative Stress
6.2. Mitophagy and Oxidative Stress
6.3. Cross-Talk between Autophagy and the Endoplasmic Reticulum (ER)-Mitochondria Axis in Oxidative Stress
7. Oxidative Stress Effect on Retinal Lipid Peroxidation
8. Oxidative Stress Induces DNA Damage and Mutations
9. Oxidative Stress, Genetic Factors, and Prevalent Retinopathies
9.1. Oxidative Stress and Glaucoma
9.2. Oxidative Stress and Age-Related Macular Degeneration (AMD)
9.3. Oxidative Stress and Diabetic Retinopathy (DR)
10. Oxidative Stress, Genetics, and Inherited Rare Diseases of the Retina
10.1. Oxidative Stress and Retinitis Pigmentosa (RP)
10.2. Oxidative Stress and Leber Hereditary Optic Neuropathy (LHON)
11. Potential Therapies for Retinal Diseases in front of Oxidative Stress
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Ivano, A.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Brüne, B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid. Redox Signal. 2005, 7, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Rivera-Del Valle, N.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M. Oxidants, antioxidants and thiol redox switches in the control of regulated cell death pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.J.K. Oxidative stress and free radicals. J. Mol. Struct. 2003, 666–667, 387–392. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Chua, S.Y.L.; Khawaja, A.P.; Morgan, J.; Strouthidis, N.; Reisman, C.; Dick, A.D.; Khaw, P.T.; Patel, P.J.; Foster, P.J.; for the UK Biobank Eye and Vision Consortium. The relationship between ambient atmospheric fine particulate matter (PM2.5) and glaucoma in a large community cohort. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4915–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, M.; Marín-Castaño, M.E. Nicotine increases the VEGF/PEDF ratio in retinal pigment epithelium: A possible mechanism for CNV in passive smokers with AMD. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3842–3853. [Google Scholar] [CrossRef] [Green Version]
- Wills, N.K.; Sadagopa Ramanujam, V.M.; Chang, J.; Kalariya, N.; Lewis, J.R.; Weng, T.-X.; van Kuijk, F.J.G.M. Cadmium accumulation in the human retina: Effects of age, gender, and cellular toxicity. Exp. Eye Res. 2008, 86, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bertram, K.M.; Baglole, C.J.; Phipps, R.P.; Libby, R.T. Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: Implications for age-related macular degeneration. Am. J. Physiol. 2009, 297, 1200–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksheeva, V.E.; Tiulina, V.V.; Tikhomirova, N.K.; Gancharova, O.S.; Komarov, S.V.; Philippov, P.P.; Zamyatnin, A.A., Jr.; Senin, I.I.; Zernii, E.Y. Suppression of light-induced oxidative stress in the retina by mitochondria-targeted antioxidant. Antioxidants 2018, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.K.; Singh, M.; Petit Homme, R.; Majumder, A.; Sandhu, H.S.; Tyagi, S.C. A hypothesis for treating inflammation and oxidative stress with hydrogen sulfide during age-related macular degeneration. Int. J. Ophthalmol. 2018, 11, 881–887. [Google Scholar] [CrossRef]
- Samardzija, M.; Todorova, V.; Gougoulakis, L.; Barben, M.; Nötzli, S.; Klee, K.; Storti, F.; Gubler, A.; Imsand, C.; Grimm, C. Light stress affects cones and horizontal cells via rhodopsin-mediated mechanisms. Exp. Eye Res. 2019, 186, 107719. [Google Scholar] [CrossRef]
- Rohowetz, L.J.; Kraus, J.G.; Koulen, P. Reactive oxygen species-mediated damage of retinal neurons: Drug development targets for therapies of chronic neurodegeneration of the retina. Int. J. Mol. Sci. 2018, 19, 3362. [Google Scholar] [CrossRef] [Green Version]
- Olchawa, M.M.; Herrnreiter, A.M.; Skumatz, C.M.B.; Zareba, M.; Sarna, T.S.; Burke, J.M. Photosensitized oxidative stress to ARPE-19 cells decreases protein receptors that mediate photoreceptor outer segment phagocytosis. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2276–2287. [Google Scholar] [CrossRef] [Green Version]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef]
