Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch?

 , and
, and
Abstract
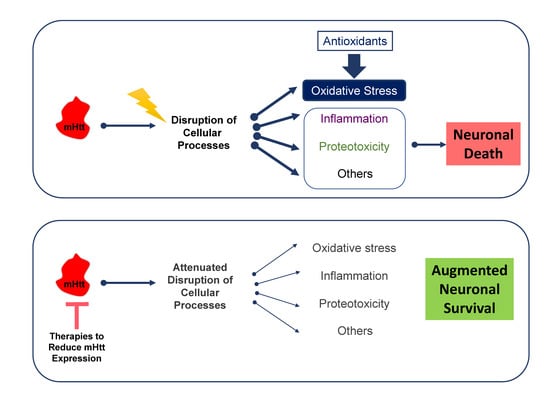
:
1. Introduction
2. Oxidative Stress
2.1. Free Radicals and Reactive Molecules
2.2. Free Radical Scavengers
3. Oxidative Stress and Huntington Disease
3.1. Mutant Huntingtin Alters Redox Homeostasis
3.2. Oxidative Stress and Neuroinflammation in HD
3.3. Oxidative Stress in Huntington Disease Patients
3.4. Oxidative Stress in Animal Models of polyQ Toxicity and Huntington Disease
3.4.1. Using Caenorhabditis elegans Models of polyQ Toxicity to Investigate Antioxidants as a Therapeutic Intervention
3.4.2. Studies Using Antioxidants to Treat Drosophila melanogaster Models of HD
3.4.3. Oxidative Stress and Antioxidant Therapies in Rodent Models of HD
3.5. Clinical Trials Using Antioxidants as a Therapeutic Intervention to Treat HD
4. Conclusions and Remarks
4.1. The Antioxidant Approach to Treat HD Is Promising but Requires Further Refinement
4.2. Do We Really Want to Remove Free Radicals, or Something Else?
4.3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative Stress and Cell Signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell Signaling: H2O2, a Necessary Evil for Cell Signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. IJMS 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Prousek, J. Fenton chemistry in biology and medicine. Pure Appl. Chem. 2007, 79, 2325–2338. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide Anion Radical (O·-2), Superoxide Dismutases, and Related Matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.E. Catalase: An old enzyme with a new role? Can. J. Biochem. Cell Biol. 1984, 62, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Ahmadinejad, F.; Geir Møller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.-S. Molecular Mechanisms behind Free Radical Scavengers Function against Oxidative Stress. Antioxidants 2017, 6, 51. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and antioxidants–quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Chen, L.; Poeggeler, B.; Manchester, L.; Reiter, R. Melatonin: A potent, endogenous hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Acuna-Castroviejo, D.; Burkhardt, S.; Karbownik, M. Melatonin: Mechanisms and actions as an antioxidant. Curr. Top. Biophys. 2000, 24, 171–184. [Google Scholar]
- Tan, D.-X.; Manchester, L.C.; Reiter, R.J.; Qi, W.-B.; Karbownik, M.; Calvo, J.R. Significance of Melatonin in Antioxidative Defense System: Reactions and Products. Neurosignals 2000, 9, 137–159. [Google Scholar] [CrossRef]
- Van Bladeren, P.J. Glutathione conjugation as a bioactivation reaction. Chem. Biol. Interact. 2000, 129, 61–76. [Google Scholar] [CrossRef]
- Beyer, R.E. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem. Cell Biol. 1992, 70, 390–403. [Google Scholar] [CrossRef]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [Green Version]
- Vonsattel, J.P.G.; DiFiglia, M. Huntington Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Illarioshkin, S.N.; Igarashi, S.; Onodera, O.; Markova, E.D.; Nikolskaya, N.N.; Tanaka, H.; Chabrashwili, T.Z.; Insarova, N.G.; Endo, K.; Ivanova-Smolenskaya, I.A.; et al. Trinucleotide repeat length and rate of progression of Huntington’s disease. Ann. Neurol. 1994, 36, 630–635. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Chiano, M.; Dodge, A.; Norbury, G.; Rosser, E.; Craufurd, D. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset of Huntington disease. Proc. Natl. Acad. Sci. USA 1997, 94, 3872–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network Organization of the Huntingtin Proteomic Interactome in Mammalian Brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Velier, J.; Kim, M.; Schwarz, C.; Kim, T.W.; Sapp, E.; Chase, K.; Aronin, N.; DiFiglia, M. Wild-Type and Mutant Huntingtins Function in Vesicle Trafficking in the Secretory and Endocytic Pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Zucker, B.; Luthi-Carter, R.; Kama, J.A.; Dunah, A.W.; Stern, E.A.; Fox, J.H.; Standaert, D.G.; Young, A.B.; Augood, S.J. Transcriptional dysregulation in striatal projection- and interneurons in a mouse model of Huntington’s disease: Neuronal selectivity and potential neuroprotective role of HAP1. Hum. Mol. Genet. 2005, 14, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthi-Carter, R. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2000, 9, 1259–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.W. Wild type huntingtin reduces the cellular toxicity of mutant huntingtin in mammalian cell models of Huntington’s disease. J. Med. Genet. 2001, 38, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Dragatsis, I.; Zeitlin, S.; Goldowitz, D. Wild-Type Huntingtin Plays a Role in Brain Development and Neuronal Survival. Mol. Neurobiol. 2003, 28, 259–276. [Google Scholar] [CrossRef]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haıëm, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.-L.; Trottier, Y. Proteases Acting on Mutant Huntingtin Generate Cleaved Products that Differentially Build Up Cytoplasmic and Nuclear Inclusions. Mol. Cell 2002, 10, 259–269. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [Green Version]
- DiGiovanni, L.F.; Mocle, A.J.; Xia, J.; Truant, R. Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum. Mol. Genet. 2016, 25, 3937–3945. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Seredenina, T.; Luthi-Carter, R. What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis. 2012, 45, 83–98. [Google Scholar] [CrossRef]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.D.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage Is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Batlevi, Y.; La Spada, A.R. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis. 2011, 43, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabofism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 1993, 43, 2689. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef]
