
Whole-Transcriptome Sequencing Reveals the Global Molecular Responses and NAC Transcription Factors Involved in Drought Stress in Dendrobium catenatum

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and RNA Sequencing
2.3. mRNA Identification and Differential Expression Analysis
2.4. Identification and Phylogenetic Tree Analysis of DcNAC Family in RJ
2.5. Expression Pattern Analysis of 32 DEDcNACs in Various Tissues
2.6. Methods of Stress Treatment, RNA Extraction, and Quantitative/Real-Time-PCR (qRT-PCR) Analysis
2.7. ROS Staining
2.8. Identification and Analysis of lncRNAs
2.9. Identification and Analysis of circRNAs
2.10. Identification and Analysis of miRNAs
2.11. ceRNA Network Construction and Analysis
3. Results
3.1. Evaluation of Tolerance to Drought Stress in D. catenatum
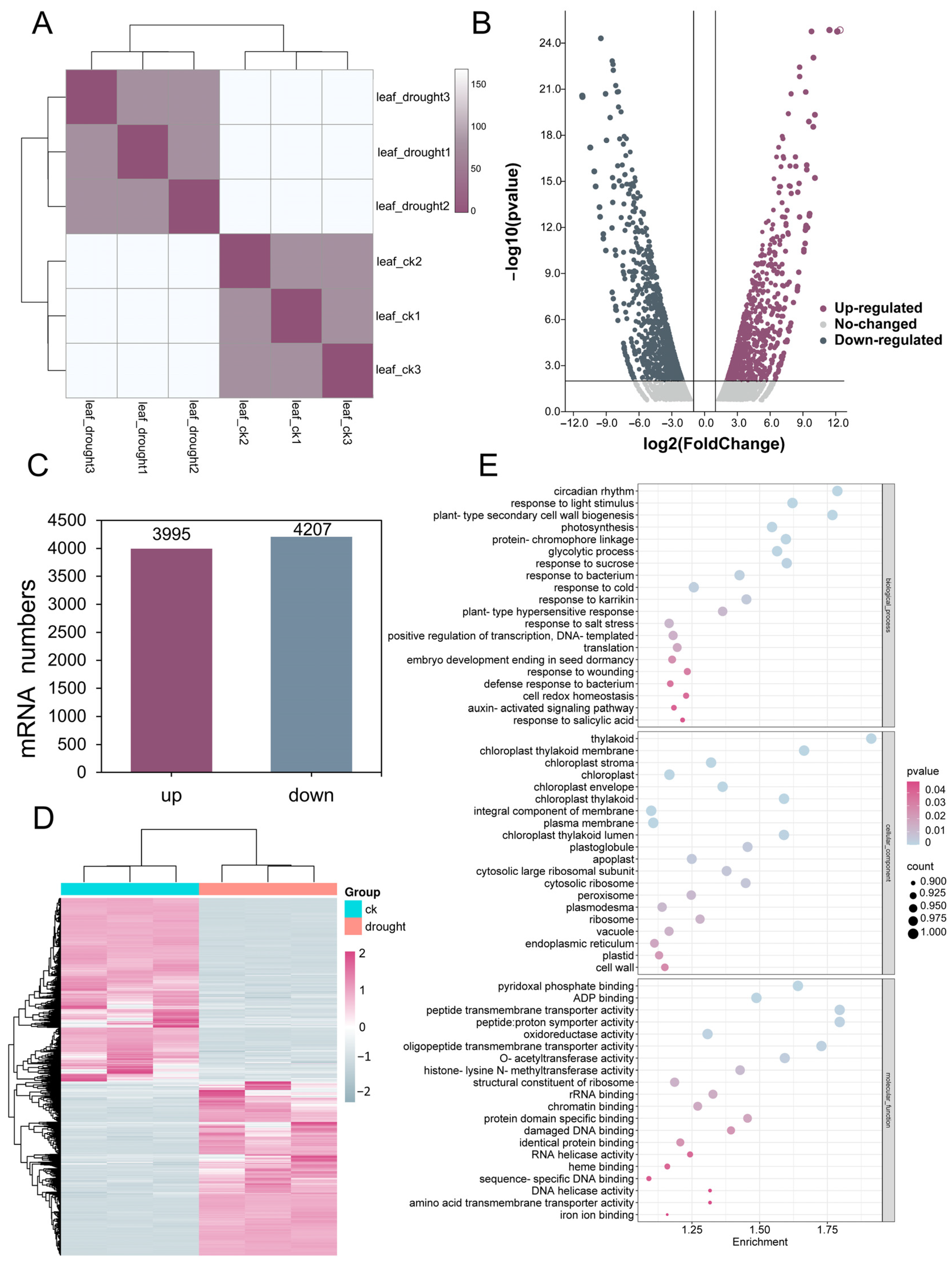
3.2. The Global Responses of mRNA to Drought Stress


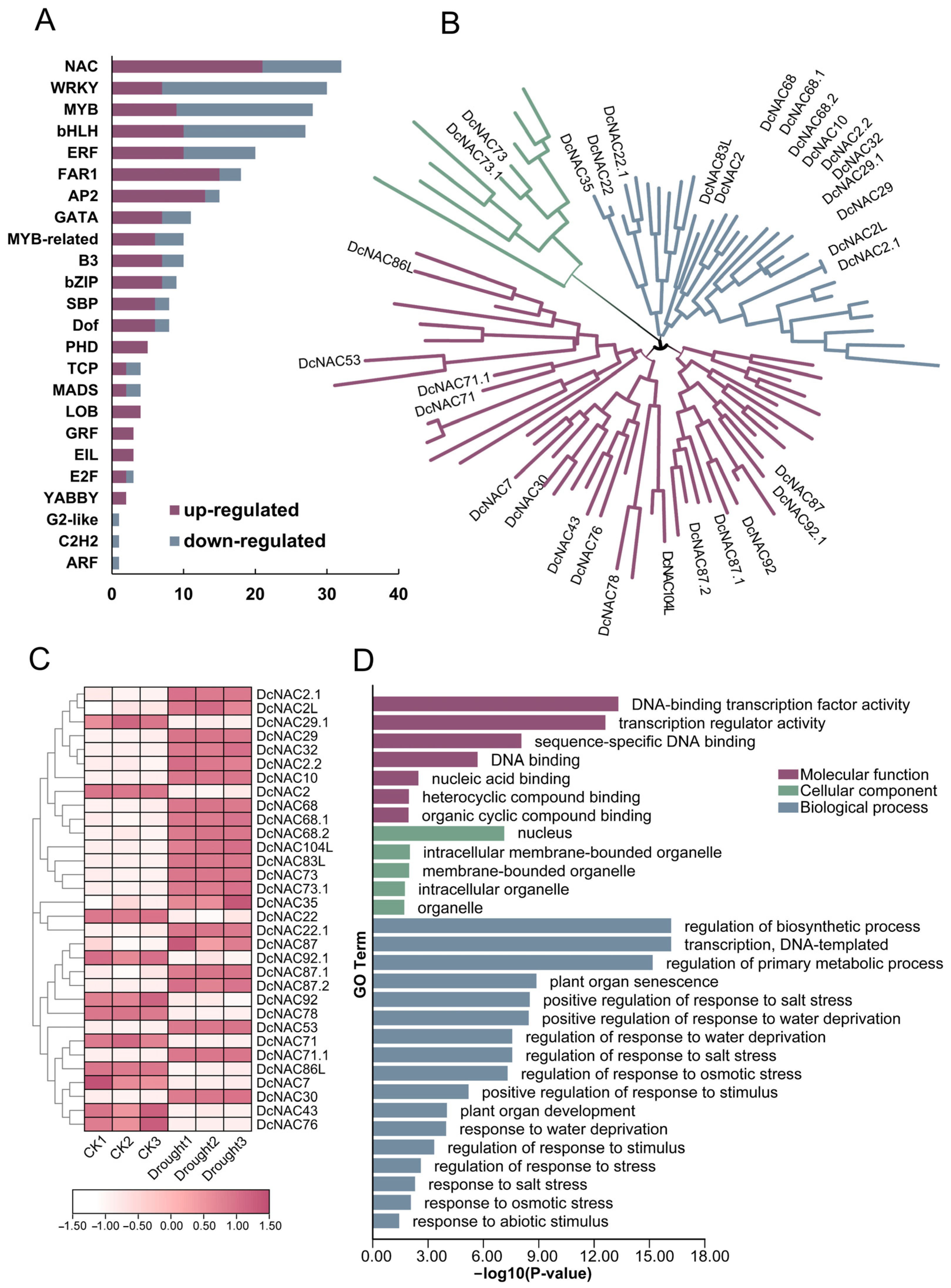
3.3. Identification and Analysis of NAC Transcription Factor Family of DEmRNAs in RJ
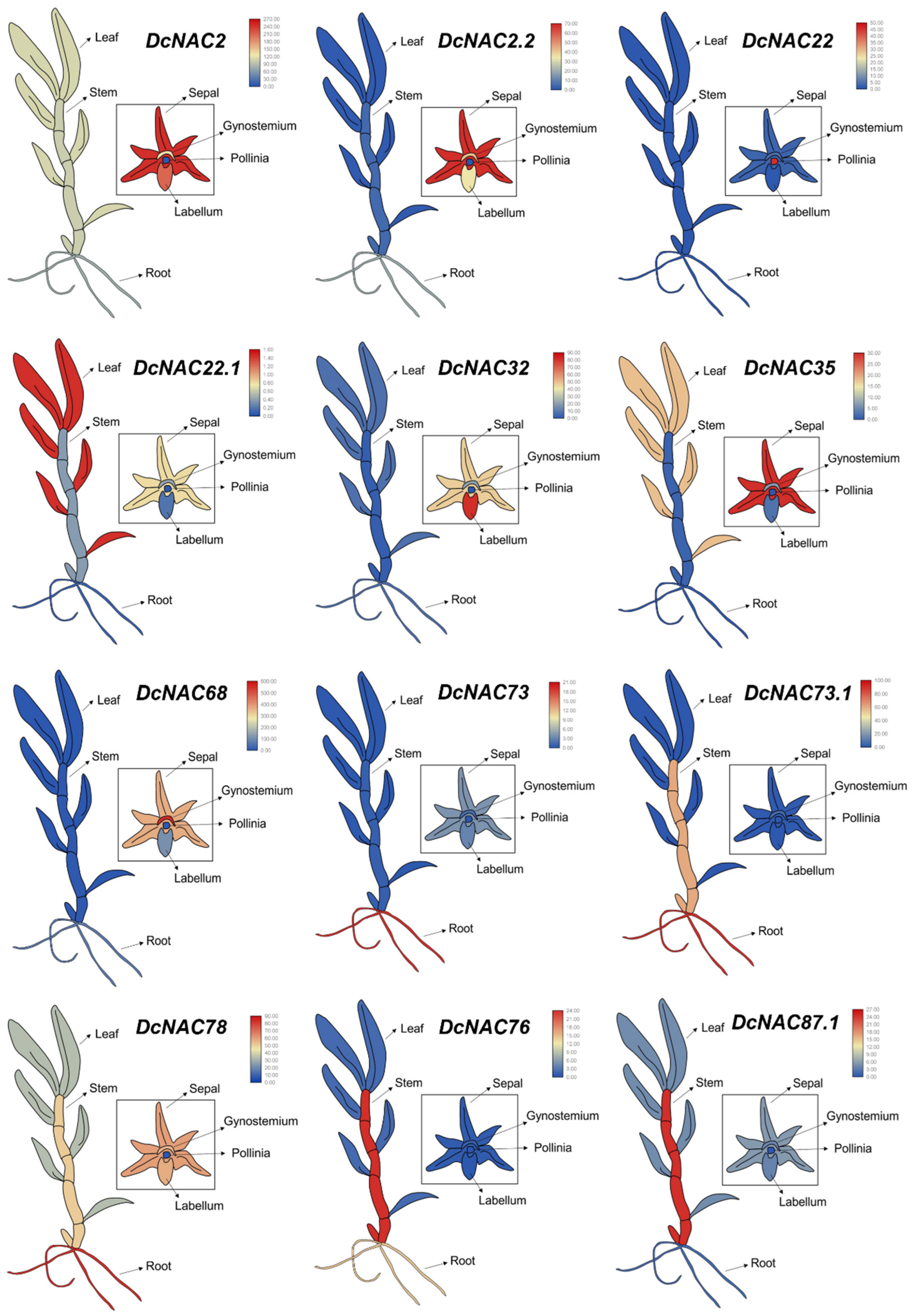
3.4. Tissue Expression Patterns of Differentially Expressed NAC Transcription Factors in RJ

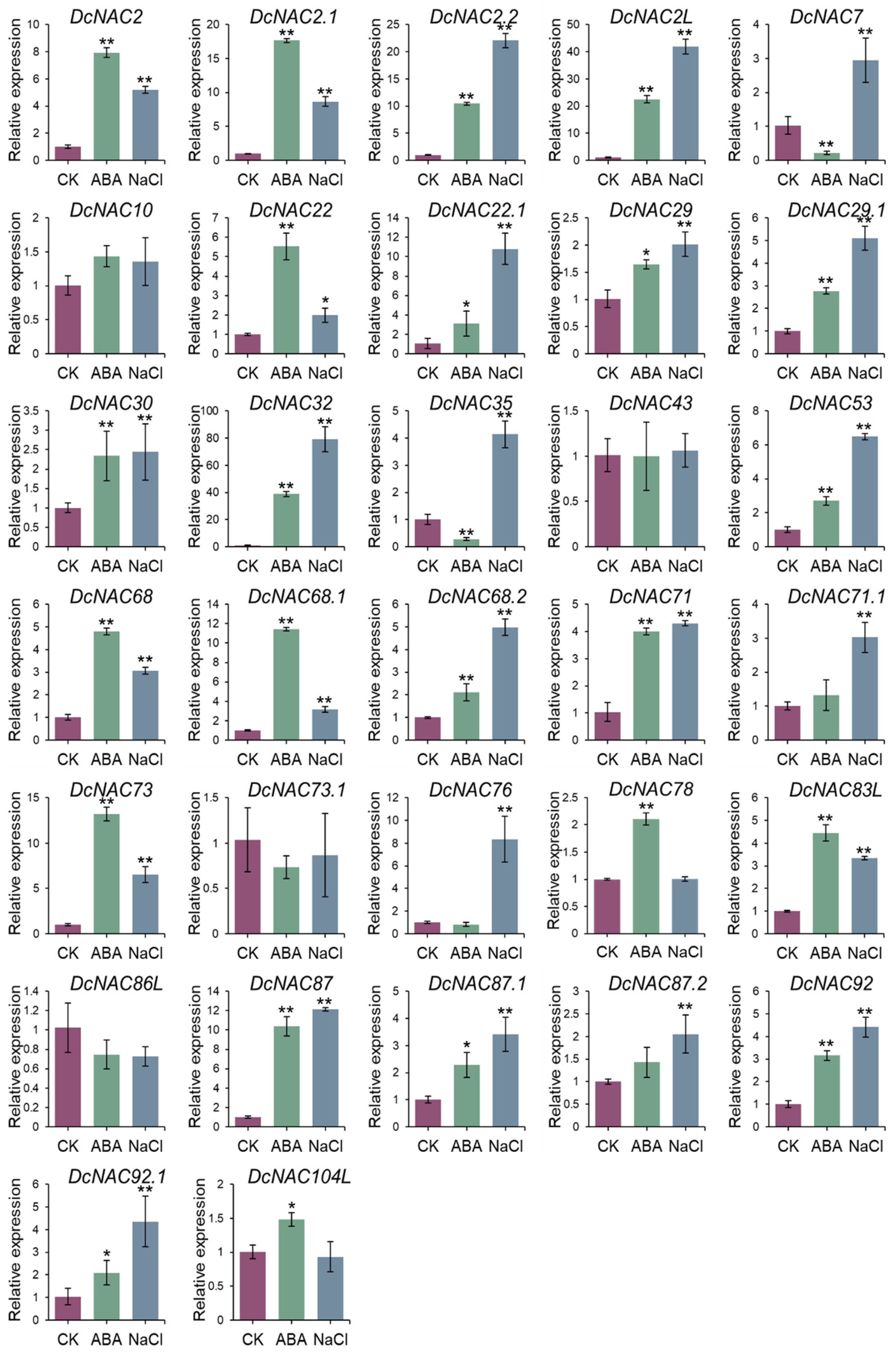
3.5. Response of DEDcNACs under ABA and NaCl Treatment

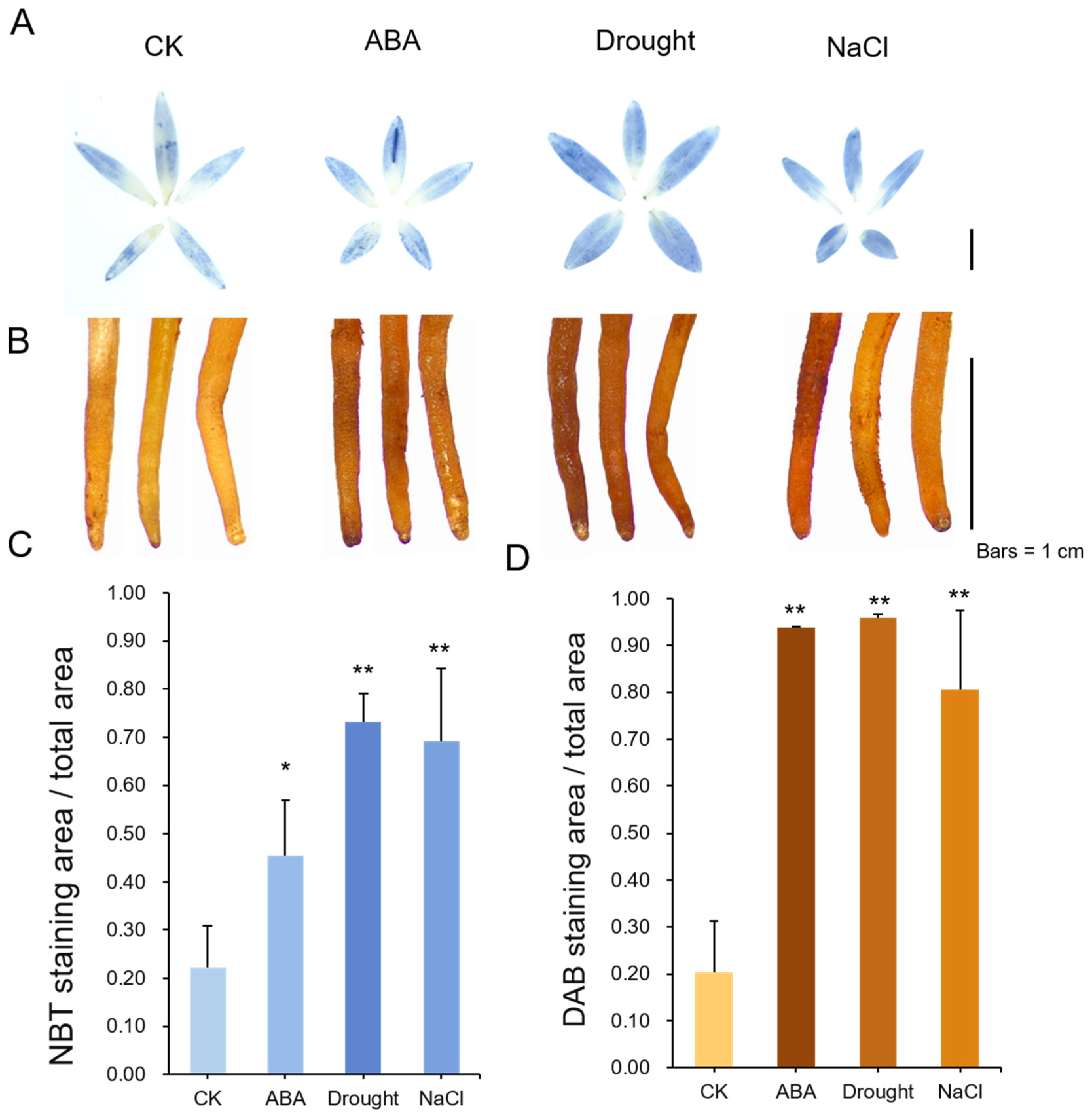
3.6. ROS Levels in RJ under Different Treatments
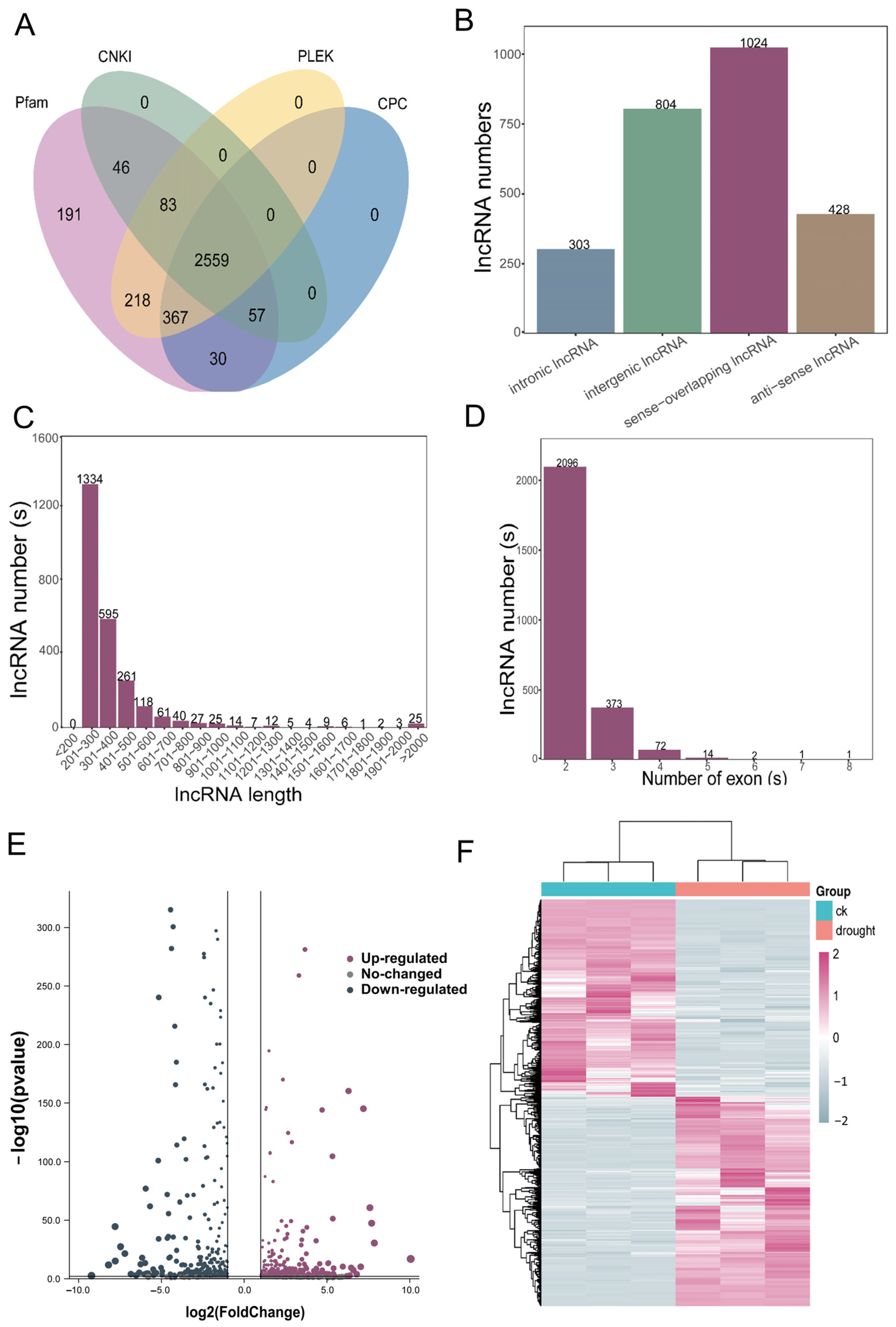
3.7. The Transcriptome-Wide Responses of lncRNA to Drought Stress
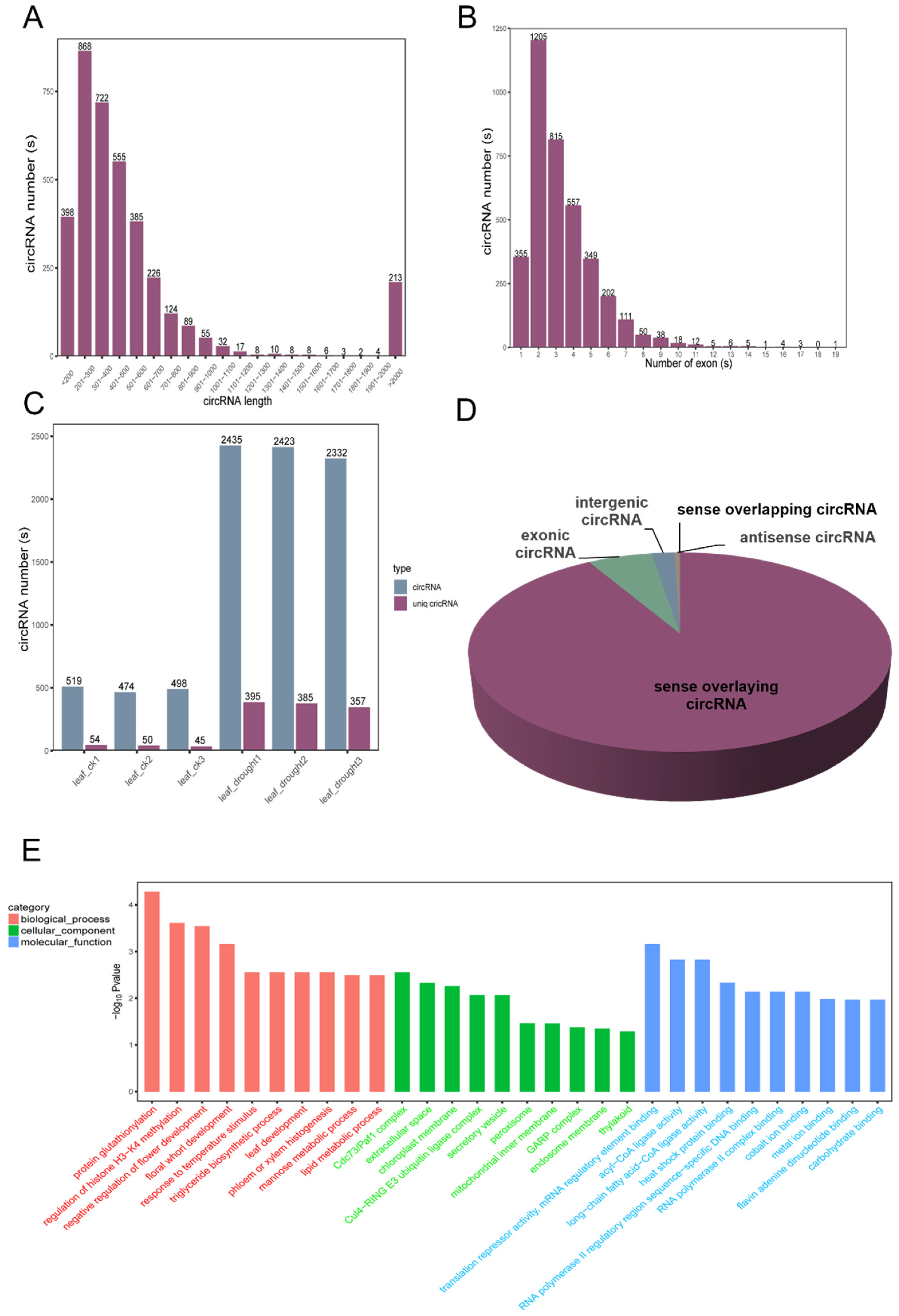
3.8. The Global Expression Patterns of circRNA to Drought Stress

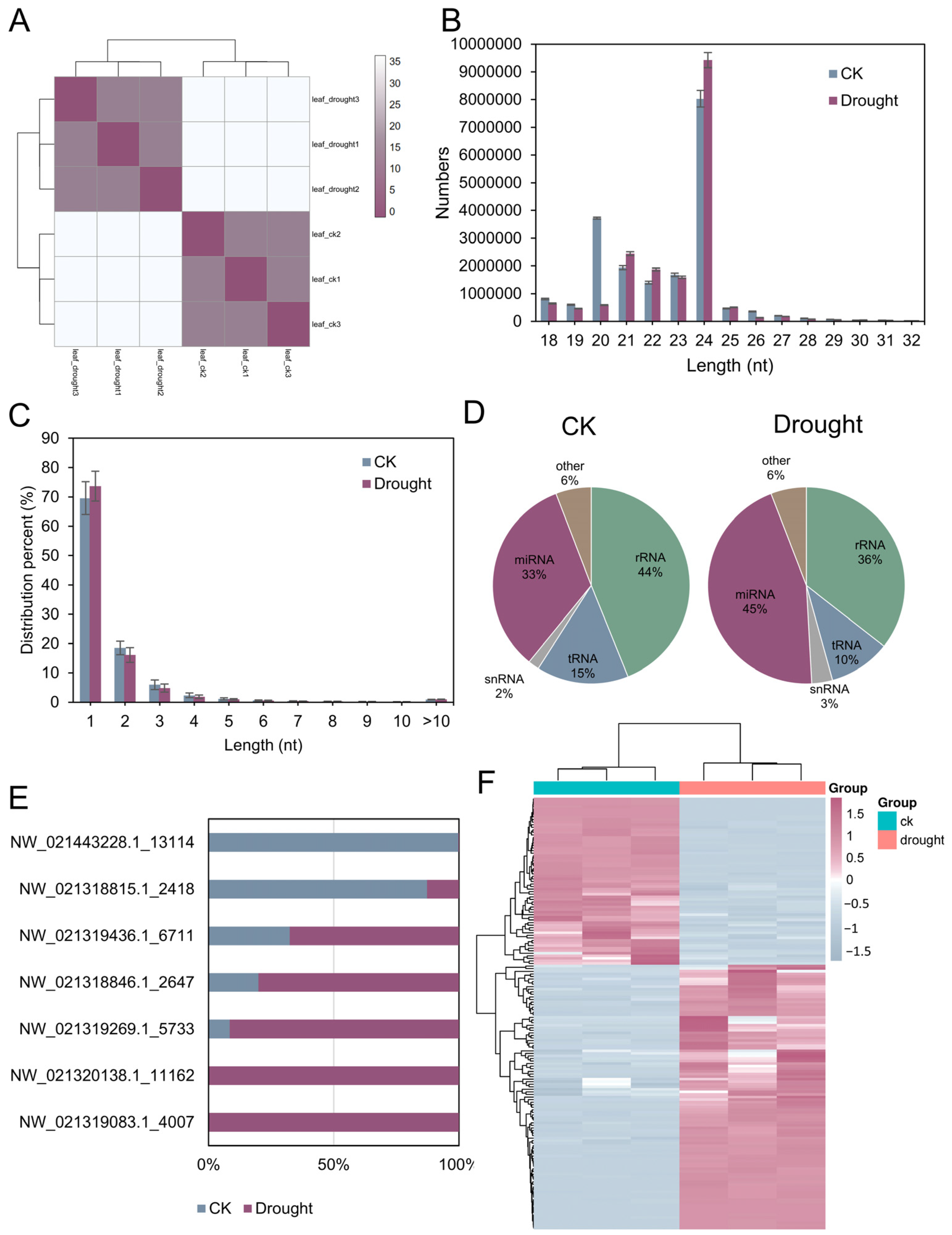
3.9. The Integrated Small RNA Response to Drought Stress in D. catenatum
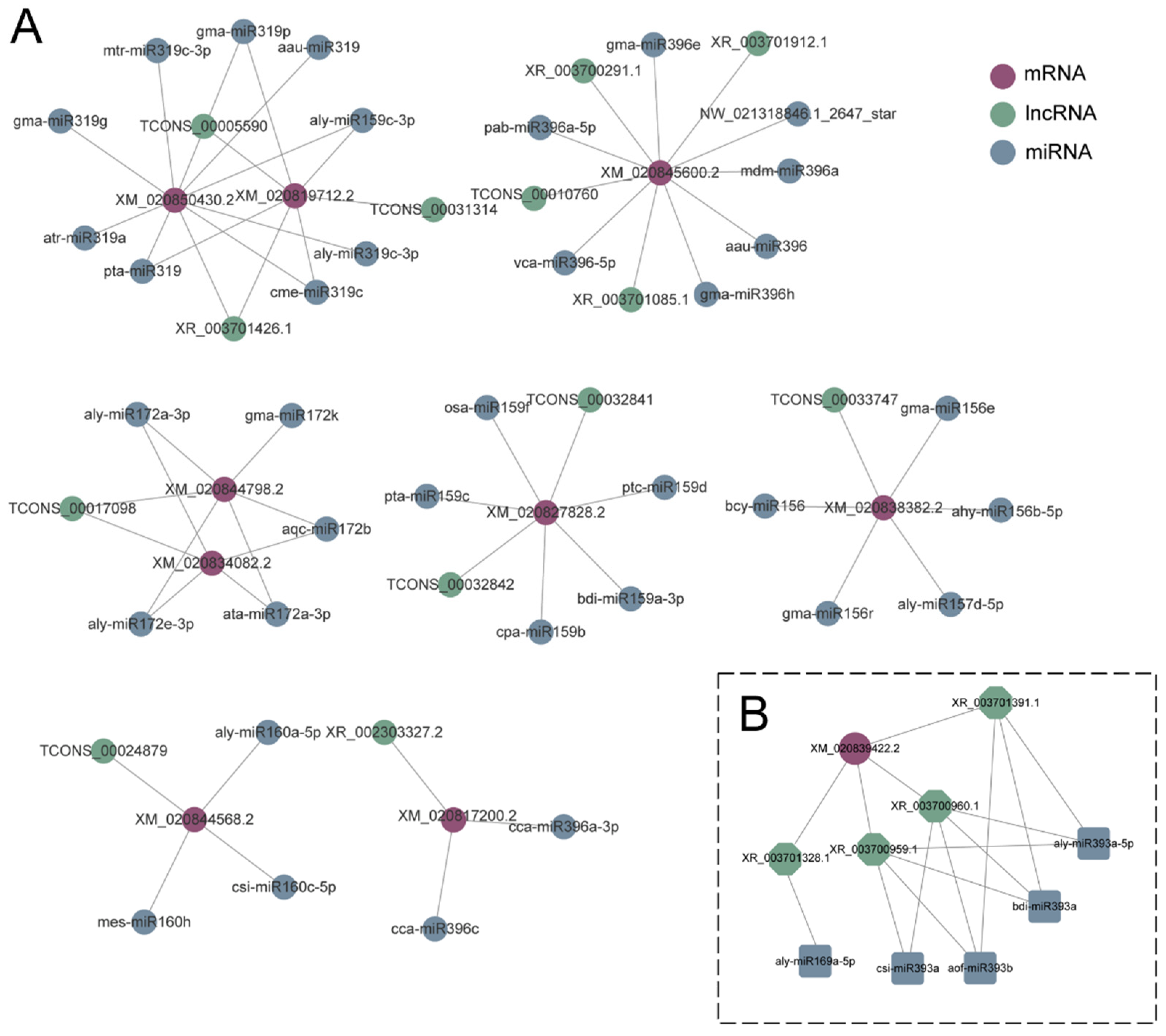
3.10. CeRNA Regulatory Network in Response to Drought Stress


4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Han, Y.; Cai, M.; Zhang, S.; Chai, J.; Sun, M.; Wang, Y.; Xie, Q.; Chen, Y.; Wang, H.; Chen, T. Genome-Wide Identification of AP2/ERF Transcription Factor Family and Functional Analysis of DcAP2/ERF#96 Associated with Abiotic Stress in Dendrobium catenatum. Int. J. Mol. Sci. 2022, 23, 13603. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, X.; Pan, X.; Jiang, Q.; Xi, Z. Exogenous Putrescine Alleviates Drought Stress by Altering Reactive Oxygen Species Scavenging and Biosynthesis of Polyamines in the Seedlings of Cabernet Sauvignon. Front. Plant Sci. 2021, 12, 767992. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, F.; Simonneau, T.; Muller, B. The Physiological Basis of Drought Tolerance in Crop Plants: A Scenario-Dependent Probabilistic Approach. Annu. Rev. Plant Biol. 2018, 69, 733–759. [Google Scholar] [CrossRef] [PubMed]
- Dinneny, J.R. Developmental Responses to Water and Salinity in Root Systems. Annu. Rev. Cell Dev. Biol. 2019, 35, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Serres, J.; Parker, J.E.; Ainsworth, E.A.; Oldroyd, G.E.D.; Schroeder, J.I. Genetic strategies for improving crop yields. Nature 2019, 575, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Li, M.; Zhao, T.; Feng, S.; Zhang, H.; Wang, L.; Han, J.; Gao, M.; Lu, K.; Chen, Q.; et al. Neofunctionalization of a polyploidization-activated cotton long intergenic non-coding RNA DAN1 during drought stress regulation. Plant Physiol. 2021, 186, 2152–2168. [Google Scholar] [CrossRef]
- Qin, T.; Zhao, H.; Cui, P.; Albesher, N.; Xiong, L. A Nucleus-Localized Long Non-Coding RNA Enhances Drought and Salt Stress Tolerance [OPEN]. Plant Physiol. 2017, 175, 1321–1336. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, Y.; Qi, X. LncRNA TCONS_00021861 is functionally associated with drought tolerance in rice (Oryza sativa L.) via competing endogenous RNA regulation. BMC Plant Biol. 2021, 21, 410. [Google Scholar] [CrossRef]
- Li, S.; Yu, X.; Lei, N.; Cheng, Z.; Zhao, P.; He, Y.; Wang, W.; Peng, M. Genome-wide identification and functional prediction of cold and/or drought-responsive lncRNAs in cassava. Sci. Rep. 2017, 7, 45981. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef]
- Waititu, J.K.; Zhang, X.; Chen, T.; Zhang, C.; Zhao, Y.; Wang, H. Transcriptome Analysis of Tolerant and Susceptible Maize Genotypes Reveals Novel Insights about the Molecular Mechanisms Underlying Drought Responses in Leaves. Int. J. Mol. Sci. 2021, 22, 6980. [Google Scholar] [CrossRef]