- Detrick, B.; Hooks, J.J. The RPE cell and the immune system. In Retinal Pigment Epithelium in Health and Disease; Klettner, A., Dithmar, S., Eds.; Springer: Cham, Switzerland, 2020; pp. 101–114. [Google Scholar] [CrossRef] [Green Version]
- Rashid, K.; Akhtar-Schäfer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A Journey Into the Retina: Müller Glia Commanding Survival and Death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic Reticulum Stress and the Unfolded Protein Responses in Retinal Degeneration. Exp. Eye Res. 2014, 125, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, J.J.; Yu, Q.; Wang, M.; Zhang, S.X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009, 583, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Li, J.; Chen, Y.; Wang, J.J.; Ratan, R.; Zhang, S.X. Activation of Endoplasmic Reticulum Stress by Hyperglycemia Is Essential for Müller Cell–Derived Inflammatory Cytokine Production in Diabetes. Diabetes 2012, 61, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cano, M.; Wang, J.J.; Li, J.; Huang, C.; Yu, Q.; Herbert, T.P.; Handa, J.T.; Zhang, S.X. Role of Unfolded Protein Response Dysregulation in Oxidative Injury of Retinal Pigment Epithelial Cells. Antioxid. Redox. Signal. 2014, 20, 2091–2106. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, J.; Zhang, S. Cigarette smoke and RPE injury: Role of oxidative stress and ER stress. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3198. [Google Scholar]
- Gorbatyuk, M.S.; Starra, C.R.; Gorbatyuk, O.S. Endoplasmic reticulum stress: New insights into the pathogenesis and treatment of retinal degenerative diseases. Prog. Retin. Eye Res. 2020, in press. [Google Scholar] [CrossRef]
- Augustine, G.J.; Chikaraishi, D.M.; Ehlers, M.D.; Einstein, G.; Fitzpatrick, D.; Hall, W.C.; Jarvis, E.; Katz, L.C.; Kauer, J.; LaMantia, A.S.; et al. Sensation and Sensory Processing in Neuroscience, 3rd ed.; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Punzo, C.; Xiong, W.; Ceplo, C.L. Loss of daylight vision in retinal degeneration: Are oxidative stress and metabolic dysregulation to blame? J. Biol. Chem. 2012, 287, 1642–1648. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.F.; Chakarova, C.F.; Abd El-Aziz, M.M.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, M.L.; Mérida, S.; Bosch-Morell, F.; Miranda, M.; Villar, V.M. Autophagy dysfunction and oxidative stress, two related mechanisms implicated in retinitis pigmentosa. Front. Physiol. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Saccà, S.S.; Roszkowska, A.M.; Izzotti, A. Environmental light and endogenous antioxidants as the main determinants of non-cancer ocular diseases. Mutat. Res. 2013, 752, 153–171. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2010, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingappan, K. NF-κB in Oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.; Kedersha, N. Stress granules. Curr. Biol. 2009, 19, R397–R398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence that ternary complex (eIF2-GTP-tRNAi Met)– deficient preinitiation complexes are core constituents. Mol. Biol. Cell 2002, 13, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courchaine, E.M.; Lu, A.; Neugebauer, K.M. Droplet organelles? EMBO J. 2016, 35, 1603–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Leeuwen, W.; Rabouille, C. Cellular stress leads to the formation of membraneless stress assemblies in eukaryotic cells. Traffic 2019, 20, 623–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namkoong, S.; Ho, A.; Woo, Y.M.; Kwak, H.; Lee, J.H. Systematic characterization of stress-induced RNA granulation. Mol. Cell 2018, 70, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.M.; Huang, D.Y.; Sekar, P.; Hsu, S.H.; Lin, W.W. Reactive oxygen species dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J. Biomed. Sci. 2019, 26, 40. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [Green Version]