- Yang, J.-L.; Weissman, L.; Bohr, V.A.; Mattson, M.P. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 2008, 7, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolobkova, Y.A.; Vigont, V.A.; Shalygin, A.V.; Kaznacheyeva, E.V. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 2017, 9, 34–46. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Hemachandra Reddy, P. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Bartzokis, G.; Lu, P.H.; Tishler, T.A.; Fong, S.M.; Oluwadara, B.; Finn, J.P.; Huang, D.; Bordelon, Y.; Mintz, J.; Perlman, S. Myelin Breakdown and Iron Changes in Huntington’s Disease: Pathogenesis and Treatment Implications. Neurochem. Res. 2007, 32, 1655–1664. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P.; Schapira, A.H.V.; Marsden, C.D.; Royal Kings and Queens Parkinson’s Disease Research Group. Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. Ann. Neurol. 1992, 32, S94–S100. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995, 14, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Reijonen, S.; Kukkonen, J.P.; Hyrskyluoto, A.; Kivinen, J.; Kairisalo, M.; Takei, N.; Lindholm, D.; Korhonen, L. Downregulation of NF-κB signaling by mutant huntingtin proteins induces oxidative stress and cell death. Cell. Mol. Life Sci. 2010, 67, 1929–1941. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 509, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. Neurodegeneration in Huntington’s disease involves loss of cystathionine γ-lyase. Cell Cycle 2014, 13, 2491–2493. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, R.K.; Hennessey, T.; Johri, A.; Tiwari, S.K.; Mishra, D.; Agarwal, S.; Kim, Y.S.; Beal, M.F. Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 3474–3488. [Google Scholar] [CrossRef] [Green Version]
- McConoughey, S.J.; Basso, M.; Niatsetskaya, Z.V.; Sleiman, S.F.; Smirnova, N.A.; Langley, B.C.; Mahishi, L.; Cooper, A.J.L.; Antonyak, M.A.; Cerione, R.A.; et al. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol. Med. 2010, 2, 349–370. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s Disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [Green Version]
- Mangiarini, L.; Sathasivam, K.; Mahal, A.; Mott, R.; Seller, M.; Bates, G.P. Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat. Genet. 1997, 15, 197–200. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.C.; Truant, R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum. Mol. Genet. 2017, 26, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-Y.; Fang, Z.-H.; Yu, Z.-X.; Wang, C.-E.; Li, S.-H.; Li, X.-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wake, H.; Fields, R.D. Physiological function of microglia. Neuron Glia Biol. 2011, 7, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting: Quantifying neurons and glia in human brain. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Ben Haim, L.; Carrillo-de Sauvage, M.-A.; Ceyzériat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease: Table 1. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [Green Version]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxidative Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Gray, M. Astrocytes in Huntington’s Disease. In Neuroglia in Neurodegenerative Diseases; Verkhratsky, A., Ho, M.S., Zorec, R., Parpura, V., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1175, pp. 355–381. ISBN 9789811399121. [Google Scholar]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon? J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef]
- Hsieh, H.-L.; Yang, C.-M. Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. BioMed Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Alam, Z.I.; Halliwell, B.; Jenner, P. No evidence for increased oxidative damage to lipids, proteins, or DNA in Huntington’s disease. J. Neurochem. 2000, 75, 840–846. [Google Scholar] [CrossRef]
- Ciancarelli, I.; De Amicis, D.; Di Massimo, C.; Di Scanno, C.; Pistarini, C.; D’Orazio, N.; Tozzi Ciancarelli, M.G. Peripheral biomarkers of oxidative stress and their limited potential in evaluation of clinical features of Huntington’s patients. Biomarkers 2014, 19, 452–456. [Google Scholar] [CrossRef]
- Duran, R.; Barrero, F.J.; Morales, B.; Luna, J.D.; Ramirez, M.; Vives, F. Oxidative stress and plasma aminopeptidase activity in Huntington’s disease. J. Neural. Transm. 2010, 117, 325–332. [Google Scholar] [CrossRef]
- Jędrak, P.; Mozolewski, P.; Węgrzyn, G.; Więckowski, M.R. Mitochondrial alterations accompanied by oxidative stress conditions in skin fibroblasts of Huntington’s disease patients. Metab. Brain Dis. 2018, 33, 2005–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejia, E.M.; Chau, S.; Sparagna, G.C.; Sipione, S.; Hatch, G.M. Reduced Mitochondrial Function in Human Huntington Disease Lymphoblasts is Not Due to Alterations in Cardiolipin Metabolism or Mitochondrial Supercomplex Assembly. Lipids 2016, 51, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Leavitt, B.R. Iron dysregulation in Huntington’s disease. J. Neurochem. 2014, 130, 328–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef] [Green Version]
- Pham, A.N.; Xing, G.; Miller, C.J.; Waite, T.D. Fenton-like copper redox chemistry revisited: Hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J. Catal. 2013, 301, 54–64. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar]
- Culetto, E. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 2000, 9, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Rudich, P.; Lamitina, T. Models and mechanisms of repeat expansion disorders: A worm’s eye view. J. Genet. 2018, 97, 665–677. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [Green Version]
- Faber, P.W.; Alter, J.R.; MacDonald, M.E.; Hart, A.C. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. USA 1999, 96, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Faber, P.W.; Voisine, C.; King, D.C.; Bates, E.A.; Hart, A.C. Glutamine/proline-rich PQE-1 proteins protect Caenorhabditis elegans neurons from huntingtin polyglutamine neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 17131–17136. [Google Scholar] [CrossRef] [Green Version]
