
Integrated Transcriptomic and Proteomic Analyses of Low-Nitrogen-Stress Tolerance and Function Analysis of ZmGST42 Gene in Maize
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatment
2.2. Transcriptomic Analysis of the Maize Leaf Samples
2.3. Proteomic Analysis of the Maize Leaf Samples
2.4. Correlation Analysis
2.5. RNA Extraction, cDNA Synthesis, and qRT-PCR Verification of RNA-Seq Results
2.6. Gene Function Analysis of ZmGST42
2.6.1. Materials and Methods
2.6.2. Phenotypic Characterization of the WT Plant and Maize Mutant under Low-N Stress
2.6.3. Physiological and Biochemical Indices Determination
3. Results
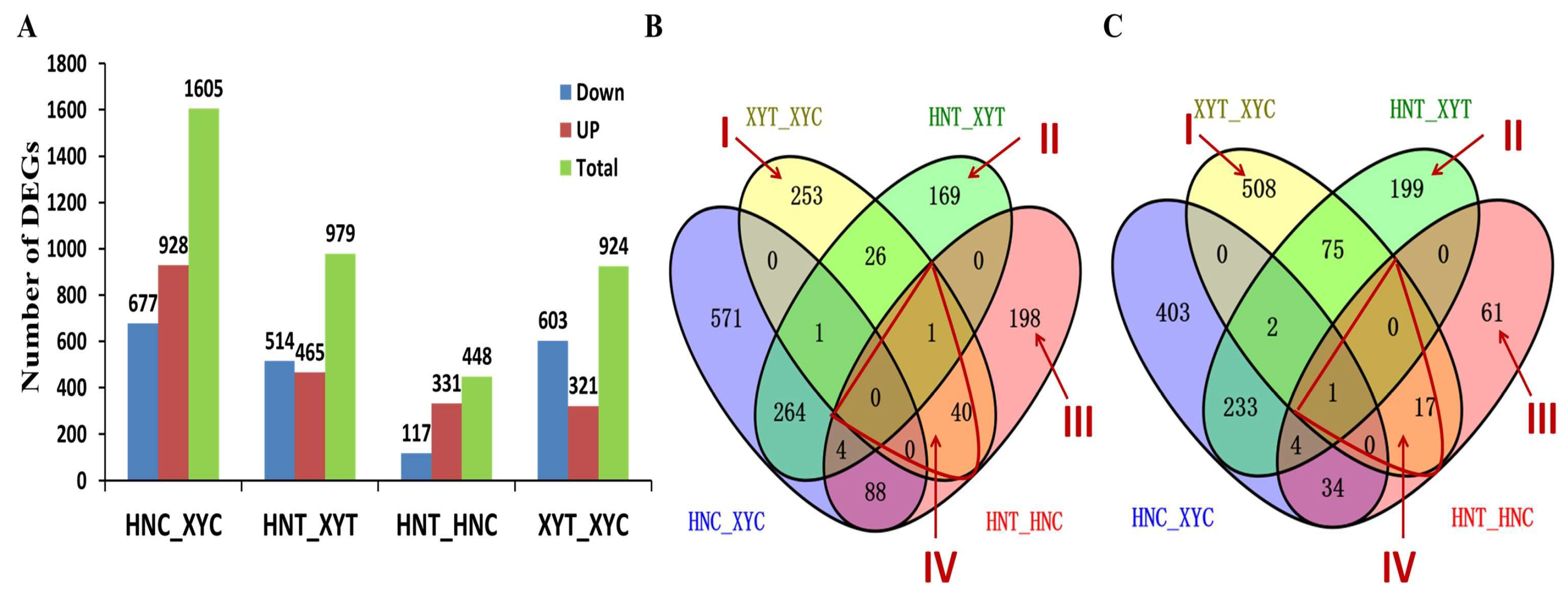
3.1. RNA-Seq Transcriptome Analysis of the Two Maize Cultivars’ Response to Low N Treatment
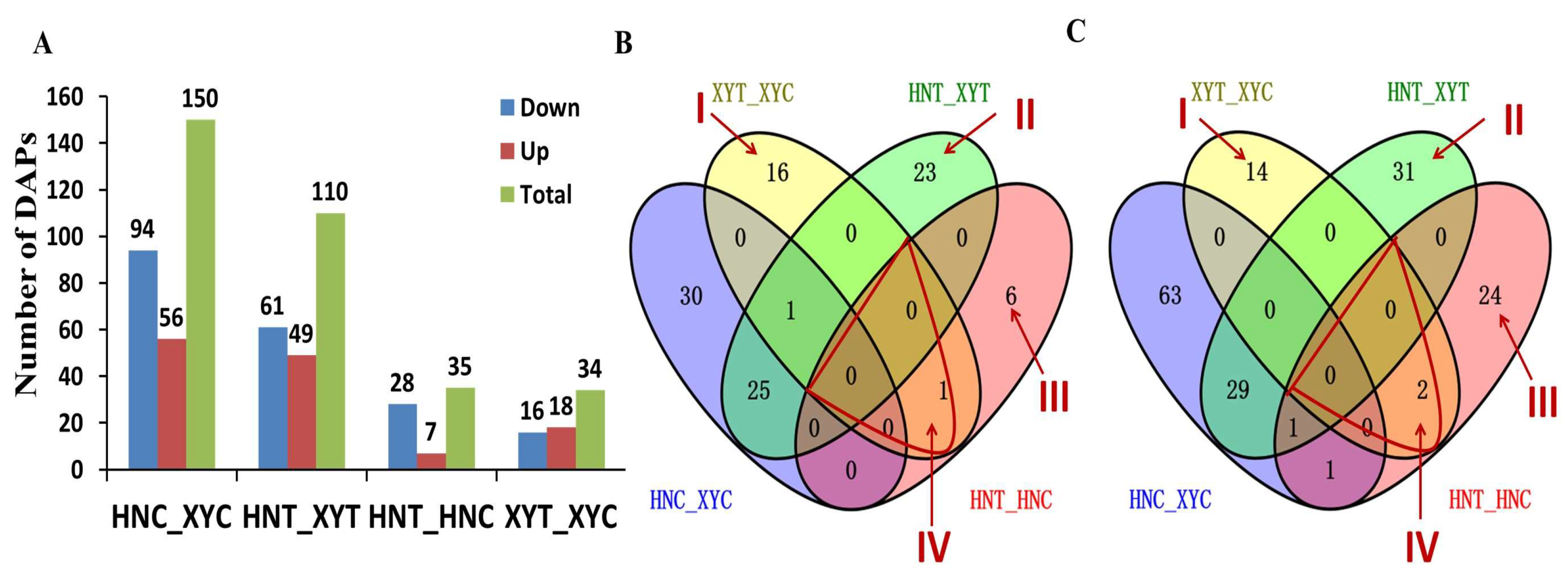
3.2. Proteomic Analysis of the Two Maize Genotypes’ Response to Low-N Treatment
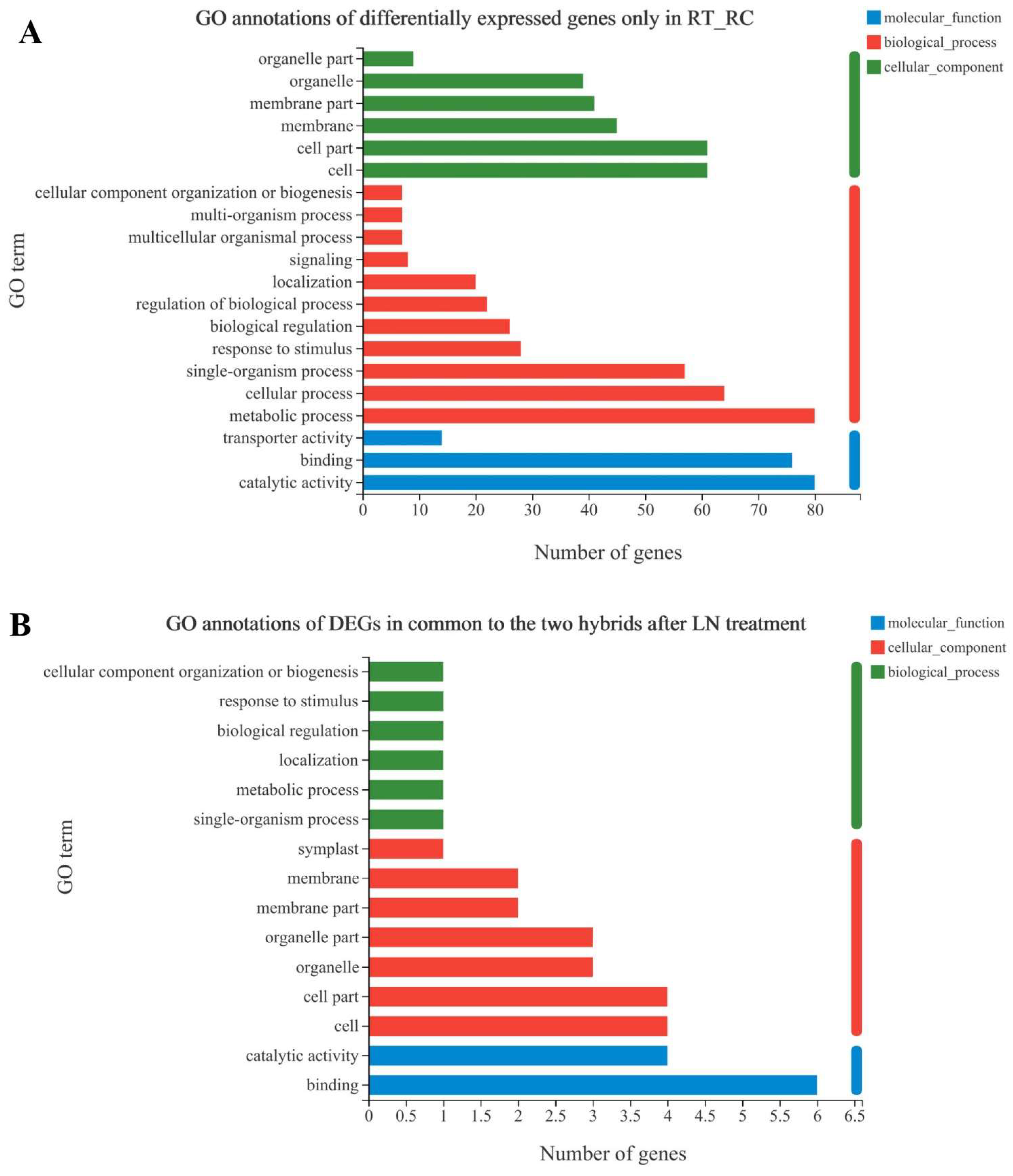
3.3. GO annotation of Specifically Expressed Genes
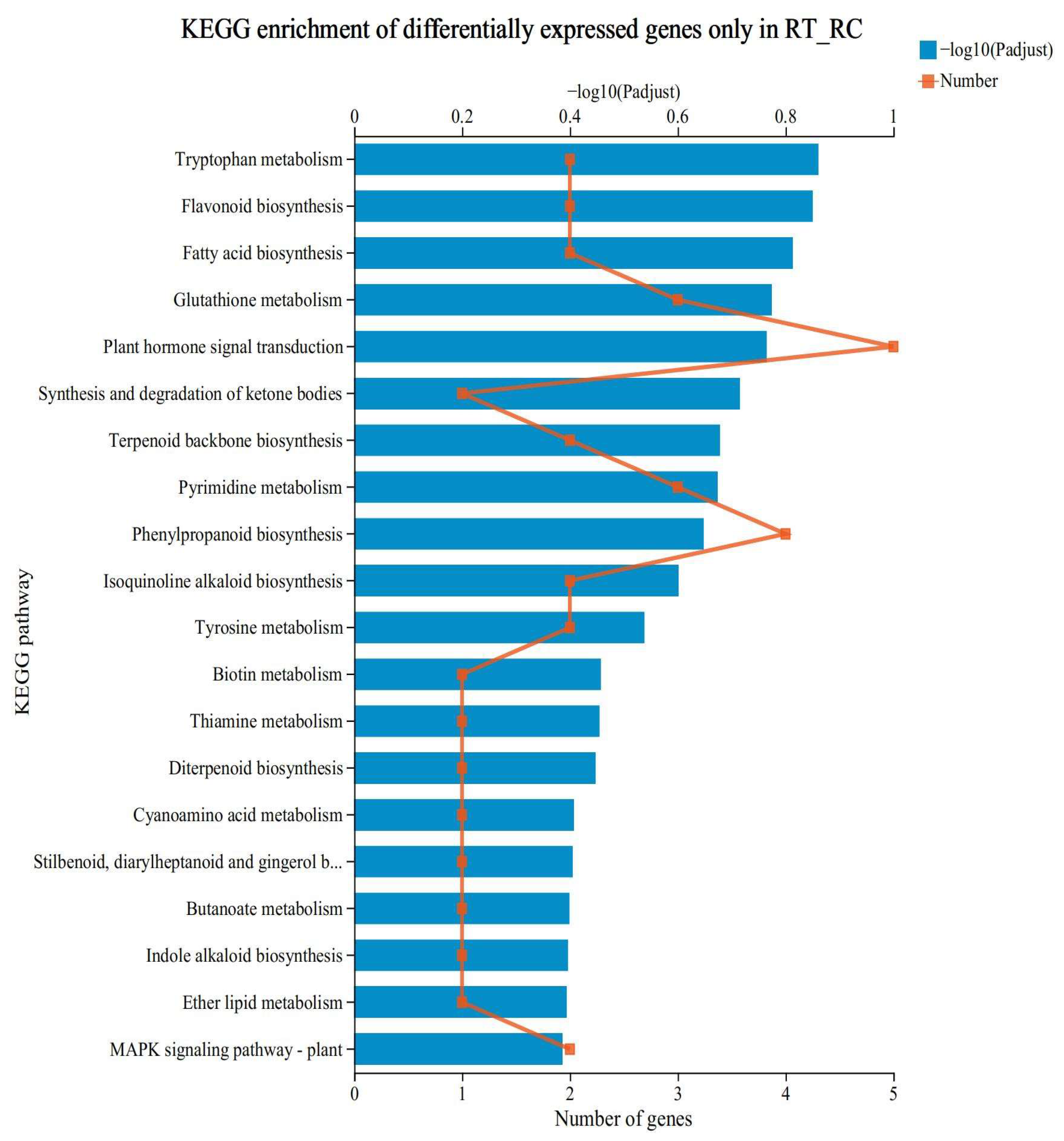
3.4. KEGG Functional Enrichment Analysis of Specifically Expressed Genes
3.5. Transcripts and Proteins Association Analysis
3.5.1. Quantitative Relationship between Transcriptome and Proteome
3.5.2. Bioinformatics Analysis of DAPs/DEGs with the Same Expression Trend
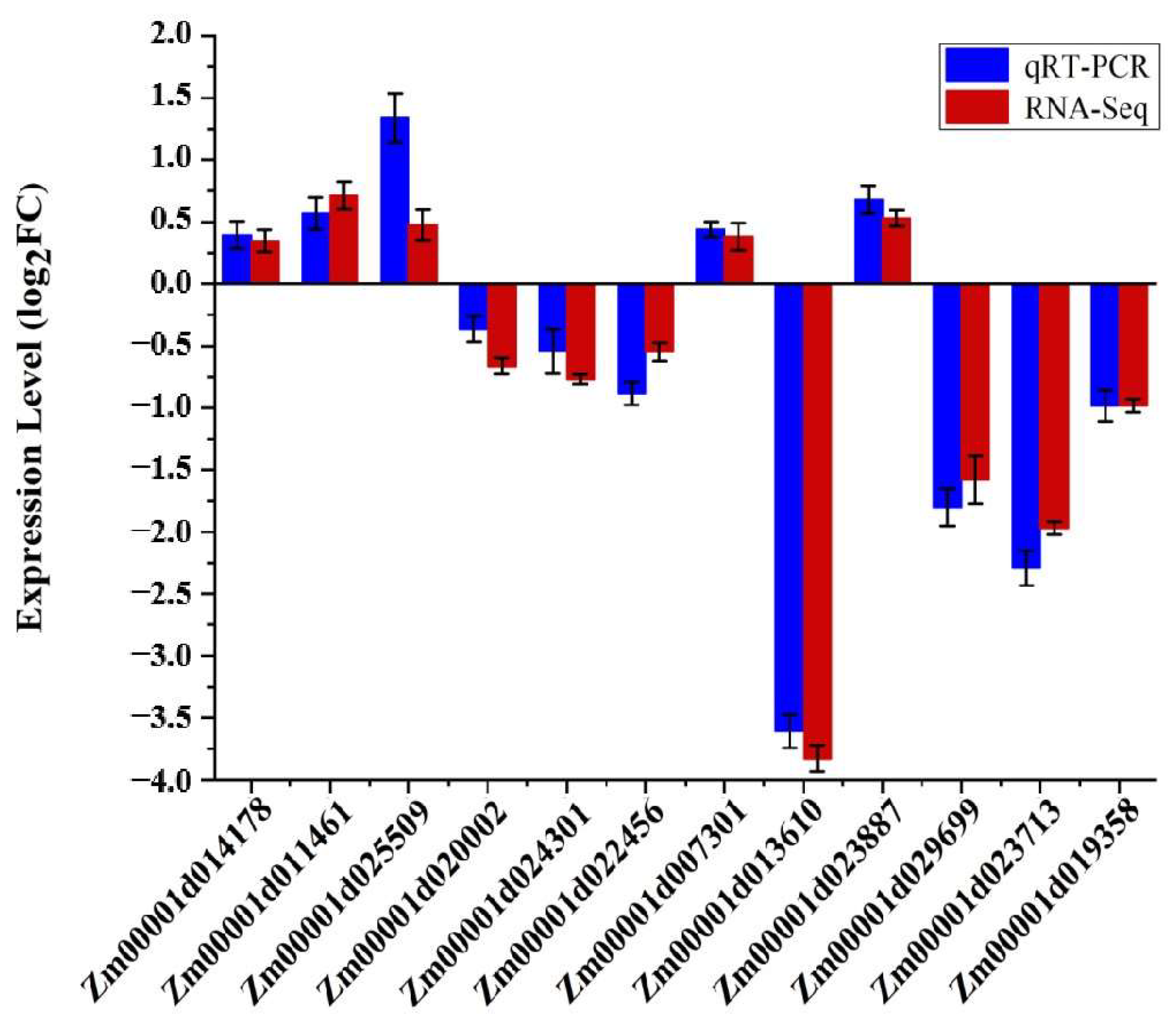
3.6. Real-Time Quantitative PCR (qRT-PCR) Analysis
3.7. Function Analysis of ZmGST42 Gene in Low-N-Stress Response
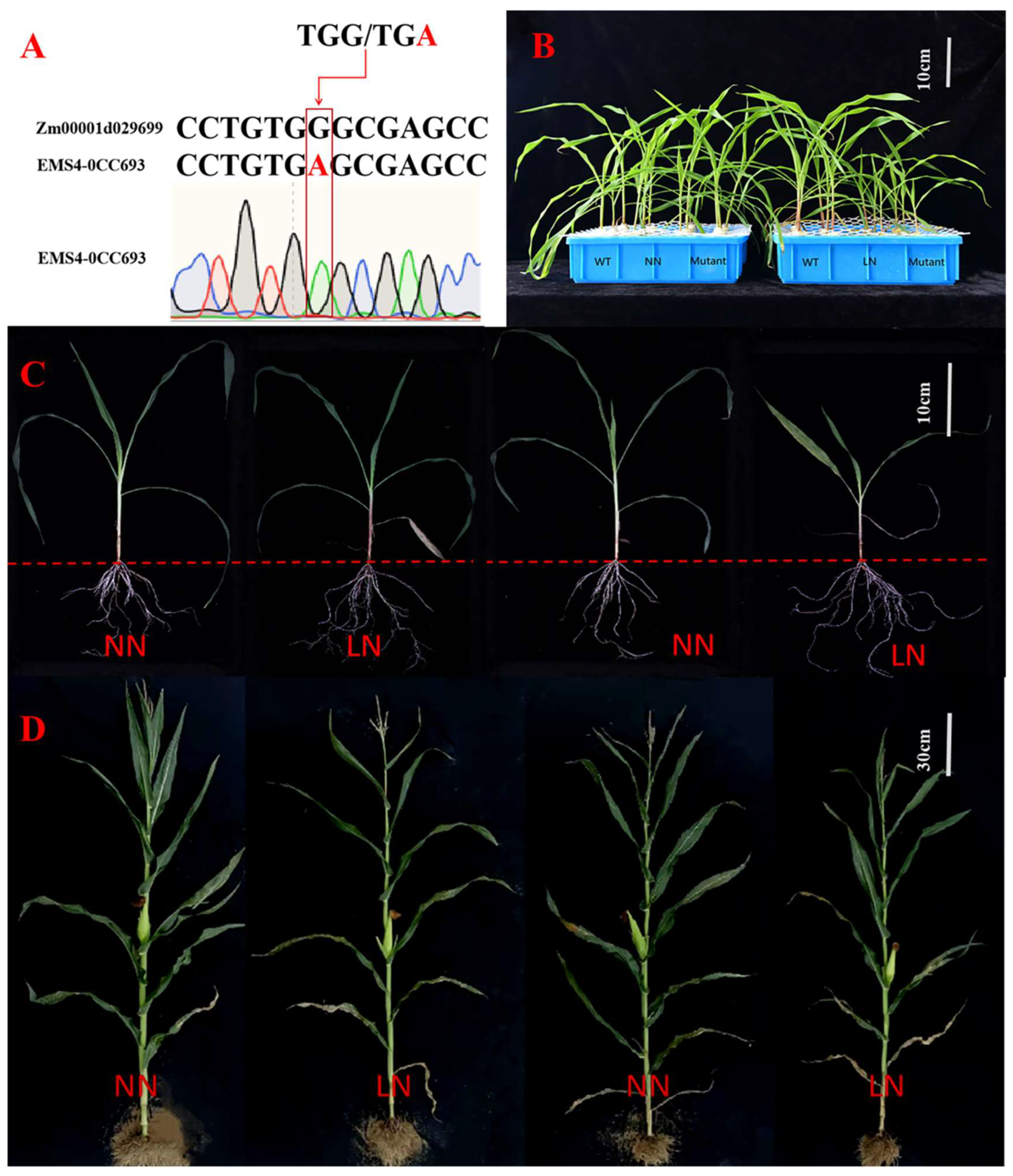
3.7.1. The Effect of Nitrogen Supply Level on Phenotypic Traits of ZmGST42 Mutant
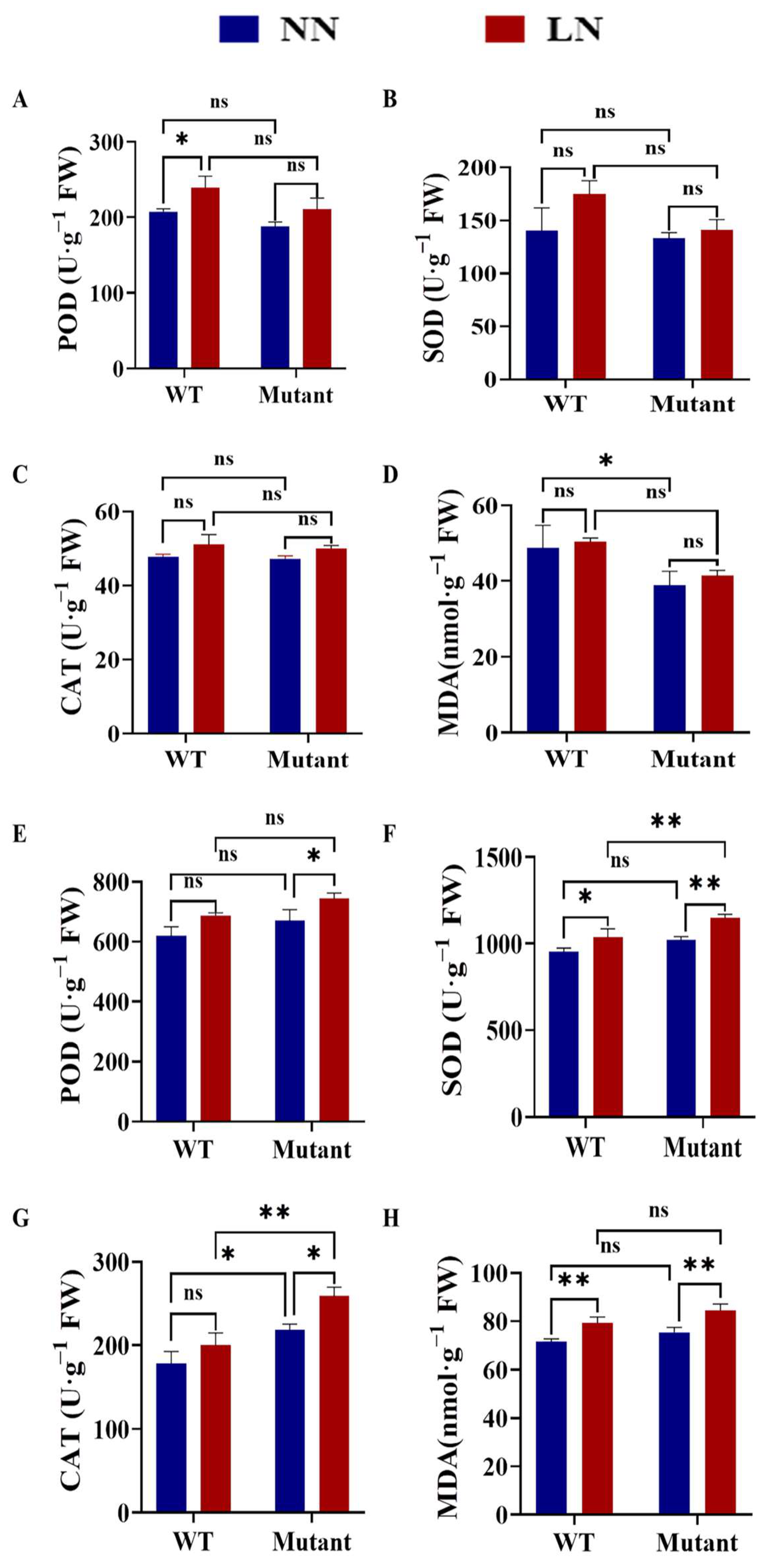
3.7.2. Effects of Different N Concentrations on the Antioxidant Enzyme Activities and MDA Content of WT and Zmgst42 Mutant Leaves
3.7.3. Effects of Different Nitrogen Concentrations on the Contents of Relevant Nitrogen Metabolizing Enzymes in ZmGST42 Leaves
4. Discussion
4.1. Low-N-Stress-Tolerance-Related DEGs and DAPs Identified in the Low-N-Tolerant Cultivar XY335
4.1.1. Genes and Proteins Related to Photosynthesis under Low-Nitrogen Stress
4.1.2. RNA Metabolism and Protein Phosphorylation-Related Proteins and Genes in Response to Low-N Stress
4.1.3. Defense-Related Proteins and Genes under Low-N Stress
4.2. Differentially Regulated DAPs and DEGs in Low-N-Sensitive Cultivar HN138
4.2.1. Antioxidant-Related Proteins and Genes under Low-Nitrogen Stress
4.2.2. Proteins and Genes Related to “Stress Response” and “Stimulus Response” under Low-N Conditions
4.2.3. Other Defense-Related Proteins and Genes in Response to Low-N Stress
4.2.4. Key Genotypic Differences in Transcriptomic and Proteomic Responses to Low N Stress
4.3. Function Analysis of ZmGST42
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, N.-Q.; Lin, H.-X. Higher Yield with Less Nitrogen Fertilizer. Nat. Plants 2020, 6, 1078–1079. [Google Scholar] [CrossRef]
- Chen, K.-E.; Chen, H.-Y.; Tseng, C.-S.; Tsay, Y.-F. Improving Nitrogen Use Efficiency by Manipulating Nitrate Remobilization in Plants. Nat. Plants 2020, 6, 1126–1135. [Google Scholar] [CrossRef]
- Ye, J.Y.; Tian, W.H.; Jin, C.W. Nitrogen in Plants: From Nutrition to the Modulation of Abiotic Stress Adaptation. Stress Biol. 2022, 2, 4. [Google Scholar] [CrossRef]
- Gheith, E.M.S.; El-Badry, O.Z.; Lamlom, S.F.; Ali, H.M.; Siddiqui, M.H.; Ghareeb, R.Y.; El-Sheikh, M.H.; Jebril, J.; Abdelsalam, N.R.; Kandil, E.E. Maize (Zea mays L.) Productivity and Nitrogen Use Efficiency in Response to Nitrogen Application Levels and Time. Front. Plant Sci. 2022, 13, 941343. [Google Scholar] [CrossRef]
- Kissel, D.E.; Bock, B.R.; Ogles, C.Z. Thoughts on Acidification of Soils by Nitrogen and Sulfur Fertilizers. Agrosystems Geosci. Environ. 2020, 3, e20060. [Google Scholar] [CrossRef]
