
New NADPH Oxidase 2 Inhibitors Display Potent Activity against Oxidative Stress by Targeting p22phox-p47phox Interactions

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
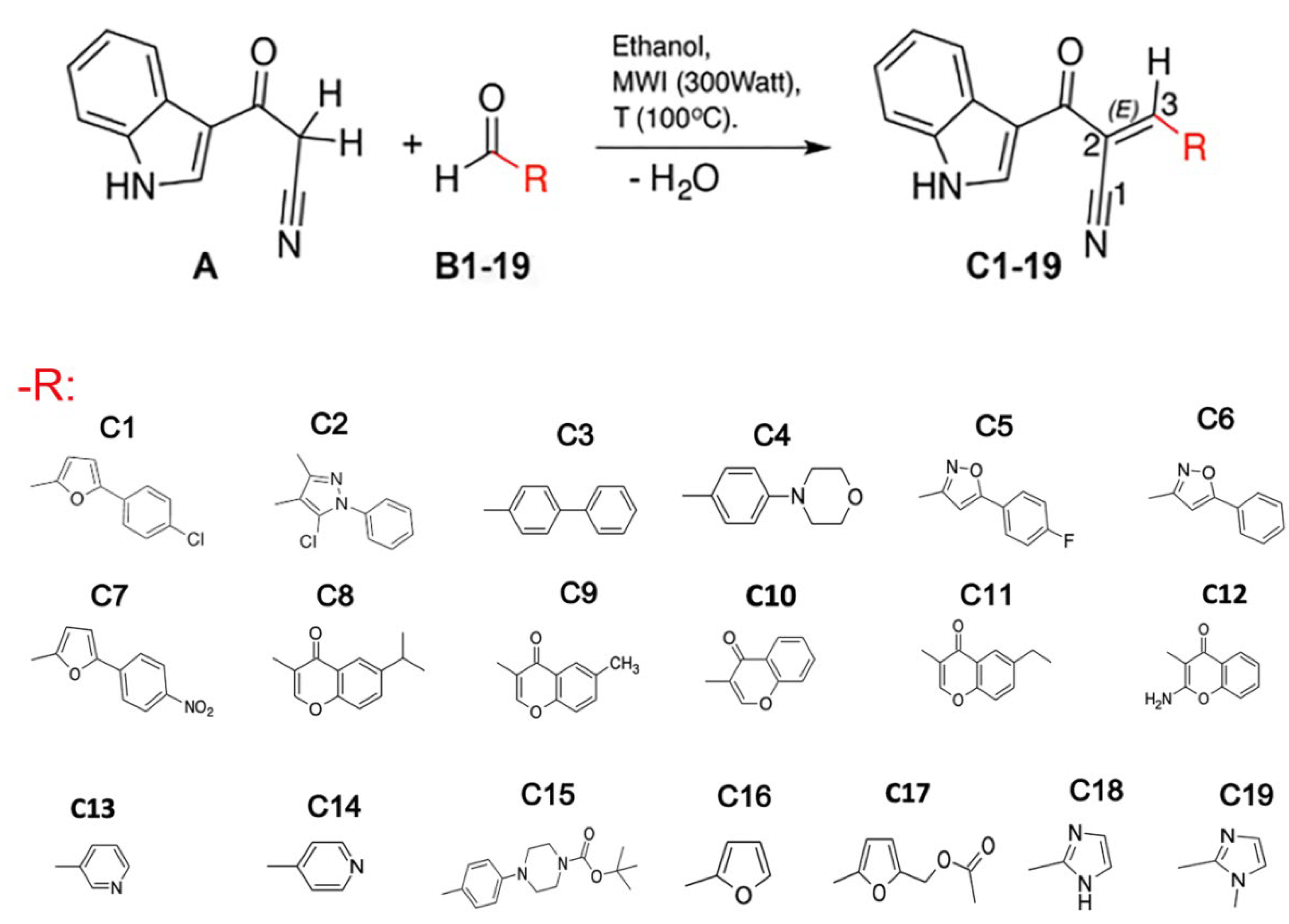
2.1. Indole Heteroaryl-Acrylonitriles Synthesis
2.2. Cells and Reagents
2.3. HL-60 Cell Differentiation
2.4. Cytotoxicity Assays