- Machiela, E.; Dues, D.J.; Senchuk, M.M.; Van Raamsdonk, J.M. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol. Dis. 2016, 96, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Thabit, S.; Handoussa, H.; Roxo, M.; El Sayed, N.S.; Cestari de Azevedo, B.; Wink, M. Evaluation of antioxidant and neuroprotective activities of Cassia fistula (L.) using the Caenorhabditis elegans model. PeerJ 2018, 6, e5159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Zhang, J.; Li, H.; Wang, Q.; Xiao, L.; Weng, H.; Zhou, X.; Ma, C.W.; Ma, F.; Hu, M.; et al. Epimedium Polysaccharide Alleviates Polyglutamine-Induced Neurotoxicity in Caenorhabditis elegans by Reducing Oxidative Stress. Rejuvenation Res. 2017, 20, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, H.; Roxo, M.; Röhrig, T.; Richling, E.; Wang, X.; Wink, M. Anti-Aging and Antioxidant Potential of Paullinia cupana var. sorbilis: Findings in Caenorhabditis elegans Indicate a New Utilization for Roasted Seeds of Guarana. Medicines 2017, 4, 61. [Google Scholar] [CrossRef]
- Mohankumar, A.; Devagi, G.; Govindan, S.; Nivitha, S.; Dallemer, F.; Kalaivani, P.; Palanisamy, S.; Prabhakaran, R. Organoruthenium(II) Complexes Ameliorates Oxidative Stress and Impedes the Age Associated Deterioration in Caenorhabditis elegans through JNK-1/DAF-16 Signalling. Sci. Rep. 2018, 8, 7688. [Google Scholar] [CrossRef]
- Singh, N.K.; Sonani, R.R.; Awasthi, A.; Prasad, B.; Patel, A.R.; Kumar, J.; Madamwar, D. Phycocyanin moderates aging and proteotoxicity in Caenorhabditis elegans. J. Appl. Phycol. 2016, 28, 2407–2417. [Google Scholar] [CrossRef]
- Xiao, L.; Li, H.; Zhang, J.; Yang, F.; Huang, A.; Deng, J.; Liang, M.; Ma, F.; Hu, M.; Huang, Z. Salidroside Protects Caenorhabditis elegans Neurons from Polyglutamine-Mediated Toxicity by Reducing Oxidative Stress. Molecules 2014, 19, 7757–7769. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shi, R.; Li, H.; Xiang, Y.; Xiao, L.; Hu, M.; Ma, F.; Ma, C.W.; Huang, Z. Antioxidant and neuroprotective effects of Dictyophora indusiata polysaccharide in Caenorhabditis elegans. J. Ethnopharmacol. 2016, 192, 413–422. [Google Scholar] [CrossRef]
- Boasquívis, P.F.; Silva, G.M.M.; Paiva, F.A.; Cavalcanti, R.M.; Nunez, C.V.; de Paula Oliveira, R. Guarana (Paullinia cupana) Extract Protects Caenorhabditis elegans Models for Alzheimer Disease and Huntington Disease through Activation of Antioxidant and Protein Degradation Pathways. Oxidative Med. Cell. Longev. 2018, 2018, 9241308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Gómez, D.; Mora-Lorca, J.A.; Sáenz-Narciso, B.; Naranjo-Galindo, F.J.; Muñoz-Lobato, F.; Parrado-Fernández, C.; Goikolea, J.; Cedazo-Minguez, Á.; Link, C.D.; Neri, C.; et al. Loss of glutathione redox homeostasis impairs proteostasis by inhibiting autophagy-dependent protein degradation. Cell Death Differ. 2019, 26, 1545–1565. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladurner, A.; Schmitt, C.A.; Schachner, D.; Atanasov, A.G.; Werner, E.R.; Dirsch, V.M.; Heiss, E.H. Ascorbate stimulates endothelial nitric oxide synthase enzyme activity by rapid modulation of its phosphorylation status. Free Radic. Biol. Med. 2012, 52, 2082–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Stadnichuk, I.N.; Krasil’nikov, P.M.; Zlenko, D.V. Cyanobacterial Phycobilisomes and Phycobiliproteins. Mikrobiologiia 2015, 84, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Wang, Y.; Qiu, Q.; Deng, H.; Yuan, L.; Li, R.; Song, D.; Li, Y.Y.; Li, D.; Wang, Z. Salidroside protects human fibroblast cells from premature senescence induced by H2O2 partly through modulating oxidative status. Mech. Ageing Dev. 2010, 131, 723–731. [Google Scholar] [CrossRef]
- Hara, C.; Kiho, T.; Tanaka, Y.; Ukai, S. Anti-inflammatory activity and conformational behavior of a branched (1→3)-β-d-glucan from an alkaline extract of dictyophora indusiata fisch. Carbohydr. Res. 1982, 110, 77–87. [Google Scholar] [CrossRef]
- Lee, I.-K.; Yun, B.-S.; Han, G.; Cho, D.-H.; Kim, Y.-H.; Yoo, I.-D. Dictyoquinazols A, B, and C, new neuroprotective compounds from the mushroom Dictyophora indusiata. J. Nat. Prod. 2002, 65, 1769–1772. [Google Scholar] [CrossRef]
- Deng, C.; Hu, Z.; Fu, H.; Hu, M.; Xu, X.; Chen, J. Chemical analysis and antioxidant activity in vitro of a β-d-glucan isolated from Dictyophora indusiata. Int. J. Biol. Macromol. 2012, 51, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, L.; Martins, C.A.; Sampaio, G.R.; Monteiro, M.P.; César, L.A.M.; Mioto, B.M.; Mori, C.S.; Mendes, T.M.N.; Ribeiro, M.L.; Arçari, D.P.; et al. Bioavailability of catechins from guaraná (Paullinia cupana) and its effect on antioxidant enzymes and other oxidative stress markers in healthy human subjects. Food Funct. 2016, 7, 2970–2978. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Nonaka, G.-I.; Chen, R.-F.; Nishioka, I. Tannins and related compounds. LXI. Isolation and structures of novel bi- and triflavanoids from the leaves of Cassia fistula L. Chem. Pharm. Bull. 1988, 36, 39–47. [Google Scholar] [CrossRef] [Green Version]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratovitski, T.; Gucek, M.; Jiang, H.; Chighladze, E.; Waldron, E.; D’Ambola, J.; Hou, Z.; Liang, Y.; Poirier, M.A.; Hirschhorn, R.R.; et al. Mutant Huntingtin N-terminal Fragments of Specific Size Mediate Aggregation and Toxicity in Neuronal Cells. J. Biol. Chem. 2009, 284, 10855–10867. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-C.M.; Yoshihara, M.; Littleton, J.T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3224–3229. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.-Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Melkani, G.C.; Trujillo, A.S.; Ramos, R.; Bodmer, R.; Bernstein, S.I.; Ocorr, K. Huntington’s Disease Induced Cardiac Amyloidosis Is Reversed by Modulating Protein Folding and Oxidative Stress Pathways in the Drosophila Heart. PLoS Genet. 2013, 9, e1004024. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Ávila, M.; Fernández Vega, A.; de la Mata, M.; Delgado Pavón, A.; de Miguel, M.; Pérez Calero, C.; Villanueva Paz, M.; Cotán, D.; et al. Coenzyme q10 therapy. Mol. Syndromol. 2014, 5, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Bahadorani, S.; Hilliker, A.J. Antioxidants cannot suppress the lethal phenotype of a Drosophila melanogaster model of Huntington’s disease. Genome 2008, 51, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Akaishi, T.; Abe, K. Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc. Natl. Acad. Sci. USA 2006, 103, 16568–16573. [Google Scholar] [CrossRef] [Green Version]