- McLaren, J.S. Crop Biotechnology Provides an Opportunity to Develop a Sustainable Future. Trends Biotechnol. 2005, 23, 339–342. [Google Scholar] [CrossRef]
- Yu, Y.; Qian, C.; Gu, W.; Li, C. Responses of Root Characteristic Parameters and Plant Dry Matter Accumulation, Distribution and Transportation to Nitrogen Levels for Spring Maize in Northeast China. Agriculture 2021, 11, 308. [Google Scholar] [CrossRef]
- Maresma, Á.; Ariza, M.; Martínez, E.; Lloveras, J.; Martínez-Casasnovas, J. Analysis of Vegetation Indices to Determine Nitrogen Application and Yield Prediction in Maize (Zea mays L.) from a Standard UAV Service. Remote Sens. 2016, 8, 973. [Google Scholar] [CrossRef]
- Li, G.; Cheng, G.; Lu, W.; Lu, D. Differences of Yield and Nitrogen Use Efficiency under Different Applications of Slow Release Fertilizer in Spring Maize. J. Integr. Agric. 2021, 20, 554–564. [Google Scholar] [CrossRef]
- Paponov, I.A.; Bondarenko, Y.V.; Neumann, G.; Engels, C. Effect of Nitrogen Supply on Individual Kernel Weight and 14C Partitioning in Kernels of Maize during Lag Phase and Grain Filling. In Plant Nutrition; Horst, W.J., Schenk, M.K., Bürkert, A., Claassen, N., Flessa, H., Frommer, W.B., Goldbach, H., Olfs, H.-W., Römheld, V., Sattelmacher, B., et al., Eds.; Springer: Dordrecht, The Netherlands, 2001; pp. 122–123. ISBN 978-0-7923-7105-2. [Google Scholar]
- Mc, R.; Wm, R.; Mej, S.; Sv, F.; Lt, Z.; Jcc, G.; Ro, D. Genotypic Variation and Relationships among Nitrogen-Use Efficiency and Agronomic Traits in Tropical Maize Inbred Lines. Genet. Mol. Res. 2017, 22, 16. [Google Scholar] [CrossRef]
- Ge, M.; Lyu, Y.-D.; Zhang, T.-F.; Zhou, L.; Lin, F.; Zhao, H. Global Transcriptome Analysis in High- and Low-Nitrogen Responsive Inbred Lines of Maize. Acta Agron. Sin. 2016, 42, 1487. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, J.; Gao, F.; Liu, P.; Zhao, B.; Zhang, J. Photosynthetic Characteristics and Chloroplast Ultrastructure of Summer Maize Response to Different Nitrogen Supplies. Front. Plant Sci. 2018, 9, 576. [Google Scholar] [CrossRef]
- Ma, L.; Huang, Z.; Li, S.; Ashraf, U.; Yang, W.; Liu, H.; Xu, D.; Li, W.; Mo, Z. Melatonin and Nitrogen Applications Modulate Early Growth and Related Physio-Biochemical Attributes in Maize Under Cd Stress. J. Soil Sci. Plant Nutr. 2021, 21, 978–990. [Google Scholar] [CrossRef]
- Li, X.; Ji, P.; Zhou, B.; Dong, W.; Zhang, L.; Xiao, K.; Yin, B.; Zhang, Y. Nitrogen Partitioning Traits and Expression Patterns of N Metabolism-Associated Genes in Maize Hybrids with Contrasting N Utilization Efficiencies. Agron. J. 2021, 113, 1439–1456. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive Oxygen Gene Network of Plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Vaish, S.; Gupta, D.; Mehrotra, R.; Mehrotra, S.; Basantani, M.K. Glutathione S-Transferase: A Versatile Protein Family. 3 Biotech 2020, 10, 321. [Google Scholar] [CrossRef]
- Xu, J.; Tian, Y.-S.; Xing, X.-J.; Peng, R.-H.; Zhu, B.; Gao, J.-J.; Yao, Q.-H. Over-Expression of AtGSTU19 Provides Tolerance to Salt, Drought and Methyl Viologen Stresses in Arabidopsis. Physiol. Plant. 2016, 156, 164–175. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Zhang, Y.; Li, C.; Gong, S.; Yan, S.; Li, G.; Hu, G.; Ren, H.; Yang, J.; et al. Comparative Transcriptome Analysis of Salt-Sensitive and Salt-Tolerant Maize Reveals Potential Mechanisms to Enhance Salt Resistance. Genes Genom. 2019, 41, 781–801. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, A.-Q.; Xing, X.-J.; Chen, L.; Fu, X.-Y.; Peng, R.-H.; Tian, Y.-S.; Yao, Q.-H. Transgenic Arabidopsis Plants Expressing Grape Glutathione S-Transferase Gene (VvGSTF13) Show Enhanced Tolerance to Abiotic Stress. Biochem. Mosc. 2018, 83, 755–765. [Google Scholar] [CrossRef]
