


The Binding of HSPA8 and Mitochondrial ALDH2 Mediates Oxygen-Glucose Deprivation-Induced Fibroblast Senescence
Abstract
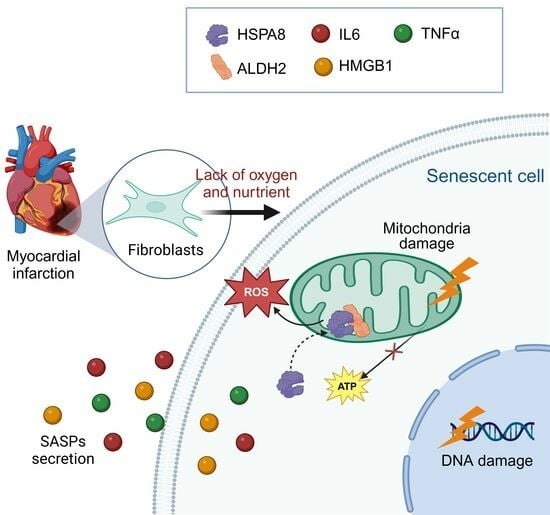
:
1. Introduction
2. Methods
2.1. Bioinformatics Analysis
2.2. Chemicals and Reagents
2.3. Cell Culture
2.4. Oxygen and Glucose Deprivation Cell Model
2.5. Quantitative Reverse Transcription PCR (RT-qPCR)
2.6. Western Blot Analysis
2.7. Senescence-Associated β-Galactosidase (SA-β-gal) Staining and Activity Assay
2.8. Cell Proliferation Assay
2.9. Measurement of Glycolysis Rate
2.10. ALDH2 Enzyme Activity Assay
2.11. Mitochondrial Membrane Potential Assay
2.12. Measurement of ROS and ATP
2.13. Co-Immunoprecipitation (Co-IP) Assay
2.14. Mass Spectrometry
2.15. Immunofluorescence Staining
2.16. Extraction of Cytoplasmic and Mitochondrial Proteins
2.17. Transfection of siRNA
2.18. Disuccinimidyl Suberate (DSS) Crosslinking
2.19. Statistical Analysis
3. Results
3.1. Identification and Functional Enrichment Analysis of DEGs in Mice CFs after MI
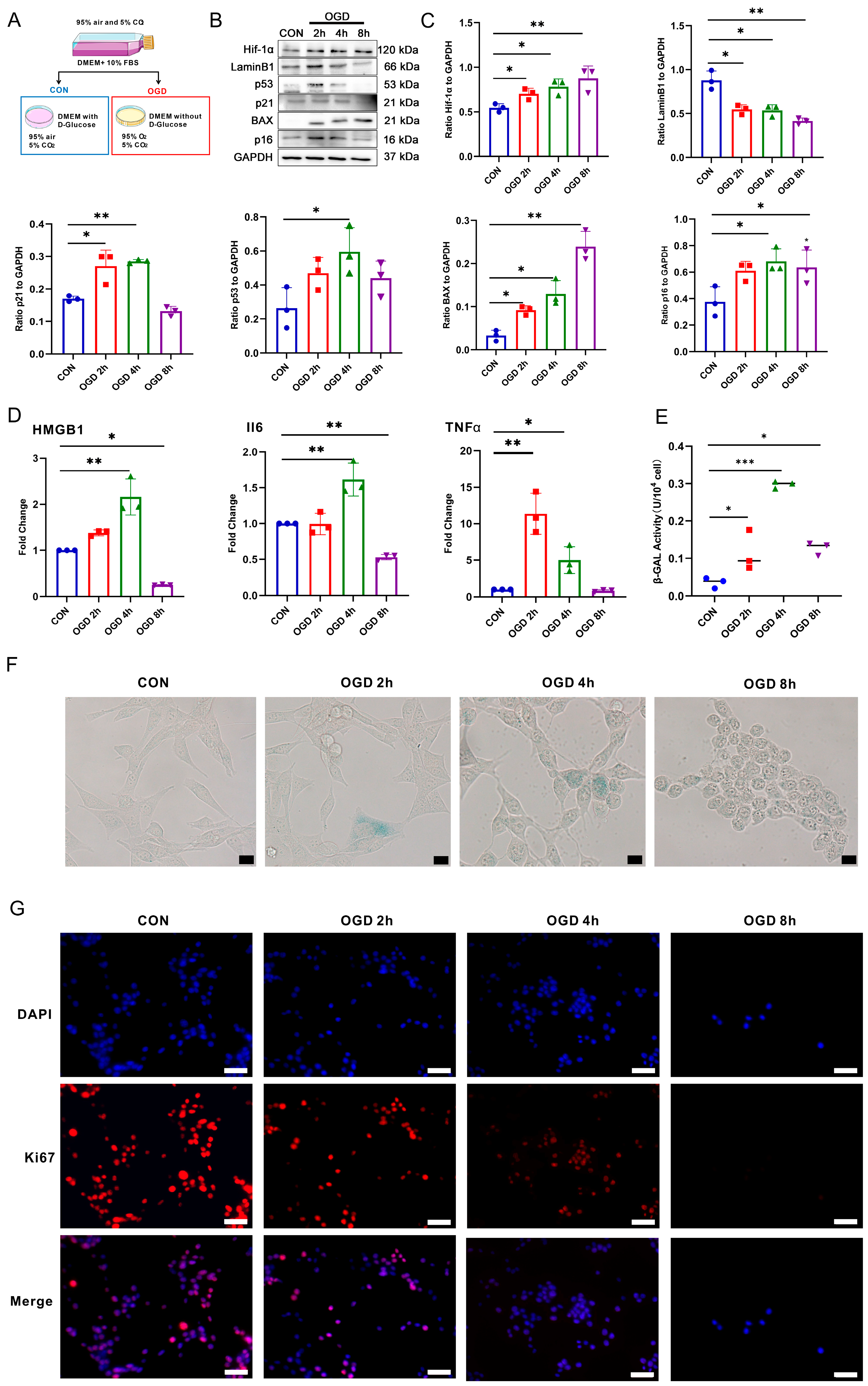
3.2. OGD Treatment-Induced Fibroblast Senescence
3.3. Bioinformatics Analysis Revealed That ALDH2 Is a Potential Regulator of Fibroblast Senescence after OGD
3.4. Activation of ALDH2 Can Inhibit Fibroblast Senescence after OGD
3.5. Activation of ALDH2 Improves Mitochondrial Damage Induced by OGD
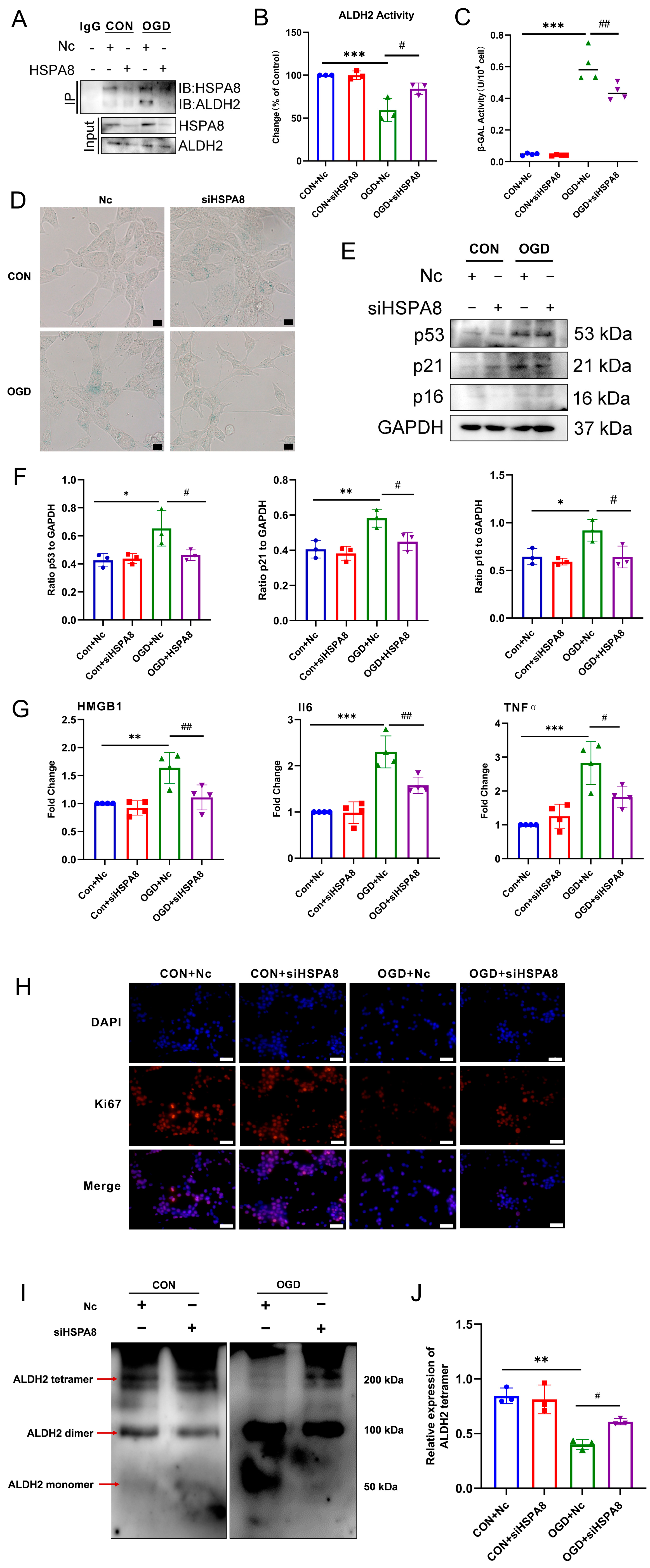
3.6. OGD-Induced HSPA8 Translocation to Mitochondria to Inhibit ALDH2 Enzyme Activity
3.7. Inhibition of HSPA8 Can Improve Fibroblast Senescence by Increasing ALDH2 Activity after OGD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Roth, G.; Mensah, G.; Johnson, C.; Addolorato, G.; Ammirati, E.; Baddour, L.; Barengo, N.; Beaton, A.; Benjamin, E.; Benziger, C.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Olson, E.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.; Luo, X.; Qi, X.; Tan, W.; Huang, G.; Acharya, A.; Smith, C.; Tallquist, M.; Neilson, E.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.; Hacker, A.; Langburt, D.; Dewar, M.; McFadden, M.; Zhang, H.; Kuzmanov, U.; Zhou, Y.; Hussain, B.; Ehsan, F.; et al. Myocardial Infarction Induces Cardiac Fibroblast Transformation within Injured and Noninjured Regions of the Mouse Heart. J. Proteome Res. 2021, 20, 2867–2881. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lee, R.; Garbern, J. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.; Desprez, P.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.; Passos, J.; Richardson, G. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech. Ageing Dev. 2022, 208, 111740. [Google Scholar] [CrossRef]