- Lamin-Samu, A.T.; Zhuo, S.; Ali, M.; Lu, G. Long non-coding RNA transcriptome landscape of anthers at different developmental stages in response to drought stress in tomato. Genomics 2022, 114, 110383. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, J.; Kan, J.; Wang, H.; Li, X.; Yang, Q.; Li, H.; Chang, Y. Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia. Genes 2018, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-C.; Zhang, J.; Zhang, D.-Y.; Nan, Y.-H.; Ge, S.; Guo, Y.-L. Identification of long noncoding natural antisense transcripts (lncNATs) correlated with drought stress response in wild rice (Oryza nivara). BMC Genom. 2021, 22, 424. [Google Scholar] [CrossRef]
- Qi, X.; Xie, S.; Liu, Y.; Yi, F.; Yu, J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 2013, 83, 459–473. [Google Scholar] [CrossRef] [PubMed]
- López-Galiano, M.J.; García-Robles, I.; González-Hernández, A.I.; Camañes, G.; Vicedo, B.; Real, M.D.; Rausell, C. Expression of miR159 Is Altered in Tomato Plants Undergoing Drought Stress. Plants 2019, 8, 201. [Google Scholar] [CrossRef]
- Balyan, S.; Kumar, M.; Mutum, R.D.; Raghuvanshi, U.; Agarwal, P.; Mathur, S.; Raghuvanshi, S. Identification of miRNA-mediated drought responsive multi-tiered regulatory network in drought tolerant rice, Nagina 22. Sci. Rep. 2017, 7, 15446. [Google Scholar] [CrossRef] [PubMed]
- Kantar, M.; Lucas, S.J.; Budak, H. miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 2011, 233, 471–484. [Google Scholar] [CrossRef]
- Liu, M.; Yu, H.; Zhao, G.; Huang, Q.; Lu, Y.; Ouyang, B. Identification of drought-responsive microRNAs in tomato using high-throughput sequencing. Funct. Integr. Genom. 2018, 18, 67–78. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Yang, J.; Huang, H.; Lu, Q.; Zhao, J.; Wang, F.; Wang, D. Genome-wide identification of drought-responsive microRNAs and their target genes in Chinese jujube by deep sequencing. Genes Genom. 2022, 45, 231–245. [Google Scholar] [CrossRef]
- Gentile, A.; Dias, L.I.; Mattos, R.S.; Ferreira, T.H.; Menossi, M. MicroRNAs and drought responses in sugarcane. Front. Plant Sci. 2015, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Q.; Xu, W.; Yao, L.; Wang, X.; Wang, H.; Xu, Y.; Li, L.; Duan, C.; Yi, Z.; et al. Identification of novel drought-responsive miRNA regulatory network of drought stress response in common vetch (Vicia sativa). Open Life Sci. 2021, 16, 1111–1121. [Google Scholar] [CrossRef]