- Kakumanu, A.; Ambavaram, M.M.R.; Klumas, C.; Krishnan, A.; Batlang, U.; Myers, E.; Grene, R.; Pereira, A. Effects of Drought on Gene Expression in Maize Reproductive and Leaf Meristem Tissue Revealed by RNA-Seq. Plant Physiol. 2012, 160, 846–867. [Google Scholar] [CrossRef]
- Sant’Ana, D.V.P.; Lefsrud, M. Tomato Proteomics: Tomato as a Model for Crop Proteomics. Sci. Hortic. 2018, 239, 224–233. [Google Scholar] [CrossRef]
- Terzi, H.; Yıldız, M. Proteomic Analysis Reveals the Role of Exogenous Cysteine in Alleviating Chromium Stress in Maize Seedlings. Ecotoxicol. Environ. Saf. 2021, 209, 111784. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, N.; Liu, S.; Dong, A.; Zenda, T.; Liu, X.; Li, J.; Duan, H. Comparative Proteomic Analysis of Two Contrasting Maize Hybrids’ Responses to Low Nitrogen Stress at the Twelve Leaf Stage and Function Verification of ZmTGA Gene. Genes 2022, 13, 670. [Google Scholar] [CrossRef]
- Hama, B.M.; Mohammed, A.A. Physiological Performance of Maize (Zea mays L.) under Stress Conditions of Water Deficit and High Temperature. Appl. Ecol. Environ. Res. 2019, 17, 1261–1278. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Wang, X.; Liu, G.; Jin, H.; Dong, A.; Yang, Y.; Duan, H. Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines. Int. J. Mol. Sci. 2019, 20, 1268. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lu, X.; Liu, J.; Ren, W.; Yang, Q.; Chai, Z.; Chen, R.; Wang, L.; Zhao, J.; Lang, Z.; Wang, H.; et al. Gene-Indexed Mutations in Maize. Mol. Plant 2018, 11, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Usandivaras, L.M.A.; Gutiérrez-Boem, F.H.; Salvagiotti, F. Contrasting Effects of Phosphorus and Potassium Deficiencies on Leaf Area Development in Maize. Crop Sci. 2018, 58, 2099–2109. [Google Scholar] [CrossRef]
- Azama, K.; Abe, S.; Sugimoto, H.; Davies, E. Lysine-Containing Proteins in Maize Endosperm: A Major Contribution from Cytoskeleton-Associated Carbohydrate-Metabolizing Enzymes. Planta 2003, 217, 628–638. [Google Scholar] [CrossRef]
- Wei, S.; Wang, X.; Shi, D.; Li, Y.; Zhang, J.; Liu, P.; Zhao, B.; Dong, S. The Mechanisms of Low Nitrogen Induced Weakened Photosynthesis in Summer Maize (Zea mays L.) under Field Conditions. Plant Physiol. Biochem. 2016, 105, 118–128. [Google Scholar] [CrossRef]
- Han, L.-B.; Song, G.-L.; Zhang, X. Preliminary Observations on Physiological Responses of Three Turfgrass Species to Traffic Stress. HortTechnology 2008, 18, 139–143. [Google Scholar] [CrossRef]
- Durak, I.; Yurtarslanl, Z.; Canbolat, O.; Akyol, Ö. A Methodological Approach to Superoxide Dismutase (SOD) Activity Assay Based on Inhibition of Nitroblue Tetrazolium (NBT) Reduction. Clin. Chim. Acta 1993, 214, 103–104. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Plumb-Dhindsa, P.; Thorpe, T.A. Leaf Senescence: Correlated with Increased Levels of Membrane Permeability and Lipid Peroxidation, and Decreased Levels of Superoxide Dismutase and Catalase. J. Exp. Bot. 1981, 32, 93–101. [Google Scholar] [CrossRef]
- Nakano, Y.; Asada, K. Hydrogen Peroxide Is Scavenged by Ascorbate-Specific Peroxidase in Spinach Chloroplasts. Plant Cell Physiol. 1981, 22, 867–880. [Google Scholar] [CrossRef]
- Shaner, D.L.; Boyer, J.S. Nitrate Reductase Activity in Maize (Zea mays L.) Leaves: II. Regulation by Nitrate Flux at Low Leaf Water Potential 1. Plant Physiol. 1976, 58, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, B.; Chu, C. Nitrogen Assimilation in Plants: Current Status and Future Prospects. J. Genet. Genomics 2022, 49, 394–404. [Google Scholar] [CrossRef]