- He, J.N.; Zhang, S.D.; Qu, Y.; Wang, H.L.; Tham, C.C.; Pang, C.P.; Chu, W.K. Rapamycin removes damaged mitochondria and protects human trabecular meshwork (TM-1) cells from chronic oxidative stress. Mol. Neurobiol. 2019, 56, 6586–6593. [Google Scholar] [CrossRef]
- Yang, X.; Pan, X.; Zhao, X.; Luo, J.; Xu, M.; Bai, D.; Hu, Y.; Liu, X.; Yu, Q.; Gao, D. Autophagy and age-related eye diseases. Biomed. Res. Int. 2019, 2019, 5763658. [Google Scholar] [CrossRef]
- Adornetto, A.; Parisi, V.; Morrone, L.A.; Corasaniti, M.T.; Bagetta, G.; Tonin, P.; Russo, R. The role of autophagy in glaucomatous optic neuropathy. Front. Cell Dev. Biol. 2020, 4, 121. [Google Scholar] [CrossRef] [Green Version]
- Mirra, S.; Marfany, G. Mitochondrial gymnastics in retinal cells: A resilience mechanism against oxidative stress and neurodegeneration. Adv. Exp. Med. Biol. 2019, 1185, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Kuang, H.Y. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy 2014, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, in press. [Google Scholar] [CrossRef]
- Boya, P. Why autophagy is good for retinal ganglion cells? Eye 2017, 31, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Hass, D.T.; Barnstable, C.J. Mitochondrial uncoupling protein 2 knock-out promotes mitophagy to decrease retinal ganglion cell death in a mouse model of glaucoma. J. Neurosci. 2019, 39, 3582–3596. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum–mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Lopilly Park, H.Y.; Kim, J.H.; Park, C. K. Different contributions of autophagy to retinal ganglion cell death in the diabetic and glaucomatous retinas. Sci. Rep. 2018, 8, 13321. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Scimone, C.; Rinaldi, C.; Aragona, P.; Briuglia, S.; D’Ascola, A.; D’Angelo, R.; Sidoti, A. Stargardt Phenotype associated with two ELOVL4 promoter variants and ELOVL4 downregulation: New possible perspective to etiopathogenesis? Investig. Ophthalmol. Vis. Sci. 2018, 59, 843–857. [Google Scholar] [CrossRef] [Green Version]
- Njie-Mbye, Y.F.; Kulkarni-Chitnis, M.; Opere, C.A.; Barrett, A.; Ohia, S.E. Lipid peroxidation: Pathophysiological and pharmacological implications in the eye. Front. Physiol. 2013, 4, 366. [Google Scholar] [CrossRef] [Green Version]
- Shibuki, H.; Katai, N.; Yodoi, J.; Uchida, K.; Yoshimura, N. Lipid Peroxidation and peroxynitrite in retinal ischemia–reperfusion injury. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3607–3614. [Google Scholar] [PubMed]
- Moiseyev, G.; Nikolaeva, O.; Chen, Y.; Farjo, K.; Takahashi, Y.; Ma, J.X. Inhibition of the visual cycle by A2E through direct interaction with RPE65 and implications in Stargardt disease. Proc. Natl. Acad. Sci. USA 2010, 107, 17551–17556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, L.; Bramanti, P.; Scimone, C.; Rinaldi, C.; Giorgianni, F.; Beranova-Giorgianni, S.; Koirala, D.; D’Angelo, R.; Sidoti, A. miRNAexpression profile of retinal pigment epithelial cells under oxidative stress conditions. FEBS Open Bio. 2018, 8, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, L.; D’Angelo, R.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; Scimone, C. Effects of A2E-induced oxidative stress on retinal epithelial cells: New insights on differential gene response and retinal dystrophies. Antioxidants 2020, 9, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, S.; Hjelmeland, L.M.; Handa, J.T. Oxidative stress–induced single-strand breaks in chromosomal telomeres of human retinal pigment epithelial cells in vitro. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2139–2144. [Google Scholar] [PubMed]
- Cai, J.; Nelson, K.C.; Wu, M.; Sternberg, P.; Jones, D.P. Oxidative damage and protection of the RPE. Prog. Retin. Eye Res. 2000, 19, 205–221. [Google Scholar] [CrossRef]