- Wang, J.; Lin, J.; Wang, H.; Li, X.; Yang, Q.; Li, H.; Chang, Y. Identification and characterization of circRNAs in Pyrus betulifolia Bunge under drought stress. PLoS ONE 2018, 13, e0200692. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, Y.; Sun, S.; Li, L.; Wang, K. Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA. Genes 2022, 13, 712. [Google Scholar] [CrossRef]
- Lyu, Z.; Zhang, G.; Song, Y.; Diao, S.; He, C.; Zhang, J. Transcriptome and DNA methylome provide insights into the molecular regulation of drought stress in sea buckthorn. Genomics 2022, 114, 110345. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Hao, Z.; Zhu, Y.; Zhang, H.; Li, G. Genome-wide identification and functional analysis of circRNAs in Zea mays. PLoS ONE 2018, 13, e0202375. [Google Scholar] [CrossRef]
- Medina, C.A.; Samac, D.A.; Yu, L.-X. Pan-transcriptome identifying master genes and regulation network in response to drought and salt stresses in Alfalfa (Medicago sativa L.). Sci. Rep. 2021, 11, 17203. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, Y.; Li, Y.; Li, X. Identification and differential analysis of noncoding RNAs in response to drought in Phyllostachys aureosulcata f. spectabilis. Front. Plant Sci. 2022, 13, 1040470. [Google Scholar] [CrossRef]
- Lachmann, A.; Clarke, D.J.B.; Torre, D.; Xie, Z.; Ma’ayan, A. Interoperable RNA-Seq analysis in the cloud. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194521. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, H.; Li, C.; Wang, Y.; Qin, X.; Meng, L.; Sun, X. Genome-Wide Analysis of the WOX Transcription Factor Genes in Dendrobium catenatum Lindl. Genes 2022, 13, 1481. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Yang, D.; Wang, Y.; Yang, Y.; Sun, X. Genome-Wide Analysis of the TCP Transcription Factor Genes in Dendrobium catenatum Lindl. Int. J. Mol. Sci. 2021, 22, 10269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cui, Z.; Li, Y.; Kang, Y.; Song, X.; Wang, J.; Zhou, Y. Genome-Wide Identification and Expression Analysis of MYB Transcription Factor Superfamily in Dendrobium catenatum. Front. Genet. 