2.5. Cardiomyocyte Isolation from Mdx Mice
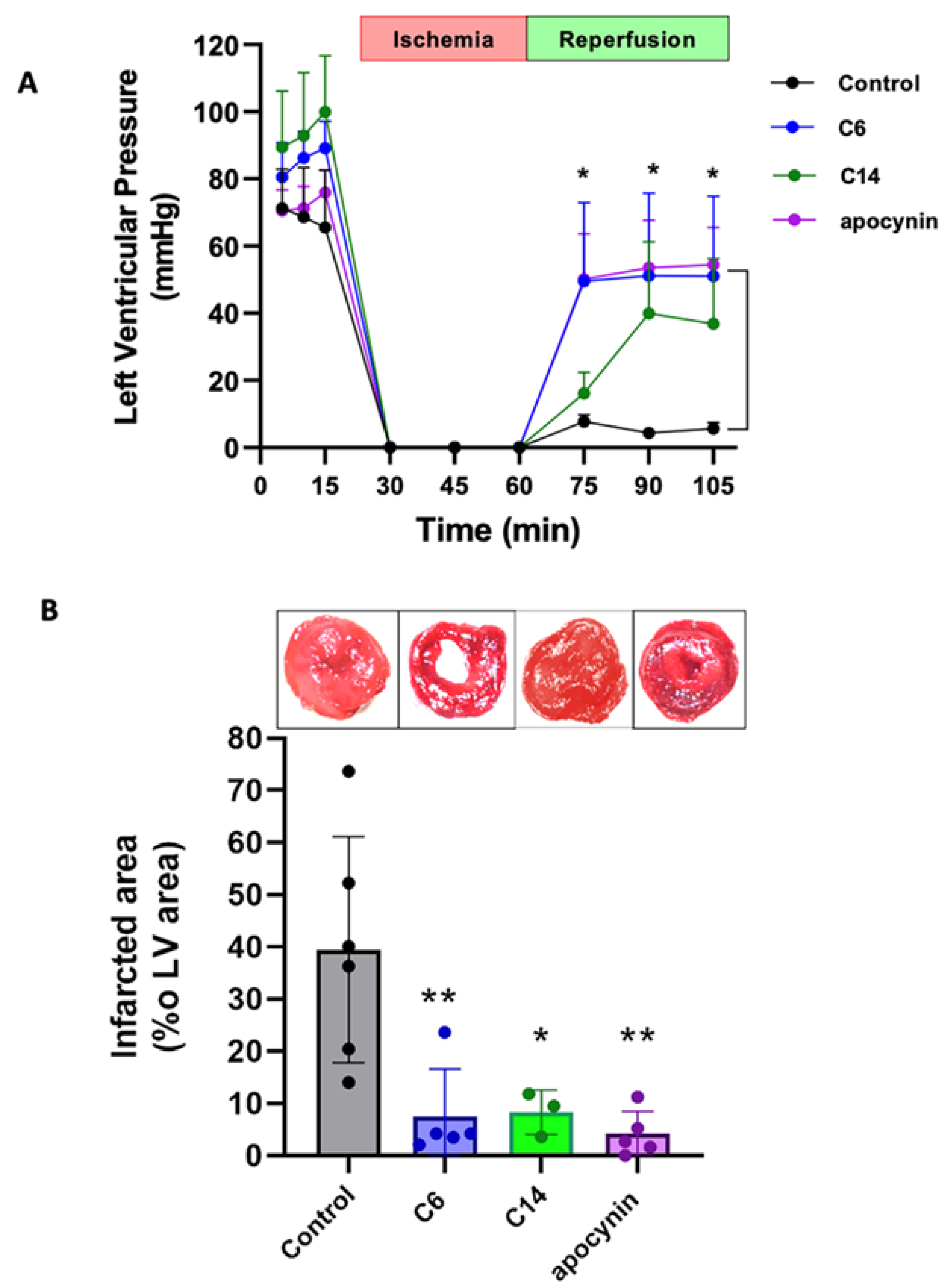
2.6. Isolated Rat Heart Preparation
2.7. Infarcted Area Calculation