- Leng, P.; Zhao, J. Transcription Factors as Molecular Switches to Regulate Drought Adaptation in Maize. Theor. Appl. Genet. 2020, 133, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, Y.; Hua, W.; Last, R.L. A New Light on Photosystem II Maintenance in Oxygenic Photosynthesis. Front. Plant Sci. 2019, 10, 975. [Google Scholar] [CrossRef]
- Agurla, S.; Gahir, S.; Munemasa, S.; Murata, Y.; Raghavendra, A.S. Mechanism of Stomatal Closure in Plants Exposed to Drought and Cold Stress. Adv. Exp. Med. Biol. 2018, 1081, 215–232. [Google Scholar] [CrossRef]
- Gjindali, A.; Herrmann, H.A.; Schwartz, J.-M.; Johnson, G.N.; Calzadilla, P.I. A Holistic Approach to Study Photosynthetic Acclimation Responses of Plants to Fluctuating Light. Front. Plant Sci. 2021, 12, 668512. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gu, Z.; Xin, D.; Hao, L.; Liu, C.; Huang, J.; Ma, B.; Zhang, H. Identification and Characterization of Putative CIPK Genes in Maize. J. Genet. Genom. 2011, 38, 77–87. [Google Scholar] [CrossRef]
- Ru, J.-N.; Hou, Z.-H.; Zheng, L.; Zhao, Q.; Wang, F.-Z.; Chen, J.; Zhou, Y.-B.; Chen, M.; Ma, Y.-Z.; Xi, Y.-J.; et al. Genome-Wide Analysis of DEAD-Box RNA Helicase Family in Wheat (Triticum aestivum) and Functional Identification of TaDEAD-Box57 in Abiotic Stress Responses. Front. Plant Sci. 2021, 12, 797276. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Jarmoskaite, I.; Lambowitz, A.M. Toward a Molecular Understanding of RNA Remodeling by DEAD-Box Proteins. RNA Biol. 2013, 10, 44–55. [Google Scholar] [CrossRef]
- Hsu, Y.-F.; Chen, Y.-C.; Hsiao, Y.-C.; Wang, B.-J.; Lin, S.-Y.; Cheng, W.-H.; Jauh, G.-Y.; Harada, J.J.; Wang, C.-S. AtRH57, a DEAD-Box RNA Helicase, Is Involved in Feedback Inhibition of Glucose-Mediated Abscisic Acid Accumulation during Seedling Development and Additively Affects Pre-Ribosomal RNA Processing with High Glucose. Plant J. 2014, 77, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhou, R.; Shi, X.; Kang, M.; Wang, H.; Chen, H. Two Thermostable Nucleases Coexisted in Staphylococcus Aureus: Evidence from Mutagenesis and In Vitro Expression. FEMS Microbiol. Lett. 2008, 284, 176–183. [Google Scholar] [CrossRef]
- Li, L.; Krishnan, M.; Baseman, J.B.; Kannan, T.R. Molecular Cloning, Expression, and Characterization of a Ca2+-Dependent, Membrane-Associated Nuclease of Mycoplasma genitalium. J. Bacteriol. 2010, 192, 4876–4884. [Google Scholar] [CrossRef]
- Wei, K.; Wang, Y.; Zhong, X.; Pan, S. Protein Kinase Structure, Expression and Regulation in Maize Drought Signaling. Mol. Breed. 2014, 34, 583–602. [Google Scholar] [CrossRef]
- Balestrini, R.; Chitarra, W.; Ghirardo, A.; Nardini, A.; Nerva, L. A Stressful Life: How Plants Cope with Multiple Biotic and Abiotic Adverse Factors. Plant Stress 2022, 5, 100095. [Google Scholar] [CrossRef]
- Mira, H.; Martínez, N.; Peñarrubia, L. Expression of a Vegetative-Storage-Protein Gene from Arabidopsis Is Regulated by Copper, Senescence and Ozone. Planta 2002, 214, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.-S.; Bae, E.-K.; Choi, Y.-I.; Noh, E.-W. Differential Expression of a Poplar Copper Chaperone Gene in Response to Various Abiotic Stresses. Tree Physiol. 2005, 25, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Barth, O.; Vogt, S.; Uhlemann, R.; Zschiesche, W.; Humbeck, K. Stress Induced and Nuclear Localized HIPP26 from Arabidopsis Thaliana Interacts via Its Heavy Metal Associated Domain with the Drought Stress Related Zinc Finger Transcription Factor ATHB29. Plant Mol. Biol. 2009, 69, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Oberschall, A.; Deak, M.; Torok, K.; Sass, L.; Vass, I.; Kovacs, I.; Feher, A.; Dudits, D.; Horvath, G.V. A Novel Aldose/Aldehyde Reductase Protects Transgenic Plants against Lipid Peroxidation under Chemical and Drought Stresses. Plant J. 2000, 24, 437–446. [Google Scholar] [CrossRef]