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The role of cellular senescence in cardiac disease: Basic biology and clinical relevance. Nat. Rev. Cardiol. 2022, 19, 250–264. [Google Scholar] [CrossRef]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.; Du, J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.; Tang, Q. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis. 2022, 13, 103–128. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.; Hildrestrand, G.; Scheffler, K.; Vinge, L.; Alfsnes, K.; Palibrk, V.; Wang, J.; Neurauter, C.; Luna, L.; Johansen, J.; et al. NEIL3-Dependent Regulation of Cardiac Fibroblast Proliferation Prevents Myocardial Rupture. Cell Rep. 2017, 18, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hao, Y.; Piao, X.; Gu, X. Aldehyde Dehydrogenase 2 as a Therapeutic Target in Oxidative Stress-Related Diseases: Post-Translational Modifications Deserve More Attention. Int. J. Mol. Sci. 2022, 23, 2682. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, C.; Lai, T.; Chang, Y.; Hwang, M.; Chang, T.; Weng, C.; Chang, P.; Chen, C.; Mochly-Rosen, D.; et al. ALDH2 deficiency induces atrial fibrillation through dysregulated cardiac sodium channel and mitochondrial bioenergetics: A multi-omics analysis. Biochim. Et Biophys. Acta Mol. Basis Dis. 2021, 1867, 166088. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Budas, G.; Churchill, E.; Disatnik, M.; Hurley, T.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Levi, R. Salvaging the Ischemic Heart: Gi-Coupled Receptors in Mast Cells Activate a PKCε/ALDH2 Pathway Providing Anti-RAS Cardioprotection. Curr. Med. Chem. 2018, 25, 4416–4431. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Cui, S.; Cui, Z.; Yang, F.; Pang, J.; Xu, F.; Chen, Y. ALDH2: A new protector against age-independent myocardial senescence. Int. J. Cardiol. 2016, 210, 38–40. [Google Scholar] [CrossRef]
- Nannelli, G.; Terzuoli, E.; Giorgio, V.; Donnini, S.; Lupetti, P.; Giachetti, A.; Bernardi, P.; Ziche, M. ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid. Med. Cell. Longev. 2018, 2018, 9765027. [Google Scholar] [CrossRef]