- Burdo, J.; Schubert, D.; Maher, P. Glutathione production is regulated via distinct pathways in stressed and non-stressed cortical neurons. Brain Res. 2008, 1189, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Pfleger, C.; Friedman, L.; Vittorino, R.; Zhao, W.; Qian, X.; Conley, L.; Ho, L.; Pasinetti, G. Potential application of grape derived polyphenols in Huntington’s disease. Transl. Neurosci. 2010, 1, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Sreejayan, N.; Rao, M.N. Nitric oxide scavenging by curcuminoids. J. Pharm. Pharmacol. 1997, 49, 105–107. [Google Scholar] [CrossRef]
- Scapagnini, G.; Caruso, C.; Calabrese, V. Therapeutic potential of dietary polyphenols against brain ageing and neurodegenerative disorders. Adv. Exp. Med. Biol. 2010, 698, 27–35. [Google Scholar] [CrossRef]
- Chongtham, A.; Agrawal, N. Curcumin modulates cell death and is protective in Huntington’s disease model. Sci. Rep. 2016, 6, 18736. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.M.; Chan, H.Y.E.; Huang, Y.; Chen, Z.Y. Green tea catechins upregulate superoxide dismutase and catalase in fruit flies. Mol. Nutr. Food Res. 2007, 51, 546–554. [Google Scholar] [CrossRef]
- Varga, J.; Dér, N.P.; Zsindely, N.; Bodai, L. Green tea infusion alleviates neurodegeneration induced by mutant Huntingtin in Drosophila. Nutr. Neurosci. 2020, 23, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef]
- Crook, Z.R.; Housman, D. Huntington’s Disease: Can Mice Lead the Way to Treatment? Neuron 2011, 69, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckel, A.W.; Tang, V.; Nuttal, D.; Gary, K.; Elder, R. Altered neuronal nitric oxide synthase expression contributes to disease progression in Huntington’s disease transgenic mice. Brain Res. 2002, 939, 76–86. [Google Scholar] [CrossRef]
- Santamaría, A.; Pérez-Severiano, F.; Rodríguez-Martínez, E.; Maldonado, P.D.; Pedraza-Chaverri, J.; Ríos, C.; Segovia, J. Comparative analysis of superoxide dismutase activity between acute pharmacological models and a transgenic mouse model of Huntington’s disease. Neurochem. Res. 2001, 26, 419–424. [Google Scholar] [CrossRef]
- Pérez-Severiano, F.; Santamaría, A.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Ríos, C.; Segovia, J. Increased formation of reactive oxygen species, but no changes in glutathione peroxidase activity, in striata of mice transgenic for the Huntington’s disease mutation. Neurochem. Res. 2004, 29, 729–733. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2923–2933. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [Green Version]
- Clifford, J.J.; Drago, J.; Natoli, A.L.; Wong, J.Y.F.; Kinsella, A.; Waddington, J.L.; Vaddadi, K.S. Essential fatty acids given from conception prevent topographies of motor deficit in a transgenic model of Huntington’s disease. Neuroscience 2002, 109, 81–88. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport 2001, 12, 3371–3373. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic Effects of Coenzyme Q10 and Remacemide in Transgenic Mouse Models of Huntington’s Disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, R.J.; Andreassen, O.A.; Jenkins, B.G.; Dedeoglu, A.; Kuemmerle, S.; Kubilus, J.K.; Kaddurah-Daouk, R.; Hersch, S.M.; Beal, M.F. Neuroprotective Effects of Creatine in a Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. 2000, 20, 4389–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elifani, F.; Amico, E.; Pepe, G.; Capocci, L.; Castaldo, S.; Rosa, P.; Montano, E.; Pollice, A.; Madonna, M.; Filosa, S.; et al. Curcumin dietary supplementation ameliorates disease phenotype in an animal model of Huntington’s disease. Hum. Mol. Genet. 2019, 28, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Karpuj, M.V.; Becher, M.W.; Springer, J.E.; Chabas, D.; Youssef, S.; Pedotti, R.; Mitchell, D.; Steinman, L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat. Med. 2002, 8, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.H.; Barber, D.S.; Singh, B.; Zucker, B.; Swindell, M.K.; Norflus, F.; Buzescu, R.; Chopra, R.; Ferrante, R.J.; Kazantsev, A.; et al. Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J. Neurochem. 2004, 91, 413–422. [Google Scholar] [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Petrasch-Parwez, E.; Lee, D.-H.; Reick, C.; Arning, L.; Saft, C.; Gold, R.; Linker, R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS ONE 2011, 6, e16172. [Google Scholar] [CrossRef]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The Melatonin MT1 Receptor Axis Modulates Mutant Huntingtin-Mediated Toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, A.A.; Wood, N.I.; Williams, P.; Wipf, P.; Morton, A.J.; McMurray, C.T. XJB-5-131-mediated improvement in physiology and behaviour of the R6/2 mouse model of Huntington’s disease is age- and sex-dependent. PLoS ONE 2018, 13, e0194580. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Jenkins, B.G.; Ferrante, K.L.; Thomas, M.; Friedlich, A.; Browne, S.E.; Schilling, G.; Borchelt, D.R.; et al. Creatine Increases Survival and Delays Motor Symptoms in a Transgenic Animal Model of Huntington’s Disease. Neurobiol. Dis. 2001, 8, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Vamos, E.; Voros, K.; Vecsei, L.; Klivenyi, P. Neuroprotective effects of L-carnitine in a transgenic animal model of Huntington’s disease. Biomed. Pharmacother. 2010, 64, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Marks, E.; Chen, J.; Moline, J.; Barrows, L.; Raisbeck, M.; Volitakis, I.; Cherny, R.A.; Chopra, V.; Bush, A.I.; et al. Altered selenium status in Huntington’s disease: Neuroprotection by selenite in the N171-82Q mouse model. Neurobiol. Dis. 2014, 71, 34–42. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Lu, G.; Barakauskas, V.E.; Barr, A.M.; Honer, W.G.; Hayden, M.R.; Leavitt, B.R. Ethyl-EPA treatment improves motor dysfunction, but not neurodegeneration in the YAC128 mouse model of Huntington disease. Exp. Neurol. 2005, 196, 266–272. [Google Scholar] [CrossRef]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laço, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative Mitochondrial-Based Protective Effects of Resveratrol and Nicotinamide in Huntington’s Disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef]