2021, 12, 714696. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Li, C.; Wang, Y.; Qin, X.; Meng, L.; Sun, X. Genome-Wide Analysis of LBD Transcription Factor Genes in Dendrobiumcatenatum. Int. J. Mol. Sci. 2022, 23, 2089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, Y.; Ding, Y.; Yu, W.; Wang, J.; Lai, H.; Zhou, Y. Identification and Expression Analysis of WRKY Gene Family in Response to Abiotic Stress in Dendrobium catenatum. Front. Genet. 2022, 13, 800019. [Google Scholar] [CrossRef]
- Yang, Q.; Xiang, W.; Li, Z.; Nian, Y.; Fu, X.; Zhou, G.; Li, L.; Zhang, J.; Huang, G.; Han, X.; et al. Genome-Wide Characterization and Expression Analysis of HD-ZIP Gene Family in Dendrobium officinale. Front. Genet. 2022, 13, 797014. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, Q.; Lyu, P.; Lin, R.; Sun, C. Identification and expression profiling of selected MADS-box family genes in Dendrobium officinale. Genetica 2019, 147, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cai, X.; Shen, Q.; Chen, X.; Xu, M.; Ye, T.; Si, D.; Wu, L.; Chen, D.; Han, Z.; et al. Genome-wide analysis of basic helix-loop-helix genes in Dendrobium catenatum and functional characterization of DcMYC2 in jasmonate-mediated immunity to Sclerotium delphinii. Front. Plant Sci. 2022, 13, 956210. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Mansoor, S.; Khan, T.; Farooq, I.; Shah, L.R.; Sharma, V.; Sonne, C.; Rinklebe, J.; Ahmad, P. Drought and global hunger: Biotechnological interventions in sustainability and management. Planta 2022, 256, 97. [Google Scholar] [CrossRef]
- Lou, D.; Lu, S.; Chen, Z.; Lin, Y.; Yu, D.; Yang, X. Molecular characterization reveals that OsSAPK3 improves drought tolerance and grain yield in rice. BMC Plant Biol. 2023, 23, 53. [Google Scholar] [CrossRef]