- Cortina, S.; Gordon, W.C.; Luki, W.J.; Bazan, N.G. Oxidative stress-induced retinal damage up-regulates DNA polymerase gamma and 8-oxoguanine-DNA-glycosylase in photoreceptor synaptic mitochondria. Exp. Eye Res. 2005, 81, 742–750. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α protects RPE cells of the aging retina against oxidative stress-induced degeneration through the regulation of senescence and mitochondrial quality control. the significance for AMD pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef] [Green Version]
- Nagar, S.; Noveral, S.M.; Trudler, D.; Lopez, K.M.; McKercher, S.R.; Han, X.; Yates, J.R.; Piña-Crespo, J.C.; Nakanishi, N.; Satoh, T.; et al. MEF2D haploinsufficiency downregulates the NRF2 pathway and renders photoreceptors susceptible to light-induced oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, E4048–E4056. [Google Scholar] [CrossRef] [Green Version]
- Tsuruma, K.; Nishimura, Y.; Kishi, S.; Shimazawa, M.; Tanaka, T.; Hara, H. SEMA4A mutations lead to susceptibility to light irradiation, oxidative stress, and ER stress in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6729–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidl, D.; Garhofer, G.; Schmetterer, L. The complex interaction between ocular perfusion pressure and ocular blood flow—Relevance for glaucoma. Exp. Eye Res. 2011, 93, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Vittitow, J.L.; Borrás, T. Expression of optineurin, a glaucoma-linked gene, is influenced by elevated intraocular pressure. Biochem. Biophys. Res. Commun. 2002, 298, 67–74. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Mederios, F.A. The pathophysiology and treatment of glaucoma. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Kroeber, M.; Ohlmann, A.; Russell, P.; Tamm, E.R. Transgenic studies on the role of optineurin in the mouse eye. Exp. Eye Res. 2006, 82, 1075–1085. [Google Scholar] [CrossRef]
- De Marco, N.; Buono, M.; Troise, F.; Diez-Roux, G. Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J. Biol. Chem. 2006, 281, 16147–16156. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I. Oxidative stress in age-related macular degeneration: NRF2 as therapeutic target. Front. Pharmacol. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of mitochondrial DNA damage in ROS-mediated pathogenesis of age-related macular degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life. Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Sitnilska, V.; Kersten, E.; Altay, L.; Schick, T.; Enders, P.; de Jong, E.K.; Langmann, Y.; Hoyng, C.B.; den Hollander, A.I.; Fauser, S. Major predictive factors for progression of early to late age-related macular degeneration. Ophthalmologica 2020, in press. [Google Scholar] [CrossRef]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.N.; Issa, P.C.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; El Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salvodelli, M.; Naud, M.C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 13873. [Google Scholar] [CrossRef] [Green Version]
- Sobrin, L.; Seddon, J.M. Nature and nurture- genes and environment- predict onset and progression of macular degeneration. Prog. Retin. Eye Res. 2014, 40, 1–15. [Google Scholar] [CrossRef]
- Mulfaul, K.; Ozaki, E.; Fernando, N.; Brennan, K.; Chirco, K.R.; Connolly, E.; Greene, C.; Maminishkis, A.; Salomon, R.G.; Linetsky, M.; et al. Toll-like receptor 2 facilitates oxidative damage-induced retinal degeneration. Cell Rep. 2020, 30, 2209–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.L.; Thompson, D.A.; et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 2015, 11, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Baird, L.; Holmström, K.M.; Meyer, C.J.; Abramov, A.Y. The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, O.