- Simpson, P.J.; Tantitadapitak, C.; Reed, A.M.; Mather, O.C.; Bunce, C.M.; White, S.A.; Ride, J.P. Characterization of Two Novel Aldo–Keto Reductases from Arabidopsis: Expression Patterns, Broad Substrate Specificity, and an Open Active-Site Structure Suggest a Role in Toxicant Metabolism Following Stress. J. Mol. Biol. 2009, 392, 465–480. [Google Scholar] [CrossRef]
- Ning, D.-L.; Liu, K.-H.; Liu, C.-C.; Liu, J.-W.; Qian, C.-R.; Yu, Y.; Wang, Y.-F.; Wang, Y.-C.; Wang, B.-C. Large-Scale Comparative Phosphoprotein Analysis of Maize Seedling Leaves during Greening. Planta 2016, 243, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wu, J.; Zheng, Q.; Jiang, Y.; Zhang, M.; Zhu, S. Genome-Wide Identification and Comparative Analysis of Alfin-Like Transcription Factors in Maize. Genes Genom. 2017, 39, 261–275. [Google Scholar] [CrossRef]
- Han, G.; Qiao, Z.; Li, Y.; Wang, C.; Wang, B. The Roles of CCCH Zinc-Finger Proteins in Plant Abiotic Stress Tolerance. Int. J. Mol. Sci. 2021, 22, 8327. [Google Scholar] [CrossRef]
- Dardanelli, M.; Angelini, J.; Fabra, A. A Calcium-Dependent Bacterial Surface Protein Is Involved in the Attachment of Rhizobia to Peanut Roots. Can. J. Microbiol. 2003, 49, 399–405. [Google Scholar] [CrossRef]
- Gucciardo, S.; Wisniewski, J.-P.; Brewin, N.J.; Bornemann, S. A Germin-Like Protein with Superoxide Dismutase Activity in Pea Nodules with High Protein Sequence Identity to a Putative Rhicadhesin Receptor. J. Exp. Bot. 2007, 58, 1161–1171. [Google Scholar] [CrossRef]
- Wong, J.H.; Balmer, Y.; Cai, N.; Tanaka, C.K.; Vensel, W.H.; Hurkman, W.J.; Buchanan, B.B. Unraveling Thioredoxin-Linked Metabolic Processes of Cereal Starchy Endosperm Using Proteomics. FEBS Lett. 2003, 547, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Coll, N.S.; Epple, P.; Dangl, J.L. Programmed Cell Death in the Plant Immune System. Cell Death Differ. 2011, 18, 1247–1256. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, H.; Hong, Y.; Liu, S.; Li, D.; Song, F. Stress-Responsive Expression, Subcellular Localization and Protein–Protein Interactions of the Rice Metacaspase Family. Int. J. Mol. Sci. 2015, 16, 16216–16241. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, X.; Wang, K.; Li, H.; Duan, X.; Tang, C.; Kang, Z. TaMCA1, a Regulator of Cell Death, Is Important for the Interaction between Wheat and Puccinia Striiformis. Sci. Rep. 2016, 6, 26946. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Niu, S.; Han, C.; Yu, J.; Li, D. Nuclear Localization of Beet Black Scorch Virus Capsid Protein and Its Interaction with Importin α. Virus Res. 2011, 155, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ge, X.; Ma, H. The Rice OsDIL Gene Plays a Role in Drought Tolerance at Vegetative and Reproductive Stages. Plant Mol. Biol. 2013, 82, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Liu, P.; Xue, Q.; Ji, C.; Qi, T.; Guo, J.; Guo, J.; Kang, Z. TaDIR1-2, a Wheat Ortholog of Lipid Transfer Protein AtDIR1 Contributes to Negative Regulation of Wheat Resistance against Puccinia striiformis f. sp. tritici. Front. Plant Sci. 2017, 8, 521. [Google Scholar] [CrossRef] [PubMed]
- Fakih, Z.; Plourde, M.B.; Germain, H. Differential Participation of Plant Ribosomal Proteins from the Small Ribosomal Subunit in Protein Translation under Stress. Biomolecules 2023, 13, 1160. [Google Scholar] [CrossRef]