- Xun, Z.; Rivera-Sánchez, S.; Ayala-Peña, S.; Lim, J.; Budworth, H.; Skoda, E.M.; Robbins, P.D.; Niedernhofer, L.J.; Wipf, P.; McMurray, C.T. Targeting of XJB-5-131 to Mitochondria Suppresses Oxidative DNA Damage and Motor Decline in a Mouse Model of Huntington’s Disease. Cell Rep. 2012, 2, 1137–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, A.; Holt, A.; Brown, C.; Cosme, C.; Wipf, P.; Gomez-Marin, A.; Castro, M.R.; Ayala-Peña, S.; McMurray, C.T. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum. Mol. Genet. 2016, 25, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, M.A.; Zhu, C.; Medvedeva, V.; Lerner, R.P.; Patassini, S.; Franich, N.R.; Maiti, P.; Frautschy, S.A.; Zeitlin, S.; Levine, M.S.; et al. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol. Neurodegener. 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, A.; Kanwal, A.; Banerjee, S.K.; Sandhir, R. Mitochondrial modulators in experimental Huntington’s disease: Reversal of mitochondrial dysfunctions and cognitive deficits. Neurobiol. Aging 2015, 36, 2186–2200. [Google Scholar] [CrossRef]
- Kašparová, S.; Sumbalová, Z.; Bystrický, P.; Kucharská, J.; Liptaj, T.; Mlynárik, V.; Gvozdjáková, A. Effect of coenzyme Q10 and vitamin E on brain energy metabolism in the animal model of Huntington’s disease. Neurochem. Int. 2006, 48, 93–99. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible nitric oxide modulation in protective effect of FK-506 against 3-nitropropionic acid-induced behavioral, oxidative, neurochemical, and mitochondrial alterations in rat brain. Drug Chem. Toxicol. 2010, 33, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Mehrotra, A.; Kamboj, S.S. Lycopene prevents 3-nitropropionic acid-induced mitochondrial oxidative stress and dysfunctions in nervous system. Neurochem. Int. 2010, 57, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-Acetylcysteine Reverses Mitochondrial Dysfunctions and Behavioral Abnormalities in 3-Nitropropionic Acid-Induced Huntington’s Disease. Neurodegener. Dis. 2012, 9, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Diwan, V.; Kaur, H.; Bhateja, D.; Singh, C.K.; Sharma, S.; Padi, S.S.V. Nicotinamide reverses behavioral impairments and provides neuroprotection in 3˗nitropropionic acid induced animal model of Huntington’s disease: Implication of oxidative stress-poly(ADP-ribose) polymerase pathway. Metab. Brain Dis. 2018, 33, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Mehrotra, A. Quercetin supplementation is effective in improving mitochondrial dysfunctions induced by 3-nitropropionic acid: Implications in Huntington’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, D.; Gangshettiwar, A. Combination of lycopene, quercetin and poloxamer188 alleviates anxiety and depression in 3-nitropropionic acid-induced Huntingtons disease in rats. J. Intercult. Ethnopharmacol. 2014, 3, 186–191. [Google Scholar] [CrossRef]
- Suganya, S.N.; Sumathi, T. Effect of rutin against a mitochondrial toxin, 3-nitropropionicacid induced biochemical, behavioral and histological alterations-a pilot study on Huntington’s disease model in rats. Metab. Brain Dis. 2017, 32, 471–481. [Google Scholar] [CrossRef]
- Bortolatto, C.F.; Jesse, C.R.; Wilhelm, E.A.; Chagas, P.M.; Nogueira, C.W. Organoselenium Bis Selenide Attenuates 3-Nitropropionic Acid-Induced Neurotoxicity in Rats. Neurotox. Res. 2013, 23, 214–224. [Google Scholar] [CrossRef]
- Tasset, I.; Pontes, A.J.; Hinojosa, A.J.; de la Torre, R.; Túnez, I. Olive oil reduces oxidative damage in a 3-nitropropionic acid-induced Huntington’s disease-like rat model. Nutr. Neurosci. 2011, 14, 106–111. [Google Scholar] [CrossRef]
- Kalonia, H.; Kumar, P.; Kumar, A. Targeting oxidative stress attenuates malonic acid induced Huntington like behavioral and mitochondrial alterations in rats. Eur. J. Pharmacol. 2010, 634, 46–52. [Google Scholar] [CrossRef]
- Miyamoto, M.; Coyle, J.T. Idebenone attenuates neuronal degeneration induced by intrastriatal injection of excitotoxins. Exp. Neurol. 1990, 108, 38–45. [Google Scholar] [CrossRef]
- Luis-García, E.R.; Limón-Pacheco, J.H.; Serrano-García, N.; Hernández-Pérez, A.D.; Pedraza-Chaverri, J.; Orozco-Ibarra, M. Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J. Biochem. Mol. Toxicol. 2017, 31, e21837. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Severiano, F.; Ríos, C.; Segovia, J. Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Res. 2000, 862, 234–237. [Google Scholar] [CrossRef]
- Schilling, G.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klivenyi, P.; Bende, Z.; Hartai, Z.; Penke, Z.; Nemeth, H.; Toldi, J.; Vecsei, L. Behaviour changes in a transgenic model of Huntington’s disease. Behav. Brain Res. 2006, 169, 137–141. [Google Scholar] [CrossRef]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.A.; Leavitt, B.R.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Slow, E.J. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genetics 2003, 12, 1555–1567. [Google Scholar] [CrossRef]
- Brocardo, P.S.; McGinnis, E.; Christie, B.R.; Gil-Mohapel, J. Time-Course Analysis of Protein and Lipid Oxidation in the Brains of Yac128 Huntington’s Disease Transgenic Mice. Rejuvenation Res. 2016, 19, 140–148. [Google Scholar] [CrossRef]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-M.; Wu, Y.-R.; Chang, K.-H. Altered Aconitase 2 Activity in Huntington’s Disease Peripheral Blood Cells and Mouse Model Striatum. IJMS 2017, 18, 2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menalled, L.B.; Sison, J.D.; Dragatsis, I.; Zeitlin, S.; Chesselet, M.-F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 2003, 465, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of Oxidant Damage in Huntington’s Disease: Translational Strategies Using Antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, E.; Guyot, M.C.; Mittoux, V.; Altairac, S.; Condé, F.; Palfi, S.; Hantraye, P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 1998, 70, 794–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrotra, A.; Sandhir, R. Mitochondrial cofactors in experimental Huntington’s disease: Behavioral, biochemical and histological evaluation. Behav. Brain Res. 2014, 261, 345–355. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxidative Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991, 11, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Kim, S.U.; McLarnon, J.G. Blockade of quinolinic acid-induced neurotoxicity by pyruvate is associated with inhibition of glial activation in a model of Huntington’s disease. Exp. Neurol. 2004, 187, 150–159. [Google Scholar] [CrossRef]