- Basu, S.; Roychoudhury, A. Expression profiling of abiotic stress-inducible genes in response to multiple stresses in rice (Oryza sativa L.) varieties with contrasting level of stress tolerance. BioMed Res. Int. 2014, 2014, 706890. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Deyholos, M.K. Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 2006, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; You, J.; Xie, K.; Xie, W.; Xiong, L. Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol. Genet. Genom. MGG 2008, 280, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Mochida, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. Genome-wide survey and expression analysis of the plant-specific NAC transcription factor family in soybean during development and dehydration stress. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2011, 18, 263–276. [Google Scholar] [CrossRef]
- Dudhate, A.; Shinde, H.; Yu, P.; Tsugama, D.; Gupta, S.K.; Liu, S.; Takano, T. Comprehensive analysis of NAC transcription factor family uncovers drought and salinity stress response in pearl millet (Pennisetum glaucum). BMC Genom. 2021, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Li, C.; Lu, X.; Zhao, X.; Yan, C.; Wang, J.; Sun, Q.; Shan, S. Comprehensive genomic characterization of NAC transcription factor family and their response to salt and drought stress in peanut. BMC Plant Biol. 2020, 20, 454. [Google Scholar] [CrossRef]
- Wang, G.; Yuan, Z.; Zhang, P.; Liu, Z.; Wang, T.; Wei, L. Genome-wide analysis of NAC transcription factor family in maize under drought stress and rewatering. Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2020, 26, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, Y.; Bowers, C.; Ma, H. Isolation, sequence analysis, and expression studies of florally expressed cDNAs in Arabidopsis. Plant Mol. Biol. 2003, 53, 545–563. [Google Scholar] [CrossRef]
- Bu, Q.; Jiang, H.; Li, C.-B.; Zhai, Q.; Zhang, J.; Wu, X.; Sun, J.; Xie, Q.; Li, C. Role of the Arabidopsis thaliana NAC transcription factors ANAC019 and ANAC055 in regulating jasmonic acid-signaled defense responses. Cell Res. 2008, 18, 756–767. [Google Scholar] [CrossRef]
- Jensen, M.K.; Kjaersgaard, T.; Nielsen, M.M.; Galberg, P.; Petersen, K.; O’Shea, C.; Skriver, K. The Arabidopsis thaliana NAC transcription factor family: Structure-function relationships and determinants of ANAC019 stress signalling. Biochem. J. 2010, 426, 183–196. [Google Scholar] [CrossRef]