- Yan, H.; Shi, H.; Hu, C.; Luo, M.; Xu, C.; Wang, S.; Li, N.; Tang, W.; Zhou, Y.; Wang, C.; et al. Transcriptome Differences in Response Mechanisms to Low-Nitrogen Stress in Two Wheat Varieties. Int. J. Mol. Sci. 2021, 22, 12278. [Google Scholar] [CrossRef]
- Nazir, M.; Pandey, R.; Siddiqi, T.O.; Ibrahim, M.M.; Qureshi, M.I.; Abraham, G.; Vengavasi, K.; Ahmad, A. Nitrogen-Deficiency Stress Induces Protein Expression Differentially in Low-N Tolerant and Low-N Sensitive Maize Genotypes. Front. Plant Sci. 2016, 7, 298. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, K.; Jha, A.K.; Yadava, P.; Pal, M.; Rakshit, S.; Singh, I. Global Gene Expression Profiling under Nitrogen Stress Identifies Key Genes Involved in Nitrogen Stress Adaptation in Maize (Zea mays L.). Sci. Rep. 2022, 12, 4211. [Google Scholar] [CrossRef]
- Yang, L.; Gao, C.; Jiang, L. Leucine-Rich Repeat Receptor-like Protein Kinase AtORPK1 Promotes Oxidative Stress Resistance in an AtORPK1-AtKAPP Mediated Module in Arabidopsis. Plant Sci. Int. J. Exp. Plant Biol. 2022, 315, 111147. [Google Scholar] [CrossRef]
- Park, H.C.; Kim, D.W.; Park, J.; Baek, D.; Yun, D.-J. AtLRRop2, an Leucine-Rich Repeat-Only Protein, Mediates Cold Stress Response in Arabidopsis Thaliana. Plant Biotechnol. Rep. 2021, 15, 641–649. [Google Scholar] [CrossRef]
- Zhan, F.; Li, B.; Jiang, M.; Qin, L.; Wang, J.; He, Y.; Li, Y. Effects of a Root-Colonized Dark Septate Endophyte on the Glutathione Metabolism in Maize Plants under Cadmium Stress. J. Plant Interact. 2017, 12, 421–428. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Hong, J.K.; Kim, H.S.; Kwak, S.-S. Overexpression of the Sweetpotato Peroxidase Gene Swpa4 Enhances Tolerance to Methyl Viologen-Mediated Oxidative Stress and Dehydration in Arabidopsis Thaliana. J. Plant Biochem. Biotechnol. 2021, 30, 215–220. [Google Scholar] [CrossRef]
- Musdal, Y.; Mannervik, B. Substrate Specificities of Two Tau Class Glutathione Transferases Inducible by 2,4,6-Trinitrotoluene in Poplar. Biochim. Biophys. Acta BBA-Gen. Subj. 2015, 1850, 1877–1883. [Google Scholar] [CrossRef]
- Ordoñez, R.A.; Savin, R.; Slafer, G.A. Genetic Variation in the Critical Specific Leaf Nitrogen Maximising Yield among Modern Maize Hybrids. Field Crops Res. 2015, 172, 99–105. [Google Scholar] [CrossRef]
- Crema, A.; Boschetti, M.; Nutini, F.; Cillis, D.; Casa, R. Influence of Soil Properties on Maize and Wheat Nitrogen Status Assessment from Sentinel-2 Data. Remote Sens. 2020, 12, 2175. [Google Scholar] [CrossRef]
- Fu, W.; Wang, Y.; Ye, Y.; Zhen, S.; Zhou, B.; Wang, Y.; Hu, Y.; Zhao, Y.; Huang, Y. Grain Yields and Nitrogen Use Efficiencies in Different Types of Stay-Green Maize in Response to Nitrogen Fertilizer. Plants 2020, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-C.; Zhu, J.-F.; Shu, Z.-Z.; Wang, W.; Yan, C.; Xu, S.-B.; Wu, D.-X.; Wang, C.-Y.; Dong, Z.; Sun, G. Physiological and Transcriptional Response to Heat Stress in Heat-Resistant and Heat-Sensitive Maize (Zea mays L.) Inbred Lines at Seedling Stage. Protoplasma 2020, 257, 1615–1637. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Cui, Y.; Li, X.; Xiao, K.; Tao, P.; Zhang, Y. Responses of Photosynthetic Characteristics and Enzyme Activity of Nitrogen Metabolism to Low Nitrogen in Maize with Different Nitrogen Tolerance. Int. J. Agric. Biol. Eng. 2020, 13, 133–143. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Gene ID 1 | Description 2 | Log2FC 3 | p Value 4 | Expr. 5 | KEGG Pathway 6 |
|---|---|---|---|---|---|---|
| 1 | Zm00001d024533 | Putative DEAD-box ATP-dependent RNA helicase family protein | 8.72 | 5.39 × 10−12 | up | Spliceosome |