2.8. Preparation of HL-60 Cell Membranes
2.9. NADPH Oxidase Activity
2.10. Cytochrome C Reduction
2.11. NADPH Oxidase Inhibition Protocol in HL-60 Cell-Derived Neutrophils
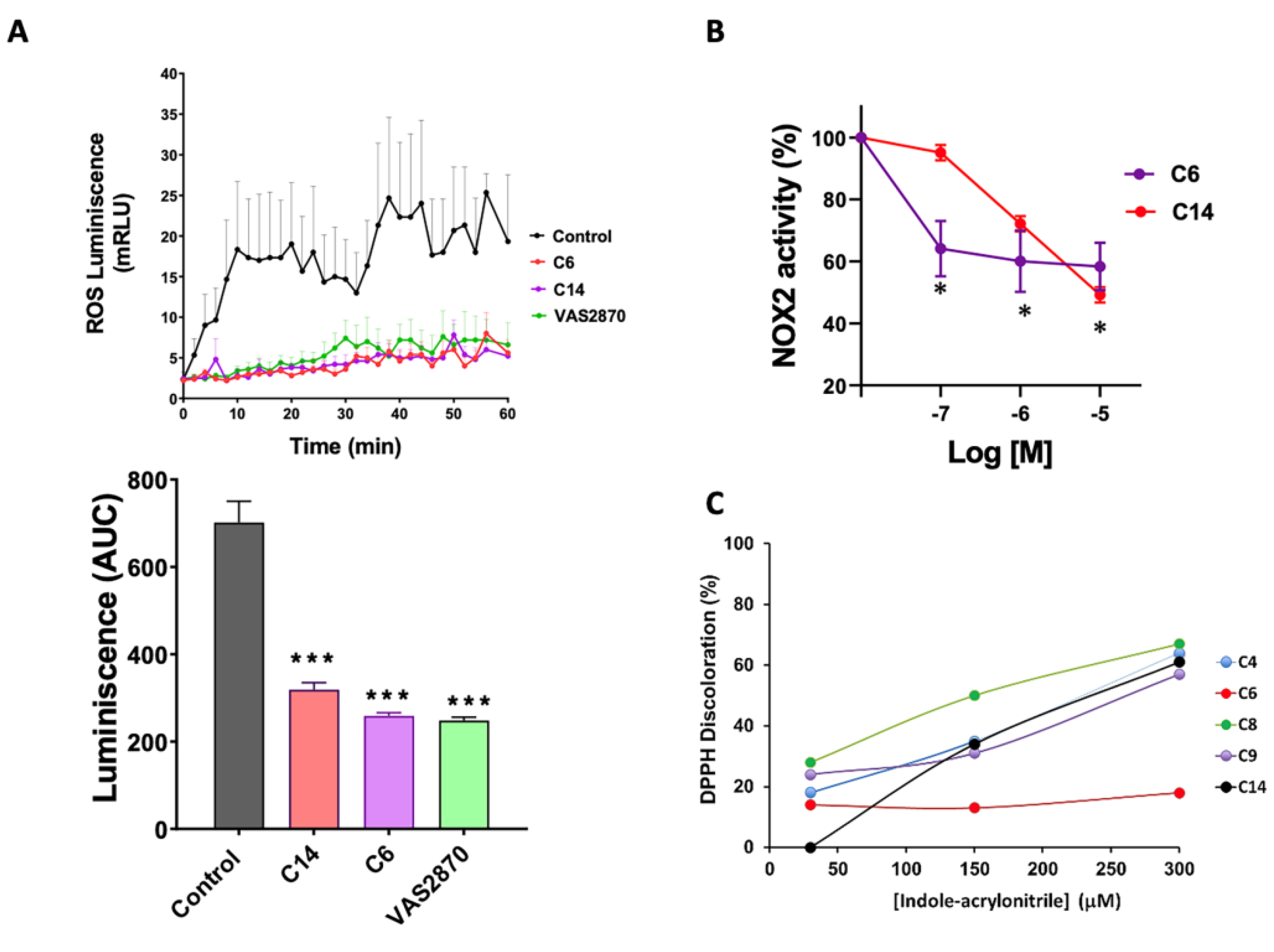
2.12. Assessment of DPPH Radical Scavenging Activity
2.13. NOX2-Mediated ROS Production Inhibition in Mdx Cardiomyocytes
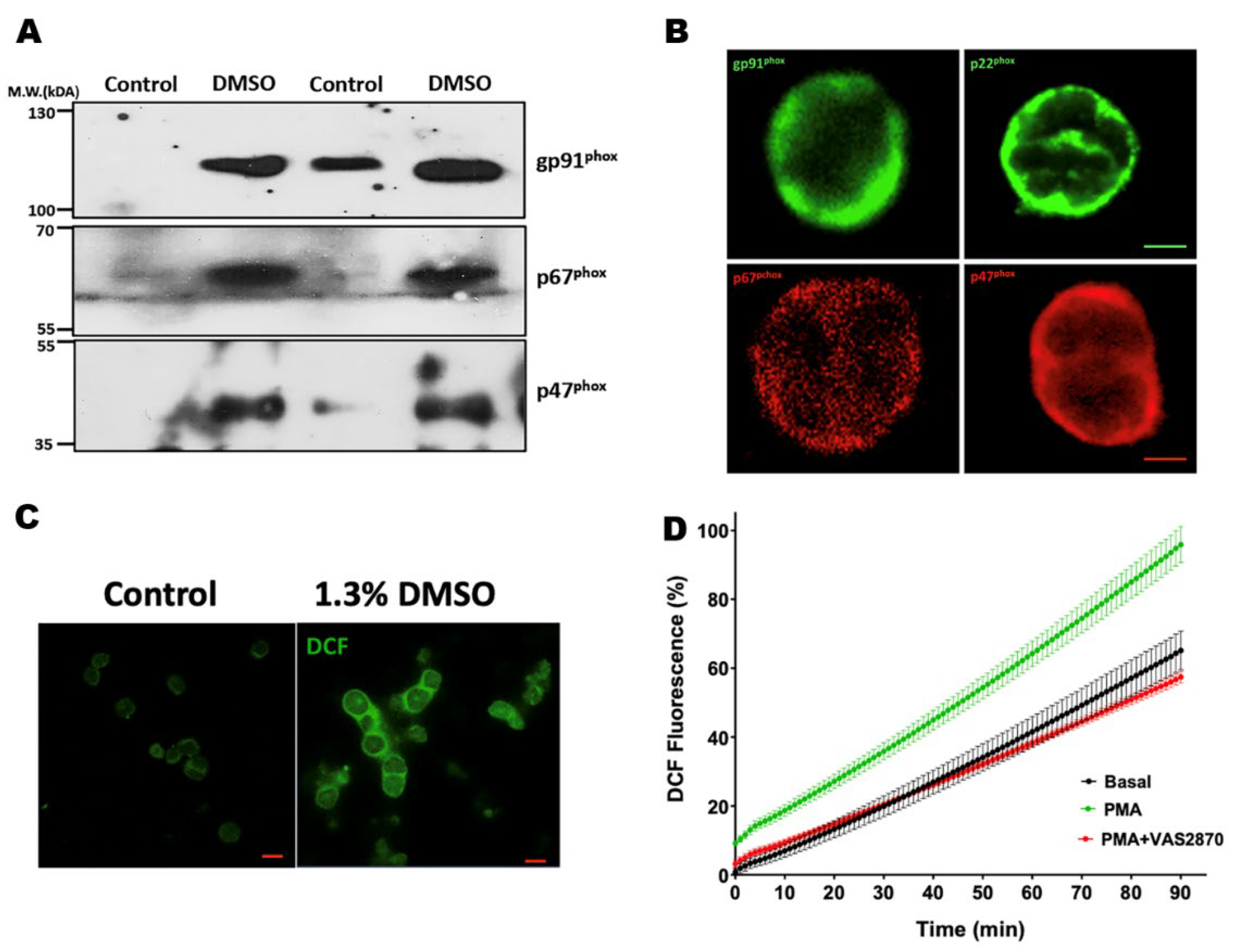