- Leipnitz, G.; Schumacher, C.; Scussiato, K.; Dalcin, K.B.; Wannmacher, C.M.D.; Wyse, A.T.D.; Dutra-Filho, C.S.; Wajner, M.; Latini, A. Quinolinic acid reduces the antioxidant defenses in cerebral cortex of young rats. Int. J. Dev. Neurosci. 2005, 23, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Bahl, J.J.; Bressler, R. The pharmacology of carnitine. Annu. Rev. Pharmacol. Toxicol. 1987, 27, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Arockia Rani, P.J.; Panneerselvam, C. Carnitine as a free radical scavenger in aging. Exp. Gerontol. 2001, 36, 1713–1726. [Google Scholar] [CrossRef]
- Cogburn, L.A.; Wilson-Placentra, S.; Letcher, L.R. Influence of pinealectomy on plasma and extrapineal melatonin rhythms in young chickens (Gallus domesticus). Gen. Comp. Endocrinol. 1987, 68, 343–356. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X. Role of CSF in the transport of melatonin. J. Pineal Res. 2002, 33, 61. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin directly scavenges hydrogen peroxide: A potentially new metabolic pathway of melatonin biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Ou, P.; Tritschler, H.J.; Wolff, S.P. Thioctic (lipoic) acid: A therapeutic metal-chelating antioxidant? Biochem. Pharmacol. 1995, 50, 123–126. [Google Scholar] [CrossRef]
- Sies, H.; Stahl, W. Lycopene: Antioxidant and biological effects and its bioavailability in the human. Proc. Soc. Exp. Biol. Med. 1998, 218, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Hanasaki, Y.; Ogawa, S.; Fukui, S. The correlation between active oxygens scavenging and antioxidative effects of flavonoids. Free Radic. Biol. Med. 1994, 16, 845–850. [Google Scholar] [CrossRef]
- Kandemir, F.M.; Ozkaraca, M.; Yildirim, B.A.; Hanedan, B.; Kirbas, A.; Kilic, K.; Aktas, E.; Benzer, F. Rutin attenuates gentamicin-induced renal damage by reducing oxidative stress, inflammation, apoptosis, and autophagy in rats. Ren. Fail. 2015, 37, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kostic, D.; Dimitrijevic, D.; Stojanović, G.; Palić, I.; Đorđević, A.; Ickovski, J. Xanthine Oxidase: Isolation, Assays of Activity, and Inhibition. J. Chem. 2015, 2015, 294858. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Gueven, N.; Woolley, K.; Smith, J. Border between natural product and drug: Comparison of the related benzoquinones idebenone and coenzyme Q10. Redox Biol. 2015, 4, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Peyser, C.E.; Folstein, M.; Chase, G.A.; Starkstein, S.; Brandt, J.; Cockrell, J.R.; Bylsma, F.; Coyle, J.T.; McHugh, P.R.; Folstein, S.E. Trial of d-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry 1995, 152, 1771–1775. [Google Scholar] [CrossRef]
- Chandra, A.; Johri, A.; Beal, M.F. Prospects for neuroprotective therapies in prodromal Huntington’s disease. Mov. Disord. 2014, 29, 285–293. [Google Scholar] [CrossRef] [Green Version]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Rosas, H.D.; Doros, G.; Gevorkian, S.; Malarick, K.; Reuter, M.; Coutu, J.P.; Triggs, T.D.; Wilkens, P.J.; Matson, W.; Salat, D.H.; et al. PRECREST: A phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology 2014, 82, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Neuroprotective effects of creatine. Amino Acids 2011, 40, 1305–1313. [Google Scholar] [CrossRef]
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.-L.; Meyers, C.M.; Nahin, R.; Rosas, H.D. The CREST-E study of creatine for Huntington disease. Neurology 2017, 89, 594–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranen, N.G.; Peyser, C.E.; Coyle, J.T.; Bylsma, F.W.; Sherr, M.; Day, L.; Folstein, M.F.; Brandt, J.; Ross, C.A.; Folstein, S.E. A controlled trial of idebenone in Huntington’s disease. Mov. Disord. 1996, 11, 549–554. [Google Scholar] [CrossRef]
- Vaddadi, K.S.; Soosai, E.; Chiu, E.; Dingjan, P. A randomised, placebo-controlled, double blind study of treatment of Huntington’s disease with unsaturated fatty acids. Neuroreport 2002, 13, 29–33. [Google Scholar] [CrossRef]
- Puri, B.K.; Bydder, G.M.; Counsell, S.J.; Corridan, B.J.; Richardson, A.J.; Hajnal, J.V.; Appel, C.; Mckee, H.M.; Vaddadi, K.S.; Horrobin, D.F. MRI and neuropsychological improvement in Huntington disease following ethyl-EPA treatment. Neuroreport 2002, 13, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K.; Leavitt, B.R.; Hayden, M.R.; Ross, C.A.; Rosenblatt, A.; Greenamyre, J.T.; Hersch, S.; Vaddadi, K.S.; Sword, A.; Horrobin, D.F.; et al. Ethyl-EPA in Huntington disease: A double-blind, randomized, placebo-controlled trial. Neurology 2005, 65, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch. Neurol. 2008, 65, 1582–1589. [Google Scholar] [CrossRef]
- The Huntington Study Group. Safety and tolerability of the free-radical scavenger OPC-14117 in Huntington’s disease. Neurology 1998, 50, 1366–1373. [Google Scholar] [CrossRef]
- McGarry, A.; McDermott, M.P.; Kieburtz, K.; Fung, W.L.A.; McCusker, E.; Peng, J.; de Blieck, E.A.; Cudkowicz, M.; Huntington Study Group 2CARE Investigators and Coordinators. Risk factors for suicidality in Huntington disease: An analysis of the 2CARE clinical trial. Neurology 2019, 92, e1643–e1651. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 664–674. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Parpura, V. Neurological and psychiatric disorders as a neuroglial failure. Period. Biol. 2014, 116, 115–124. [Google Scholar]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective Targeting of a Redox-active Ubiquinone to Mitochondria within Cells Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Genrikhs, E.E.; Stelmashook, E.V.; Popova, O.V.; Kapay, N.A.; Korshunova, G.A.; Sumbatyan, N.V.; Skrebitsky, V.G.; Skulachev, V.P.; Isaev, N.K. Mitochondria-targeted antioxidant SkQT1 decreases trauma-induced neurological deficit in rat and prevents amyloid-β-induced impairment of long-term potentiation in rat hippocampal slices. J. Drug Target. 2015, 23, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Vachey, G.; Déglon, N. CRISPR/Cas9-Mediated Genome Editing for Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 463–481. [Google Scholar] [CrossRef]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Skotte, N.H.; Southwell, A.L.; Østergaard, M.E.; Carroll, J.B.; Warby, S.C.; Doty, C.N.; Petoukhov, E.; Vaid, K.; Kordasiewicz, H.; Watt, A.T.; et al. Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: Providing a therapeutic option for all Huntington disease patients. PLoS ONE 2014, 9, e107434. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: SiRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Mestre, T.A. Recent advances in the therapeutic development for Huntington disease. Parkinsonism Relat. Disord. 2019, 59, 125–130. [Google Scholar] [CrossRef]