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, M.; Thimmalappula, R.; Fujihara, M.; Nagai, N.; Sporn, M.; Wang, A.L.; Neufeld, A.H.; Biswal, S.; Handa, J.T. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vis. Res. 2010, 50, 652–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, M.; Nagai, N.; Sussan, T.E.; Biswal, S.; Handa, J.T. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS ONE 2008, 3, e3119. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Olvera-Montaño, C.; Castellanos-González, J.A.; Navarro-Partida, J.; Cardona-Muñoz, E.G.; López-Contreras, A.K.; Roman-Pintos, L.M.; Robles-Rivera, R.R.; Rodríguez-Carrizalez, A.D. Oxidative stress as the main target in diabetic retinopathy pathophysiology. J. Diabetes Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is there excess oxidative stress and damage in eyes of patients with Retinitis Pigmentosa? Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachsel-Moncho, L.; Benlloch-Navarro, S.; Fernández-Carbonell, A.; Ramírez-Lamelas, D.T.; Olivar, T.; Silvestre, D.; Poch, E.; Miranda, M. Oxidative stress and autophagy-related changes during retinal degeneration and development. Cell Death Dis. 2018, 9, 812. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef]
- Kroeger, H.; Chiang, W.C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case–control study in a Sicilian population. Mol. Biol. Rep. 2018, 45, 1349–1355. [Google Scholar] [CrossRef]
- Tuson, M.; Garanto, A.; Gonzàlez-Duarte, R.; Marfany, G. Overexpression of CERKL, a gene responsible for retinitis pigmentosa in humans, protects cells from apoptosis induced by oxidative stress. Mol. Vis. 2009, 15, 168–180. [Google Scholar]
- Fathinajafabadi, A.; Pérez-Jiménez, E.; Riera, M.; Knecht, E.; Gonzàlez-Duarte, R. CERKL, a retinal disease gene, encodes an mRNA-binding protein that localizes in compact and untranslated mRNPs associated with microtubules. PLoS ONE 2014, 9, e87898. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, L.; Zhang, J.; Huang, M.; Wong, F.; Liu, X.; Liu, F.; Cui, X.; Yang, G.; Chen, J.; et al. CERKL interacts with mitochondrial TRX2 and protects retinal cells from oxidative stress-induced apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Tuson, M.; Marfany, G.; Gonzàlez-Duarte, R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive Retinitis Pigmentosa (RP26). Am. J. Hum. Genet. 2004, 74, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garanto, A.; Riera, M.; Pomares, P.; Permanyer, J.; de Castro-Miró, M.; Sava, F.; Abril, J.F.; Marfany, G.; Gonzàlez-Duarte, R. High transcriptional complexity of the retinitis pigmentosa CERKL gene in human and mouse. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5202–5214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, Y.; Luo, H.; Zhang, K. Leber hereditary optic neuropathy and oxidative stress. Proc. Natl. Acad. Sci. USA 2012, 109, 19882–19883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelam, K.; Goenadi, C.J.; Lun, K.; Yip, C.C.; Eong, K.-G.A. Putative protective role of lutein and zeaxanthin in diabetic retinopathy. Br. J. Ophthalmol. 2017, 101, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Milani, A.; Basirnejad, M.; Shahbazi, S.; Bolhassani, A. Carotenoids: Biochemistry, pharmacology and treatment. Br. J. Pharmacol. 2017, 174, 1290–1324. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Cho, E.; Willett, W.C.; Sastry, S.M.; Schaumberg, D.A. Intakes of lutein, zeaxanthin, and other carotenoids and age-related macular degeneration during 2 decades of prospective follow-up. JAMA Ophthalmol. 2015, 133, 1415–1424. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, K.; Liu, R.; Pan, J.; Zhang, L.; Lu, X. Vitamins and mineral supplements for retinitis pigmentosa. J. Ophthalmol. 2019, 2019. [Google Scholar] [CrossRef]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, J.D.; Culotta, V.C. Battles with iron: Manganese in oxidative stress protection. J. Biol. Chem. 2012, 287, 13541–13548. [Google Scholar] [CrossRef] [Green Version]