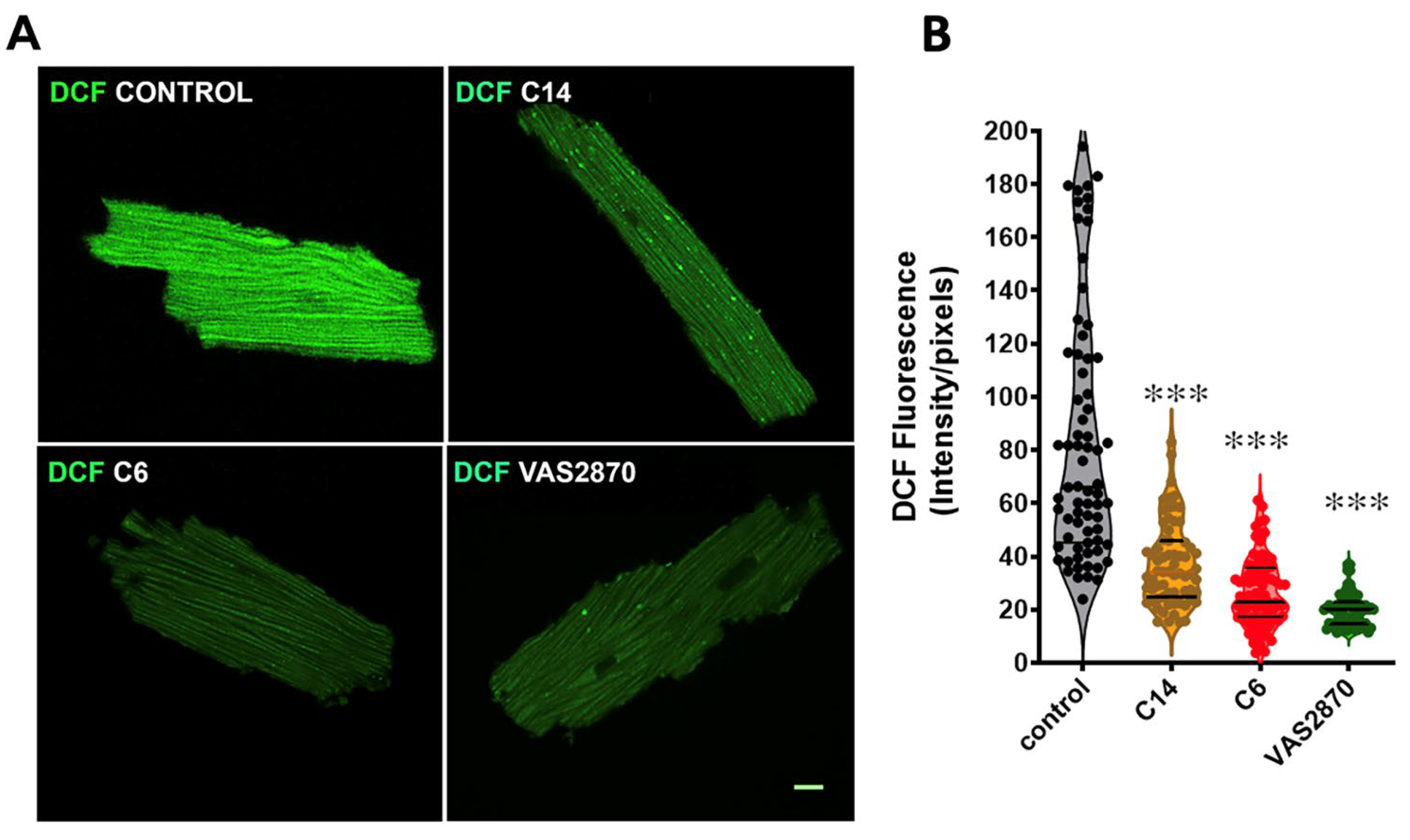
2.14. Immunofluorescence and Confocal Microscopy
2.15. Preparation of Protein Extracts, and Quantification of Total Proteins