- Sukiran, N.L.; Ma, J.C.; Ma, H.; Su, Z. ANAC019 is required for recovery of reproductive development under drought stress in Arabidopsis. Plant Mol. Biol. 2019, 99, 161–174. [Google Scholar] [CrossRef]
- Chen, D.; Chai, S.; McIntyre, C.L.; Xue, G.-P. Overexpression of a predominantly root-expressed NAC transcription factor in wheat roots enhances root length, biomass and drought tolerance. Plant Cell Rep. 2018, 37, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 174. [Google Scholar] [CrossRef]
- Mao, H.; Li, S.; Chen, B.; Jian, C.; Mei, F.; Zhang, Y.; Li, F.; Chen, N.; Li, T.; Du, L.; et al. Variation in cis-regulation of a NAC transcription factor contributes to drought tolerance in wheat. Mol. Plant 2022, 15, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Y.; Kim, S.Y.; Hyeon, D.Y.; Kim, D.H.; Dong, T.; Park, Y.; Jin, J.B.; Joo, S.-H.; Kim, S.-K.; Hong, J.C.; et al. The Arabidopsis NAC transcription factor ANAC096 cooperates with bZIP-type transcription factors in dehydration and osmotic stress responses. Plant Cell 2013, 25, 4708–4724. [Google Scholar] [CrossRef]
- Chen, K.; Guo, Y.; Song, M.; Liu, L.; Xue, H.; Dai, H.; Zhang, Z. Dual role of MdSND1 in the biosynthesis of lignin and in signal transduction in response to salt and osmotic stress in apple. Hortic. Res. 2020, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Shin, G.; Kim, M.C.; Shen, M.; Lee, S.Y.; Yun, D.-J. Histone Deacetylase HDA9 With ABI4 Contributes to Abscisic Acid Homeostasis in Drought Stress Response. Front. Plant Sci. 2020, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Malladi, A.; van Iersel, M.W. Physiological and molecular responses to drought in Petunia: The importance of stress severity. J. Exp. Bot. 2012, 63, 6335–6345. [Google Scholar] [CrossRef]
- Hasan, M.M.; Alabdallah, N.M.; Alharbi, B.M.; Waseem, M.; Yao, G.; Liu, X.-D.; Abd El-Gawad, H.G.; El-Yazied, A.A.; Ibrahim, M.F.M.; Jahan, M.S.; et al. GABA: A Key Player in Drought Stress Resistance in Plants. Int. J. Mol. Sci. 2021, 22, 10136. [Google Scholar] [CrossRef]
- Du, Q.; Zhao, M.; Gao, W.; Sun, S.; Li, W.-X. microRNA/microRNA* complementarity is important for the regulation pattern of NFYA5 by miR169 under dehydration shock in Arabidopsis. Plant J. Cell Mol. Biol. 2017, 91, 22–33. [Google Scholar] [CrossRef]