- Vishwanathan, R.; Chung, M.; Johnson, E.J. A systematic review on zinc for the prevention and treatment of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3985–3998. [Google Scholar] [CrossRef]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Rabaza, V.; López-Pedrajas, R.; Almansa, I. Progesterone, lipoic acid, and sulforaphane as promising antioxidants for retinal diseases: A review. Antioxidants 2019, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, N.A.; Patlolla, J.M.R.; Zheng, L.; Agbaga, M.-P.; Tran, J.T.A.; Wicker, L.; Kasus-Jacobi, A.; Elliott, M.H.; Rao, C.V.; Anderson, R.E. Curcumin protects retinal cells from light-and oxidant stress-induced cell death. Free Radic. Biol. Med. 2009, 46, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Farajipour, H.; Rahimian, S.; Taghizadeh, M. Curcumin: A new candidate for retinal disease therapy? J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Zhang, X.; Tohari, A.M.; Marcheggiani, F. Therapeutic potential of co-enzyme Q10 in retinal diseases. Curr. Med. Chem. 2017, 24, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Neves Dátilo, M.; Ramos Sant’Ana, M.; Pedron Formigari, G.; Brito Rodrigues, P.; Pereira de Moura, L.; Sanchez Ramos da Silva, A.; Rochete Ropelle, E.; Rodrigo Pauli, J.; Esper Cintra, D. Omega-3 from flaxseed oil protects obese mice against diabetic retinopathy through GPR120 receptor. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Willett, W.C. w-3 Intake and visual acuity in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 2012, 130, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Downie, L.E.; Vingrys, A.J. Oral omega-3 supplementation lowers intraocular pressure in normotensive adults. Transl. Vis. Sci. Technol. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala-Vila, A.; Díaz-López, A.; Valls-Pedret, C.; Cofán, M.; García-Layana, A.; Lamuela-Raventós, R.M.; Castañer, O.; Zanon-Moreno, V.; Martinez-Gonzalez, M.A.; Toledo, E.; et al. Prevención con Dieta Mediterránea (PREDIMED) Investigators. Dietary marine ω-3 fatty acids and incident sight-threatening retinopathy in middle-aged and older individuals with type 2 diabetes: Prospective investigation from the PREDIMED trial. JAMA Ophthalmol. 2016, 134, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Namekata, K.; Kimura, A.; Guo, X.; Azuchi, Y.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis. 2015, 6, e1720–e1729. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.M.; Wu, Z.Z.; Huang, Y.T.; Hsieh, C.W.; Wung, B.S. Lycopene inhibits ICAM-1 expression and NF-κB activation by Nrf2-regulated cell redox state in human retinal pigment epithelial cells. Life Sci. 2016, 155, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amero, K.K.; Kondkar, A.A.; Chalam, K.V. Resveratrol and ophthalmic diseases. Nutrients 2016, 8, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, T.; González, M.V.; Roehl, I.; Wright, N.; Pañeda, C.; Jiménez, A.I. In vitro and in vivo efficacy of SYL040012, a novel siRNA compound for treatment of glaucoma. Mol. Ther. 2014, 22, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatrai, K.P.; Xin, H.; Nguyen, V.; Szarka, S.; Blazics, B.; Prokai, L.; Koulen, P. 17β-Estradiol eye drops protect the retinal ganglion cell layer and preserve visual function in an in vivo model of glaucoma. Mol. Pharm. 2013, 10, 3253–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpkins, J.W.; Wang, J.; Wang, X.; Perez, E.; Prokai, L.; Dykens, J.A. Mitochondria play a central role in estrogen-induced neuroprotection. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 69–83. [Google Scholar] [CrossRef]
- Gu, L.; Kwong, J.M.K.; Yadegari, D.; Yu, F.; Caprioli, J.; Piri, N. The effect of celastrol on the ocular hypertension-induced degeneration of retinal ganglion cells. Neurosci. Lett. 2018, 670, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Hu, Y.; Ma, J.X. Anti-inflammatory and antioxidant effects of SERPINA3K in the retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3943–3952. [Google Scholar] [CrossRef]