- Leavitt, B.R.; Tabrizi, S.J. Antisense oligonucleotides for neurodegeneration. Science 2020, 367, 1428–1429. [Google Scholar] [CrossRef]
- Portnoff, A.D.; Stephens, E.A.; Varner, J.D.; DeLisa, M.P. Ubiquibodies, synthetic E3 ubiquitin ligases endowed with unnatural substrate specificity for targeted protein silencing. J. Biol. Chem. 2014, 289, 7844–7855. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Manrique, R.P.; Farina, F.; Cambon, K.; Sequedo, M.D.; Parker, A.J.; Millan, J.M.; Weiss, A.; Deglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Narayan Dey, P.; Chen, C.; Monteiro, O.; et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife 2018, 7, e38744. [Google Scholar] [CrossRef]
- Hervas, D.; Fornes-Ferrer, V.; Gomez-Escribano, A.P.; Sequedo, M.D.; Peiro, C.; Millan, J.M.; Vazquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [Green Version]
- Braubach, P.; Orynbayev, M.; Andronache, Z.; Hering, T.; Landwehrmeyer, G.B.; Lindenberg, K.S.; Melzer, W. Altered Ca(2+) signaling in skeletal muscle fibers of the R6/2 mouse, a model of Huntington’s disease. J. Gen. Physiol. 2014, 144, 393–413. [Google Scholar] [CrossRef]
- Joviano-Santos, J.V.; Santos-Miranda, A.; Botelho, A.F.M.; de Jesus, I.C.G.; Andrade, J.N.; de Oliveira Barreto, T.; Magalhães-Gomes, M.P.S.; Valadão, P.A.C.; Cruz, J.D.S.; Melo, M.M.; et al. Increased oxidative stress and CaMKII activity contribute to electro-mechanical defects in cardiomyocytes from a murine model of Huntington’s disease. FEBS J. 2019, 286, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, F.B.; Wild, E.J. Clinical Trials Corner: September 2017. JHD 2017, 6, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, F.B.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: August 2018. JHD 2018, 7, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, F.B.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: February 2018. JHD 2018, 7, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, F.B.; Quinn, L.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: January 2019. JHD 2019, 8, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, F.B.; Ferreira, J.J.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: June 2019. JHD 2019, 8, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| polyQ Model 2 | Compound | Effect | Reference |
|---|---|---|---|
| 40Q::YFP in muscle cells (AM141) | α-lipoic acid | No effect in number of inclusion bodies | [115] |
| Cassia fistula extract | Decreased number of inclusion bodies | [116] | |
| Epigallocatechin gallate | No effect in number of inclusion bodies | [115] | |
| Epimedium brevicornum acidic polysaccharide EbPS-A1 | No effect in number of inclusion bodies | [117] | |
| Guarana extract | Decreased number of inclusion bodies | [118] | |
| Ruthenium (II) complexes | Decreased number of inclusion bodies | [119] | |
| Phycocyanin | Decreased number of inclusion bodies | [120] | |
| Salidroside | No effect in number of inclusion bodies | [121] | |
| Vitamin C | No effect in number of inclusion bodies | [115] | |
| Htn150Q in ASH neurons (HA759) | Dictiophora indusiata polysaccharides | Attenuated polyQ-mediated chemosensory behaviour dysfunction | [122] |
| Epimedium brevicornum acidic polysaccharide EbPS-A1 | Increased survival rate under stress-induced conditions, reduced ROS levels and lipid peroxidation and increased SOD and CAT; attenuated polyQ-mediated chemosensory behaviour dysfunction | [117] | |
| Guarana extract | Attenuated polyQ-mediated chemosensory behaviour dysfunction | [123] | |
| Ruthenium (II) complexes | Increased neuronal survival and improved chemosensory behaviour | [119] | |
| Salidroside | Decreased lipid peroxidation and ROS and increased antioxidant enzyme activity; attenuated polyQ-mediated chemosensory behaviour dysfunction and increased neuronal survival | [121] |
| HD Model 2 | Compound | Effect | Reference |
|---|---|---|---|
| Htt-128Q | α-tocopherol and coenzyme Q10 | No increase in survival rate and pupal mortality | [141] |
| Httex1p 93Q | Curcumin | Reduced neuronal loss in photoreceptors and improved polyQ-induced motor neuronal dysfunction | [150] |
| Fisetin and resveratrol | Increased photoreceptor neuronal survival and rescued neuronal degeneration | [144,146] | |
| Green tea | Reduced mHtt-induced neurodegeneration, increased lifespan, inhibited aggregation, prevented photoreceptor degeneration and improved motor function | [152,153] | |
| Grape seed polyphenolic extract (GSPE) | Positive impact on longevity | [147] |
| HD Model 2 | Compound | Effect | Reference |
|---|---|---|---|
| R6/1 mice | N-Acetylcysteine | Reversed depressed-like behaviours, decreased oxidative damage markers in mitochondria and delayed onset of motor problems | [159,160] |
| Linoleic acid, Q-linolenic acid, eicosapentaenoic acid, docosahexaenoic acid, K-lipoic acid and d-K-tocopherol | Improved survival rate and motor performance | [161] | |
| R6/2 mice | α-lipoic acid | Modest improvement in survival | [162] |
| Coenzyme Q10 | Increased survival, delayed appearance of motor deficits, reduced body weight loss, cerebral atrophy and intranuclear inclusions of mHtt | [163] | |
| Creatine | Reduced mHtt aggregates, improved survival of animals and delayed mHtt-induced atrophy of striatal neurons | [164] | |
| Curcumin | Improved motor performance, activation of pro-survival pathways and maintenance of gastro-intestinal homeostasis | [165] | |
| Cystamine | Extended lifespan and reduced motor deficits | [166,167] | |