| 2 | ZemaCp020 | atpA | 6.74 | 2.23 × 10−5 | up | Oxidative phosphorylation; Photosynthesis |
| 3 | Zm00001d030822 | Cycloartenol synthase | 6.52 | 4.98 × 10−5 | up | ------ |
| 4 | Zm00001d022356 | DEAD-box ATP-dependent RNA helicase 57 | 5.55 | 5.90 × 10−4 | up | ------ |
| 5 | Zm00001d029012 | Leucine-rich repeat protein kinase family protein | 1.34 | 4.25 × 10−2 | up | ------ |
| 6 | Zm00001d016691 | Copper transport protein CCH | 1.27 | 4.76 × 10−2 | up | ------ |
| 7 | Zm00001d052242 | ATP synthase subunit delta chloroplastic | 1.26 | 4.18 × 10−2 | up | Photosynthesis; oxidative phosphorylation |
| 8 | Zm00001d005996 | PSI reaction center subunit V | 1.24 | 1.73 × 10−2 | up | Photosynthesis |
| 9 | ENSRNA049465879 | Eukaryotic small subunit ribosomal RNA | −9.17 | 1.9 × 10−2 | Down | ------ |
| 10 | Zm00001d024751 | Peroxidase 64 | −7.83 | 4.51 × 10−2 | Down | Phenylpropanoid biosynthesis |
| 11 | Zm00001d020938 | bZIP TF family protein | −5.86 | 2.89 × 10−3 | Down | Plant hormone signal transduction |
| 12 | Zm00001d042292 | SAUR-like auxin-responsive protein family | −5.71 | 1.23 × 10−3 | Down | Plant hormone signal transduction |
| 13 | Zm00001d029696 | GST U16 | −1.74 | 8.04 × 10−7 | Down | Glutathione metabolism |
| 14 | Zm00001d004664 | Cytochrome P450 94B3 | −1.03 | 2.85 × 10−3 | Down | ----- |
| 15 | Zm00001d052543 | bZIP TF family protein | −1.42 | 1.87 × 10−5 | Down | Plant hormone signal transduction |
| 16 | Zm00001d028816 | PRP 10 | −1.88 | 4.04 × 10−5 | Down | ------ |
| 17 | Zm00001d020780 | GST23 | −1.13 | 7.11 × 10−5 | Down | Glutathione metabolism |
| 18 | Zm00001d040702 | Guaiacol peroxidase1 | −1.8 | 6.90 × 10−4 | Down | Phenylpropanoid biosynthesis |
| No. | Gene ID 1 | Description 2 | Log2FC 3 | p Value 4 | Expr. 5 | KEGG Pathway 6 |
|---|---|---|---|---|---|---|
| 1 | Zm00001d027580 | Mitochondrial outer membrane protein porin 4 | 7.83 | 4.52 × 10−2 | up | ------ |
| 2 | Zm00001d049006 | PHD finger protein ALFIN-LIKE 6 | 6.59 | 2.36 × 10−5 | up | ------ |
| 3 | Zm00001d035336 | Peroxidase superfamily protein | 6.42 | 3.36 × 10−5 | up | Phenylpropanoid biosynthesis |
| 4 | Zm00001d037976 | FAM91A1-like protein | 5.68 | 2.76 × 10−2 | up | ------ |
| 5 | Zm00001d044526 | Ran BP2/NZF zinc finger-like superfamily protein | 2.75 | 1.35 × 10−6 | Up | ------ |
| 6 | Zm00001d023299 | Zinc finger CCCH domain-containing protein 46 | 2.49 | 6.80 × 10−4 | Up | ------ |
| 7 | Zm00001d011123 | Zinc finger protein | 2.08 | 1.36 × 10−5 | Up | ------ |
| 8 | Zm00001d045393 | Ran BP2/NZF zinc finger-like superfamily protein | 1.65 | 1.71 × 10−4 | Up | ------ |
| 9 | Zm00001d021732 | Cellulose synthase-like protein D3 | 1.51 | 3.47 × 10−6 | Up | ------ |
| 10 | Zm00001d038252 | potassium channel2 | 1.1 | 1.57 × 10−5 | Up | ------ |
| 11 | Zm00001d011316 | ------ | −8.50 | 3.92 × 10−4 | Down | ------ |
| 12 | Zm00001d038619 | ------ | −5.85 | 2.15 × 10−2 | Down | ------ |
| 13 | Zm00001d052131 | UPF0614 C14orf102-like protein | −5.71 | 8.04 × 10−4 | Down | ------ |
| 14 | Zm00001d002144 | Protein EPIDERMAL PATTERNING FACTOR 2 | −5.22 | 3.670 × 10−3 | Down | MAPK signaling pathway—plant |
| No. | Gene ID 1 | Description 2 | Log2FC 3 | p Value 4 | Expr. 5 | KEGG Pathway 6 |
|---|---|---|---|---|---|---|
| 1 | Zm00001d029012 | Leucine-rich repeat protein kinase family protein | 0.42 | 4.25 × 10−2 | up | ------ |
| 2 | Zm00001d013034 | 40S ribosomal protein S2-1 | 0.38 | 1.90 × 10−2 | up | Ribosome |
| 3 | Zm00001d016691 | Copper transport protein CCH | 0.35 | 4.76 × 10−2 | up | ------ |
| 4 | Zm00001d052242 | ATP synthase subunit delta chloroplastic | 0.34 | 4.18 × 10−2 | up | Photosynthesis; Oxidative phosphorylation |