- Xue, L.; Zhu, W.; Yang, F.; Dai, S.; Han, Z.; Xu, F.; Guo, P.; Chen, Y. Appropriate dose of ethanol exerts anti-senescence and anti-atherosclerosis protective effects by activating ALDH2. Biochem. Biophys. Res. Commun. 2019, 512, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Chiong, M.; Lavandero, S.; Klionsky, D.; Ren, J. Mitophagy in cardiovascular diseases: Molecular mechanisms, pathogenesis, and treatment. Trends Mol. Med. 2022, 28, 836–849. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Chen, X.; Chen, Y.; Pang, J.; Chen, Q.; Liu, Q.; Xue, H.; Zeng, Y.; Xiao, J.; Mi, J.; et al. Epigenetic modulation of Drp1-mediated mitochondrial fission by inhibition of S-adenosylhomocysteine hydrolase promotes vascular senescence and atherosclerosis. Redox Biol. 2023, 65, 102828. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef]
- Tan, X.; Chen, Y.; Zou, S.; Wang, W.; Zhang, N.; Sun, Z.; Xian, W.; Li, X.; Tang, B.; Wang, H.; et al. ALDH2 attenuates ischemia and reperfusion injury through regulation of mitochondrial fusion and fission by PI3K/AKT/mTOR pathway in diabetic cardiomyopathy. Free Radic. Biol. Med. 2023, 195, 219–230. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, B.; Zhang, J.; He, D.; Zhang, Q.; Pan, C.; Yuan, Q.; Shi, Y.; Tang, H.; Xu, F.; et al. ALDH2 (Aldehyde Dehydrogenase 2) Protects Against Hypoxia-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2303–2319. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Xu, D.; Xi, Z.; Wu, M.; Nik Nabil, W.; Zhang, J.; Sui, H.; Fu, W.; Zhou, H.; Lao, Y.; et al. In VitroThe Natural Compound Oblongifolin C Exhibits Anticancer Activity by Inhibiting HSPA8 and Cathepsin B. Front. Pharmacol. 2020, 11, 564833. [Google Scholar] [CrossRef] [PubMed]
- Valek, L.; Heidler, J.; Scheving, R.; Wittig, I.; Tegeder, I. Nitric oxide contributes to protein homeostasis by S-nitrosylations of the chaperone HSPA8 and the ubiquitin ligase UBE2D. Redox Biol. 2019, 20, 217–235. [Google Scholar] [CrossRef]
- Dokladny, K.; Myers, O.; Moseley, P. Heat shock response and autophagy—cooperation and control. Autophagy 2015, 11, 200–213. [Google Scholar] [CrossRef]
- Liu, T.; Daniels, C.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef]