- Du, Y.; Tang, J.; Li, G.; Berti-Mattera, L.; Lee, C.A.; Bartkowski, D.; Gale, D.; Monahan, J.; Niesman, M.R.; Alton, G.; et al. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2158–2164. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Kelley, M.R.; Edwards, P.; Gao, H.; Qiao, X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014, 2, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Charkoudian, L.K.; Dentchev, T.; Lukinova, N.; Wolkow, N.; Dunaief, J.L.; Franz, K.J. Author information Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide. J. Inorg. Biochem. 2008, 102, 2130–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mian, F.; Kang, C.; Lee, S.; Choi, J.H.; Bae, S.S.; Kim, S.H.; Kim, Y.H.; Ryu, S.H.; Suh, P.G.; Kim, J.S.; et al. Cleavage of focal adhesion kinase is an early marker and modulator of oxidative stress-induced apoptosis. Chem. Biol. Interact. 2008, 171, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Toro, A.L.; Barber, A.J.; Dennis, M.D. REDD1 activates a ROS-generating feedback loop in the retina of diabetic mice. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.J.; Chao, Y.M.; Liu, N.C.; Chan, J.Y.H. Differential effects of bevacizumab, ranibizumab and aflibercept on cell viability, phagocytosis and mitochondrial bioenergetics of retinal pigment epithelial cell. Acta Ophthalmol. 2015, 93, 631–643. [Google Scholar] [CrossRef]
- Jiang, W.; Tang, L.; Zeng, J.; Chen, B. Adeno-associated virus mediated SOD gene therapy protects the retinal ganglion cells from chronic intraocular pressure elevation induced injury via attenuating oxidative stress and improving mitochondrial dysfunction in a rat model. Am. J. Transl. Res. 2016, 8, 799–810. [Google Scholar]
- Berkowitz, B.A.; Kern, T.S.; Bissig, D.; Patel, P.; Bhatia, A.; Kefalov, V.J.; Roberts, R. Systemic retinaldehyde treatment corrects retinal oxidative stress, rod dysfunction, and impaired visual performance in diabetic mice. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6294–6303. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Fernández de la Cámara, C.; Salom, D.; Sequedo, M.D.; Hervás, D.; Marín-Lambíes, C.; Aller, E.; Jaijo, T.; Díaz-Llopis, M.; Millán, J.M.; Rodrigo, R. Altered antioxidant-oxidant status in the aqueous humor and peripheral blood of patients with retinitis pigmentosa. PLoS ONE 2013, 8, e74223. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Fernández de la Cámara, C.; Olivares-González, L.; Hervás, D.; Salom, D.; Millán, J.M.; Rodrigo, R. Infliximab reduces Zaprinast-induced retinal degeneration in cultures of porcine retina. J. Neuroinflammation 2014, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Siddam, A.D.; Barnum, C.E.; Janga, S.C.; Lachke, S.A. RNA-binding proteins in eye development and disease: Implication of conserved RNA granule components. Wiley Interdiscip. Rev. RNA. 2016, 7, 527–557. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Agent | Function | Retinal Dystrophy |
|---|---|---|
| Xanthophylls | Useful in protecting the retina from OS and ameliorating oxidative stress states | DR, AMD and RP [106,107,108,109] |
| Zinc and Manganese | Zinc supplements exhibit antioxidant and anti-inflammatory activities, delaying oxidative processes in the long term, and manganese is a cofactor of many antioxidant agents as SODs. | DR and AMD (https://clinicaltrials.gov/) [110,111,112] |
| Alpha-lipoic acid | Essential for mitochondrial function and also induces NRF2 binding to antioxidant response elements. which is important to slow down | DR, glaucoma, AMD, RP [113,114,115] |
| Curcumin | Increases the transcription of antioxidant enzymes and induces the activation of NRF2 helping to decrease oxidative stress in the retina. | Glaucoma, DR and AMD [116,117] |