| Deferoxamine | Improved behavioural performance | [168] | |
| Dimethylfumarate | Extended lifespan, beneficial effects on motor behaviour and preserved neuronal morphology | [169] | |
| Grape seed polyphenolic extract (GSPE) | Increased lifespan and improved motor performance | [147] | |
| Melatonin | Decreased mortality rate and delayed HD onset | [170] | |
| XJB-5-131 | Prevented body weight loss and improved motor performance | [171] | |
| N171-82Q mice | Creatine | Improved survival rate, delayed motor dysfunction and body weight loss with reduced aggregated mHtt | [172] |
| l-carnitine | Increased survival rate, reduced number of aggregates and improved motor performance | [173] | |
| Selenium derivatives | Reduced mHtt aggregates and decreased oxidized glutathione levels | [174] | |
| YAC128 mice | Dimethylfumarate | Beneficial effects on motor behaviour and preserved neuronal morphology | [169] |
| Ethyl-eicosapentaenoic acid | Modest improvement of motor function | [175] | |
| Resveratrol | Improved motor performance and learning | [176] | |
| Hdh(CAG)150/(CAG)150 mice | XJB-5-131 | Prevented body weight loss, mitochondrial dysfunction and oxidative damage, and improved motor performance | [177,178] |
| CAG140 knock-in mice | Curcumin | Ameliorated number of aggregates and partial improvement of motor function | [179] |
| 3-NP-induced rats | α-lipoic acid | Decreased mitochondrial swelling and reduced cognitive impairment | [180] |
| α-tocopherol and coenzyme Q10 | Failed to achieve restoration of decline in mitochondrial production of energy | [181] | |
| FK-506 | Prevented body weight loss, ameliorated locomotor activity, restored antioxidant enzyme activity and attenuated mitochondrial dysfunction | [182] | |
| Lycopene | Ameliorated mitochondrial function and prevented locomotor and memory deficits | [183] | |
| N-Acetylcysteine | Prevented mitochondrial dysfunction, behavioural deficits and neuronal death and attenuated lipid peroxidation | [184] | |
| Nicotinamide | Prevented increase in nitrite levels and malondialdehyde and depletion of GSH, improved motor performance and impeded neuronal death in striatum | [185] | |
| Quercetin | Reversed mitochondrial dysfunction, prevented decrease in antioxidant enzyme activity and improved motor performance; combination with lycopene alleviated anxiety and depression | [186,187] | |
| Rutin | Protected against body weight loss, restored antioxidant enzyme activity and improved motor and memory performance | [188] | |
| Selenium derivatives | Prevented body weight loss, induced better motor performance and restored CAT levels, reduced mHtt aggregates and decreased oxidized glutathione levels | [189] | |
| Virgin olive oil | Powerful brain antioxidant | [190] | |
| Manolic acid-induced rats | α-tocopherol | Reversed body weight loss, disrupted motor coordination and oxidative stress by preserving mitochondrial function | [191] |
| Quinolinic acid-induced rats | Idebenone | Did not induce neuroprotection | [192] |
| Sulforaphane | Prevented mitochondrial dysfunction | [193] |
| Compound | Mechanism | Study Design | No. of Patients (Test/Control) | Results | ClinicalTrials.Gov Identifier (Acronym) | Ref. |
|---|---|---|---|---|---|---|
| A-tocopherol (3000 IU daily) | Antioxidant | Double-blind, placebo-controlled | 40/33 | No effect on neurologic and neuropsychiatric symptoms Therapeutic effect on neurologic symptoms for patients in early HD | Not registered | [227] |
| Coenzyme Q10 (600 mg, 1200 mg, 2400 mg daily) | Rise in energy and antioxidant | Double blind, placebo-controlled | 45/45 | Well tolerated No significant slowing of functional decline in early HD | NCT00920699 (PREQUEL) | [228] |
| Coenzyme Q10 (2400 mg daily) | Rise in energy and antioxidant | Double blind, placebo-controlled | 224/240 | Futility analysis failed to show likelihood of benefit of CoQ 2400 mg/day | NCT00608881 (2CARE) | [229,239] |
| Creatine (8 g daily) | Rise in energy and antioxidant | Double blind, placebo-controlled | 32/32 | Restored regular levels of 8-OHdG in serum | Not registered | [100] |
| Creatine (30 g daily) | Rise in energy and antioxidant | Open-label | Not determined | Slowed ongoing cortical atrophy | Not registered | [231] |
| Creatine (10–30 g daily) | Rise in energy and antioxidant | Double blind, placebo-controlled | 47/17 | Safe and tolerable Neuroimaging demonstrated treatment-related slowing of cortical and striatal atrophy | NCT00592995 (PRECREST) | [230] |
| Creatine (40 g daily) | Rise in energy and antioxidant | Double blind, placebo-controlled | 275/278 | Creatine is not useful to treat HD | NCT00712426 CREST-E | [232] |
| Idebenone (90 mg 3 times a day) | Rise in energy and antioxidant | Double blind, placebo-controlled | 50/50 | No differences between groups; larger numbers needed for higher statistical power | Not registered | [233] |
| Fatty acids (8 g HUFAs daily) | Antioxidant | Double blind, placebo-controlled | 10/7 | Improved motor function | Not registered | [234] |
| Fatty acids (2 g ethyl-EPA daily) | Antioxidant | Pilot study, double blind, placebo-controlled | 7/4 | Improved motor function and MRI changes | Not registered | [235] |
| Fatty acids (2 g ethyl-EPA daily) | Antioxidant | Double blind, placebo-controlled | 67/68 | Improved motor function | Not registered | [236] |
| Fatty acids (1 g ethyl-EPA daily) | Antioxidant | Double blind, placebo-controlled | 150/150 | Ethyl-EPA was not beneficial | NCT00146211 TREND-HD | [237] |
| OPC-14117 (60 mg, 120 mg, 240 mg daily) | Antioxidant | Double blind, placebo-controlled | 32/32 | No effect | Not registered | [238] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono-Yagüe, J.; Gómez-Escribano, A.P.; Millán, J.M.; Vázquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. https://doi.org/10.3390/antiox9070577
Bono-Yagüe J, Gómez-Escribano AP, Millán JM, Vázquez-Manrique RP. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants. 2020; 9(7):577. https://doi.org/10.3390/antiox9070577
Chicago/Turabian StyleBono-Yagüe, José, Ana Pilar Gómez-Escribano, José María Millán, and Rafael Pascual Vázquez-Manrique. 2020. "Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch?" Antioxidants 9, no. 7: 577. https://doi.org/10.3390/antiox9070577