- Qiu, F.; Yuan, Y.; Luo, W.; Gong, Y.; Zhang, Z.; Liu, Z.; Gao, L. Asiatic acid alleviates ischemic myocardial injury in mice by modulating mitophagy- and glycophagy-based energy metabolism. Acta Pharmacol. Sin. 2022, 43, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.; Vagnozzi, R.; Maliken, B.; Sargent, M.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Gambino, V.; De Michele, G.; Venezia, O.; Migliaccio, P.; Dall’Olio, V.; Bernard, L.; Minardi, S.; Della Fazia, M.; Bartoli, D.; Servillo, G.; et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell 2013, 12, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, T.; Sun, Y.; Men, L.; Gu, Y.; Zhang, S.; Chen, X. Proteomic Analysis Reveals the Effect of Trichostatin A and Bone Marrow-Derived Dendritic Cells on the Fatty Acid Metabolism of NIH3T3 Cells under Oxygen-Glucose Deprivation Conditions. J. Proteome Res. 2021, 20, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris Consortium; Overall coordination; Logistical coordination. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Qin, Y.; Li, D.; Cai, N.; Wu, J.; Jiang, L.; Jie, L.; Zhou, Z.; Xu, J.; Wang, H. Inhibition of PDE4 protects neurons against oxygen-glucose deprivation-induced endoplasmic reticulum stress through activation of the Nrf-2/HO-1 pathway. Redox Biol. 2020, 28, 101342. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Gu, Y.; Hui, W.; Yang, X.; Liu, Y.; Chen, X. Oxygen-Glucose Deprivation Promoted Fibroblast Senescence and Collagen Expression via IL11. Int. J. Mol. Sci. 2022, 23, 12090. [Google Scholar] [CrossRef]
- Yao, P.; Zhang, Z.; Liu, H.; Jiang, P.; Li, W.; Du, W. p53 protects against alcoholic fatty liver disease via ALDH2 inhibition. EMBO J. 2023, 42, e112304. [Google Scholar] [CrossRef]
- Alessio, N.; Aprile, D.; Cappabianca, S.; Peluso, G.; Di Bernardo, G.; Galderisi, U. Different Stages of Quiescence, Senescence, and Cell Stress Identified by Molecular Algorithm Based on the Expression of Ki67, RPS6, and Beta-Galactosidase Activity. Int. J. Mol. Sci. 2021, 22, 3102. [Google Scholar] [CrossRef]
- Plumier, J.; Robertson, H.; Currie, R. Differential accumulation of mRNA for immediate early genes and heat shock genes in heart after ischaemic injury. J. Mol. Cell. Cardiol. 1996, 28, 1251–1260. [Google Scholar] [CrossRef]
- Zou, N.; Ao, L.; Cleveland, J.; Yang, X.; Su, X.; Cai, G.; Banerjee, A.; Fullerton, D.; Meng, X. Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2805–H2813. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Xu, Z.; Huang, W. Cellular Senescence Affects Cardiac Regeneration and Repair in Ischemic Heart Disease. Aging Dis. 2021, 12, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.; Frangogiannis, N. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Narita, M. Transmitting senescence to the cell neighbourhood. Nat. Cell Biol. 2013, 15, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lv, L.; Liang, R.; Guo, W.; Liao, Z.; Chen, Y.; Zhu, K.; Huang, R.; Zhao, H.; Pu, Q.; et al. miR-486 improves fibrotic activity in myocardial infarction by targeting SRSF3/p21-Mediated cardiac myofibroblast senescence. J. Cell. Mol. Med. 2022, 26, 5135–5149. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.; Espinoza-Pérez, C.; Rimassa-Taré, C.; Machuca, V.; Bustos, J.; Vallejos, M.; Vargas, H.; Díaz-Araya, G. Senescent cardiac fibroblasts: A key role in cardiac fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166642. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, J.; Xu, Y.; Mou, F.; Shan, X.; Wang, Q.; Liu, B.; Ning, K.; Liu, J.; Wang, Y.; et al. Notoginsenoside R1-loaded mesoporous silica nanoparticles targeting the site of injury through inflammatory cells improves heart repair after myocardial infarction. Redox Biol. 2022, 54, 102384. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, T.; Kim, Y.; Kim, C.; Ko, S.; Kim, B. Senolytic Therapy for Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 11967. [Google Scholar] [CrossRef]
- Feng, H.; Mou, S.; Li, W.; Zhang, N.; Zhou, Z.; Ding, W.; Bian, Z.; Liao, H. Resveratrol Inhibits Ischemia-Induced Myocardial Senescence Signals and NLRP3 Inflammasome Activation. Oxid. Med. Cell. Longev. 2020, 2020, 2647807. [Google Scholar] [CrossRef]
- Sun, X.; Gao, R.; Li, W.; Zhao, Y.; Yang, H.; Chen, H.; Jiang, H.; Dong, Z.; Hu, J.; Liu, J.; et al. Alda-1 treatment promotes the therapeutic effect of mitochondrial transplantation for myocardial ischemia-reperfusion injury. Bioact. Mater. 2021, 6, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ding, Z.; Yin, P.; Wu, J.; Hu, K.; Sun, A.; Zou, Y.; Ge, J. Hypertrophic preconditioning cardioprotection after myocardial ischaemia/reperfusion injury involves ALDH2-dependent metabolism modulation. Redox Biol. 2021, 43, 101960. [Google Scholar] [CrossRef]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yücel, Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef] [PubMed]