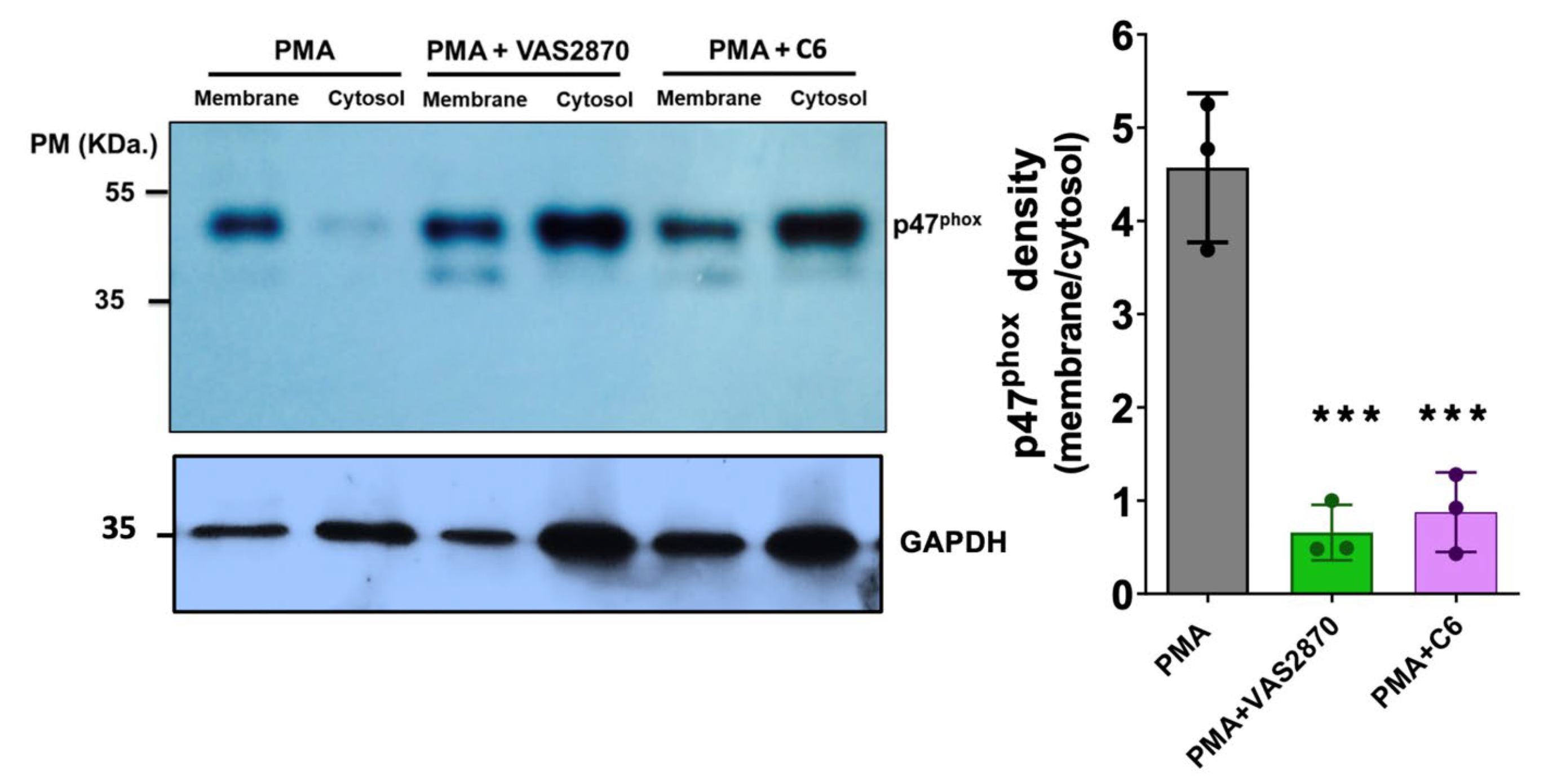
2.16. SDS-PAGE and Western Blotting
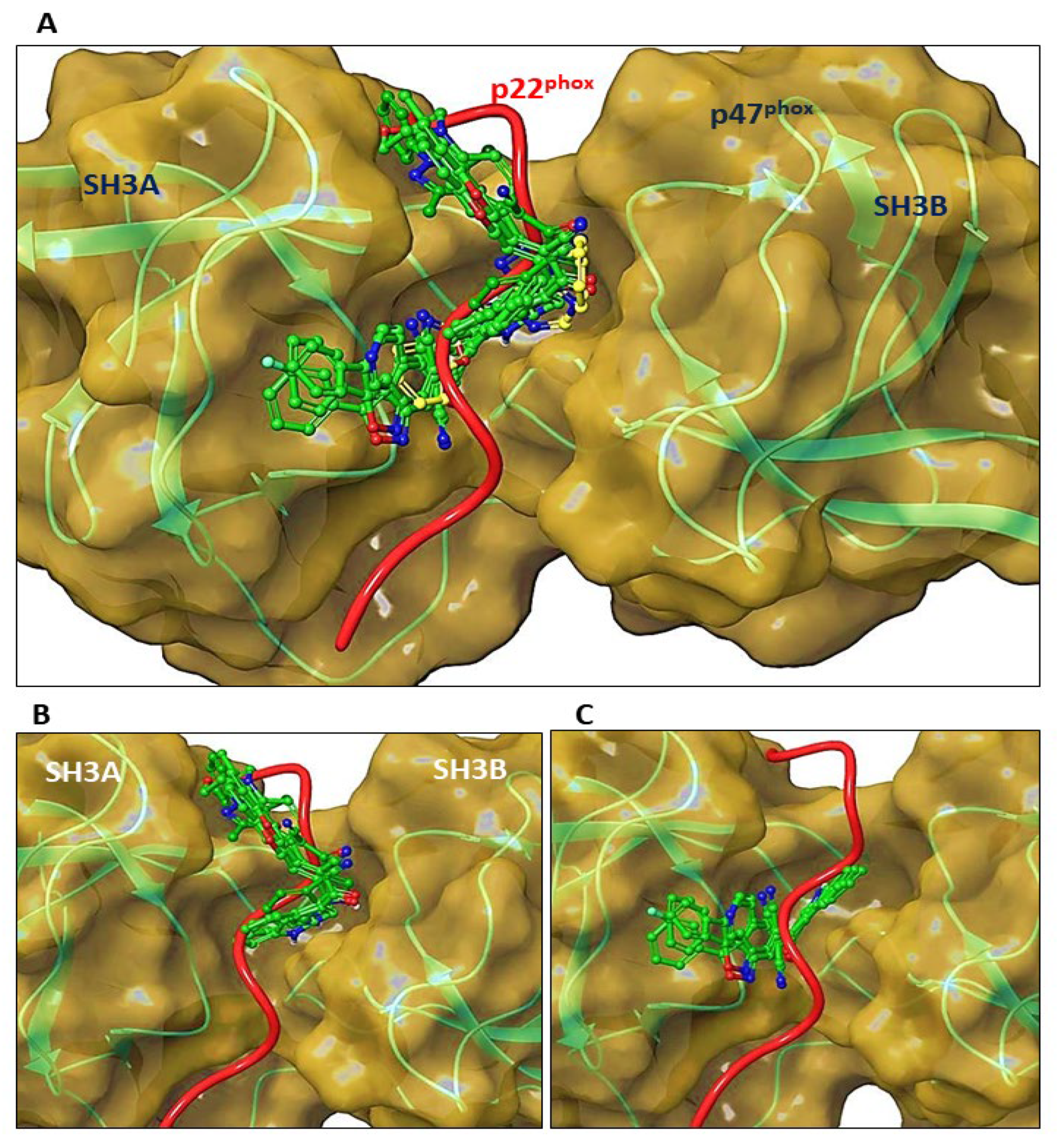
2.17. Computational Docking
2.18. Statistical Analysis