- Yuan, W.; Suo, J.; Shi, B.; Zhou, C.; Bai, B.; Bian, H.; Zhu, M.; Han, N. The barley miR393 has multiple roles in regulation of seedling growth, stomatal density, and drought stress tolerance. Plant Physiol. Biochem. PPB 2019, 142, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, B.A.; Arshad, M.; Gruber, M.Y.; Kohalmi, S.E.; Hannoufa, A. The interplay between miR156/SPL13 and DFR/WD40-1 regulate drought tolerance in alfalfa. BMC Plant Biol. 2019, 19, 434. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; He, J.; Ping, Y.; Guo, J.; Hou, N.; Cao, F.; Li, X.; Geng, D.; Wang, S.; Chen, P.; et al. The positive feedback regulatory loop of miR160-Auxin Response Factor 17-HYPONASTIC LEAVES 1 mediates drought tolerance in apple trees. Plant Physiol. 2022, 188, 1686–1708. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, N.; Wang, H.; Kasahara, H.; Liu, J.; Macpherson, C.; Machida, Y.; Kamiya, Y.; Hannah, M.A.; Chua, N.-H. IAA-Ala Resistant3, an evolutionarily conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell 2012, 24, 3590–3602. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Han, Y.; Zeng, Q.; Wang, C.; Wang, H.; Zhang, J.; Cai, M.; Lu, J.; Chen, T. Whole-Transcriptome Sequencing Reveals the Global Molecular Responses and NAC Transcription Factors Involved in Drought Stress in Dendrobium catenatum. Antioxidants 2024, 13, 94. https://doi.org/10.3390/antiox13010094
Zhang S, Han Y, Zeng Q, Wang C, Wang H, Zhang J, Cai M, Lu J, Chen T. Whole-Transcriptome Sequencing Reveals the Global Molecular Responses and NAC Transcription Factors Involved in Drought Stress in Dendrobium catenatum. Antioxidants. 2024; 13(1):94. https://doi.org/10.3390/antiox13010094
Chicago/Turabian StyleZhang, Siqi, Yuliang Han, Qinzong Zeng, Chenchang Wang, Huizhong Wang, Juncheng Zhang, Maohong Cai, Jiangjie Lu, and Tao Chen. 2024. "Whole-Transcriptome Sequencing Reveals the Global Molecular Responses and NAC Transcription Factors Involved in Drought Stress in Dendrobium catenatum" Antioxidants 13, no. 1: 94. https://doi.org/10.3390/antiox13010094