- Martini, H.; Passos, J. Cellular senescence: All roads lead to mitochondria. FEBS J. 2023, 290, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guan, T.; Shafiq, K.; Yu, Q.; Jiao, X.; Na, D.; Li, M.; Zhang, G.; Kong, J. Mitochondrial dysfunction in aging. Ageing Res. Rev. 2023, 88, 101955. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ju, P.; Kou, W.; Zhai, M.; Zeng, Y.; Maimaitiaili, N.; Shi, Y.; Xu, X.; Zhao, Y.; Jian, W.; et al. Macrophage-Specific NLRC5 Protects From Cardiac Remodeling Through Interaction With HSPA8. JACC Basic Transl. Sci. 2023, 8, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Nagasawa, Y.; Obara, Y.; Ishii, K.; Takagi, D.; Ono, K. Molecular identification of HSPA8 as an accessory protein of a hyperpolarization-activated chloride channel from rat pulmonary vein cardiomyocytes. J. Biol. Chem. 2019, 294, 16049–16061. [Google Scholar] [CrossRef]
- Nie, T.; Tao, K.; Zhu, L.; Huang, L.; Hu, S.; Yang, R.; Xu, P.; Mao, Z.; Yang, Q. Chaperone-mediated autophagy controls the turnover of E3 ubiquitin ligase MARCHF5 and regulates mitochondrial dynamics. Autophagy 2021, 17, 2923–2938. [Google Scholar] [CrossRef]
- Li, Y.; Xue, Y.; Xu, X.; Wang, G.; Liu, Y.; Wu, H.; Li, W.; Wang, Y.; Chen, Z.; Zhang, W.; et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019, 38, e98786. [Google Scholar] [CrossRef]
- Rodriguez-Zavala, J.; Weiner, H. Structural aspects of aldehyde dehydrogenase that influence dimer-tetramer formation. Biochemistry 2002, 41, 8229–8237. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Accession | Species | Forward | Reverse |
|---|---|---|---|---|
| TNFα | NM_001278601.1 | Mouse | GGACTAGCCAGGAGGGAGAACAG | GCCAGTGAGTGAAAGGGACAGAAC |
| IL6 | NM_001314054.1 | Mouse | CTTCTTGGGACTGATGCTGGTGAC | TCTGTTGGGAGTGGTATCCTCTGTG |
| HMGB1 | NM_010439.4 | Mouse | AGGCTGACAAGGCTCGTTATGAAAG | GGGCGGTACTCAGAACAGAACAAG |
| GAPDH | NM_001411841.1 | Mouse | GGCAAATTCAACGGCACAGTCAAG | TCGCTCCTGGAAGATGGTGATGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hui, W.; Song, T.; Yu, L.; Chen, X. The Binding of HSPA8 and Mitochondrial ALDH2 Mediates Oxygen-Glucose Deprivation-Induced Fibroblast Senescence. Antioxidants 2024, 13, 42. https://doi.org/10.3390/antiox13010042
Hui W, Song T, Yu L, Chen X. The Binding of HSPA8 and Mitochondrial ALDH2 Mediates Oxygen-Glucose Deprivation-Induced Fibroblast Senescence. Antioxidants. 2024; 13(1):42. https://doi.org/10.3390/antiox13010042
Chicago/Turabian StyleHui, Wenting, Tongtong Song, Ling Yu, and Xia Chen. 2024. "The Binding of HSPA8 and Mitochondrial ALDH2 Mediates Oxygen-Glucose Deprivation-Induced Fibroblast Senescence" Antioxidants 13, no. 1: 42. https://doi.org/10.3390/antiox13010042