| 5 | Zm00001d035854 | Alpha-dioxygenase 1 | 0.33 | 4.14 × 10−2 | up | alpha-Linolenic acid metabolism |
| 6 | Zm00001d005996 | Photosystem I reaction center subunit V | 0.31 | 1.73 × 10−2 | up | Photosynthesis |
| 7 | ZemaCp051 | psbH | 0.28 | 1.64 × 10−2 | up | Photosynthesis |
| 8 | Zm00001d042308 | 60S ribosomal protein L13 | 0.27 | 1.26 × 10−2 | up | Ribosome |
| 9 | Zm00001d042555 | Zinc-binding dehydrogenase family protein | 0.27 | 2.11 × 10−2 | up | ------ |
| 10 | Zm00001d029194 | cytochrome P450 family 81 subfamily D polypeptide 8 | 0.26 | 3.20 × 10−5 | up | ------ |
| 11 | Zm00001d003311 | Probable 2-oxoglutarate-dependent dioxygenase AOP1 | −1.03 | 4.42 × 10−2 | down | ------ |
| 12 | Zm00001d050457 | Protein DETOXIFICATION 27 | −0.35 | 2.47 × 10−2 | down | ------ |
| 13 | Zm00001d022060 | NAD(P)-binding Rossmann-fold superfamily protein | −0.50 | 7.03 × 10−3 | down | ------ |
| 14 | Zm00001d031666 | Probable aldo-keto reductase 4 | −0.30 | 1.33 × 10−2 | down | ------ |
| 15 | Zm00001d039732 | Staphylococcal-like nuclease CAN1 | −0.32 | 4.32 × 10−3 | down | ------ |
| No. | Gene ID 1 | Description 2 | Log2FC 3 | p Value 4 | Expr. 5 | KEGG Pathway 6 |
|---|---|---|---|---|---|---|
| 1 | Zm00001d033061 | Protein DETOXIFICATION 40 | 0.27 | 9.34 × 10−3 | up | ------ |
| 2 | Zm00001d025857 | 60S ribosomal protein L14-1 | 0.30 | 3.65 × 10−3 | up | Ribosome |
| 3 | Zm00001d004466 | Protein transport protein Sec24-like | 0.43 | 1.57 × 10−2 | up | PPER |
| 4 | Zm00001d046569 | Protein kinase superfamily protein with Phox/Bem1p domain | 0.43 | 4.43 × 10−2 | up | ------ |
| 5 | Zm00001d018954 | Rhicadhesin receptor | 0.37 | 7.77 × 10−3 | up | ------ |
| 6 | Zm00001d002000 | lipoxygenase6 | 0.28 | 3.35 × 10−2 | up | Linoleic acid metabolism |
| 7 | Zm00001d004089 | PRP1 | −0.68 | 1.59 × 10−2 | down | ------ |
| 8 | Zm00001d038911 | Non-specific lipid-transfer protein 2 | −0.60 | 3.92 × 10−2 | down | ------ |
| 9 | Zm00001d042143 | glucan endo-13-beta-glucosidase homolog1 | −0.50 | 9.34 × 10−3 | down | ------ |
| 10 | Zm00001d020780 | glutathione transferase23 | −0.45 | 4.76 × 10−2 | down | Glutathione metabolism |
| 11 | Zm00001d021119 | Beta-glucosidase 11 | 0.82 | 4.2 × 10−2 | down | ------ |
| 12 | Zm00001d004089 | Pathogenesis-related protein 1 | 0.63 | 1.59 × 10−2 | down | ------ |
| No. | Gene ID 1 | Description 2 | Log2FC 3 | p Value 4 | Expr. 5 | KEGG Pathway 6 |
|---|---|---|---|---|---|---|
| 1 | Zm00001d038929 | ATP synthase 2 | 1.36 | 0.0041 | up | Oxidative phosphorylation |
| 2 | Zm00001d043166 | UDP-glycosyltransferase 87A1 | 0.81 | 0.0319 | down | ------ |
| 3 | Zm00001d048630 | ---- | 0.24 | 0.0190 | down | ------ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zenda, T.; Liu, S.; Dong, A.; Wang, Y.; Liu, X.; Wang, N.; Duan, H. Integrated Transcriptomic and Proteomic Analyses of Low-Nitrogen-Stress Tolerance and Function Analysis of ZmGST42 Gene in Maize. Antioxidants 2023, 12, 1831. https://doi.org/10.3390/antiox12101831
Li J, Zenda T, Liu S, Dong A, Wang Y, Liu X, Wang N, Duan H. Integrated Transcriptomic and Proteomic Analyses of Low-Nitrogen-Stress Tolerance and Function Analysis of ZmGST42 Gene in Maize. Antioxidants. 2023; 12(10):1831. https://doi.org/10.3390/antiox12101831
Chicago/Turabian StyleLi, Jiao, Tinashe Zenda, Songtao Liu, Anyi Dong, Yafei Wang, Xinyue Liu, Nan Wang, and Huijun Duan. 2023. "Integrated Transcriptomic and Proteomic Analyses of Low-Nitrogen-Stress Tolerance and Function Analysis of ZmGST42 Gene in Maize" Antioxidants 12, no. 10: 1831. https://doi.org/10.3390/antiox12101831