| Ubiquinone or coenzyme Q10 | Inhibits the formation of free radicals reducing the effects of oxidative stress. Moreover, it activates eNOS and mitochondrial OXPHOS decreasing blood pressure. | AMD, RP, DR and glaucoma [118] |
| Omega 3 | Deficient consumption of omega-3 contributes to the degeneration of the retina. Additionally, PREDIMED (Prevention with Mediterranean Diet) is a clinical trial that followed for 6 years individuals with diabetes mellitus type 2. Patients whose diet included omega-3 polyunsaturated fatty acids, showed a 48% decreased incidence in DR. | Glaucoma, RP and DR [119,120,121,122] |
| Spermidine | Acts as a ROS scavenger by sequestering singlet oxygen and reactive species and reduced RGC death. | Glaucoma [123] |
| Lycopene | Decreases NF-κB activation and ROS production in RPE cells, while increasing Nrf2 and GSH levels. | AMD [124] |
| Resveratrol | Shows antiproliferative, antiangiogenic, antioxidant, endothelial, anti-inflammatory, antiplatelet, and neurogenic activity. | Glaucoma, cataract, AMD and DR [125] |
| Agent | Function | Retinal Dystrophy |
|---|---|---|
| Selective siRNAs (SYL040012) | Target specific β-receptors and have been demonstrated to decrease by 50% intraocular pressure 4 days after ocular administration, thus concomitantly reducing oxidative stress in RGCs. | Glaucoma [126] |
| 17β-estradiol | Inhibits ROS production, preserves ATP production and mitochondrial membrane potential, and decreases cellular and mitochondrial calcium loading. Moreover, estrogen’s neuroprotective effects have been tested in RGCs in an in vivo model of glaucoma | Glaucoma [127,128] |
| Celastrol | Intravitreal injection reduces IOP protecting RGC from death by activating the cellular antioxidant defense system, attenuating of microglial activation, inhibiting of tumor necrosis factor (TNF)-alpha, and nitric oxide synthase production. | Glaucoma [129] |
| SERPINA3K | Intravitreal injection of this serine protease inhibitor demonstrated to block the expression of VEGF and TNF-α. Moreover, it suppresses ROS production and upregulate manganese superoxide dismutase and glutathione levels. | DR [130] |
| PHA666859 | Blocks the p38 MAPK pathway, which plays a role in inflammatory processes. | DR [131] |
| E3330 | Exhibits RPE-protective effects downregulating intracellular ROS by attenuating the levels of NF-κB as well as the secretion of monocyte chemoattractant protein-1. | AMD [132] |
| BSIH | Iron favors ROS formation in the retina, for this reason, BSIH which is a protochelator is a potential therapeutic molecule. BSIH involves iron sequestration that occurs only when the cells are stressed by hydrogen peroxide. | AMD [133] |
| NAC (N-acetyl-l-cysteine) | Inhibits focal adhesion kinase, a kinase whose phosphorylation, induced by ROS, cause apoptosis. | DR [134,135] |
| Ranibizumab and aflibercept | Present a therapeutic effect against acrolein-induced oxidative cytotoxicity in human ARPE-19 cells via an increase in mitochondrial bioenergetics. | AMD [136] |
| AAV-SOD2 | gene therapy by overexpression of SOD2 introduced by adeno-associated viruses (AAV-SOD2) is able to attenuate oxidative stress and improve mitochondrial dysfunction of RGC and optic nerve secondary to glaucoma. | Glaucoma [137] |
| Retinoids | Systemically administration reduces retinal OS and correct rod dysfunction. | DR [138] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
B. Domènech, E.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. https://doi.org/10.3390/antiox9040347
B. Domènech E, Marfany G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants. 2020; 9(4):347. https://doi.org/10.3390/antiox9040347
Chicago/Turabian StyleB. Domènech, Elena, and Gemma Marfany. 2020. "The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies" Antioxidants 9, no. 4: 347. https://doi.org/10.3390/antiox9040347