3. Results
3.1. Compounds
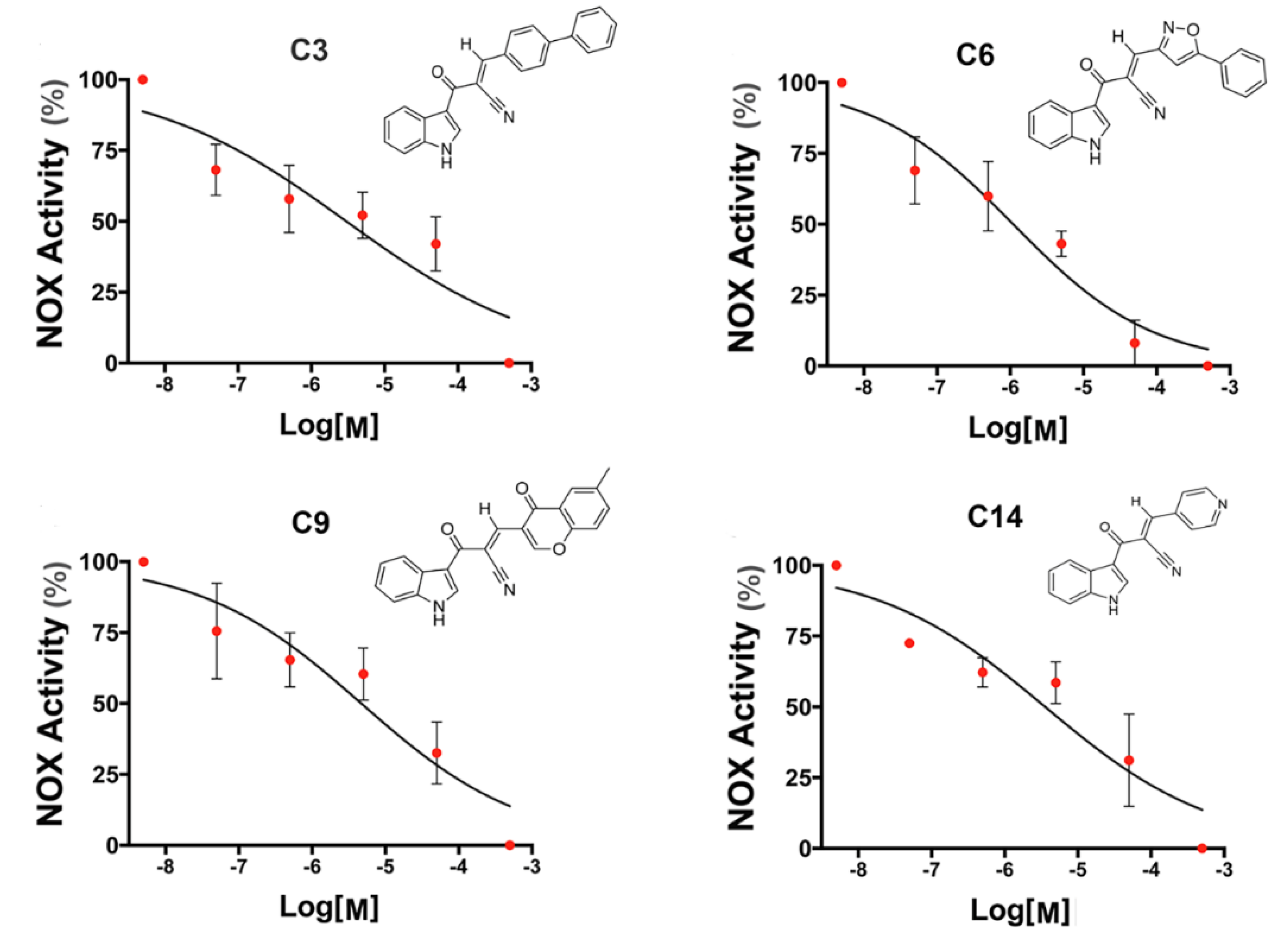
3.2. NADPH Oxidase Inhibitory Activity of Heteroarylacrylonitrile Derivatives in HL-60 Cells
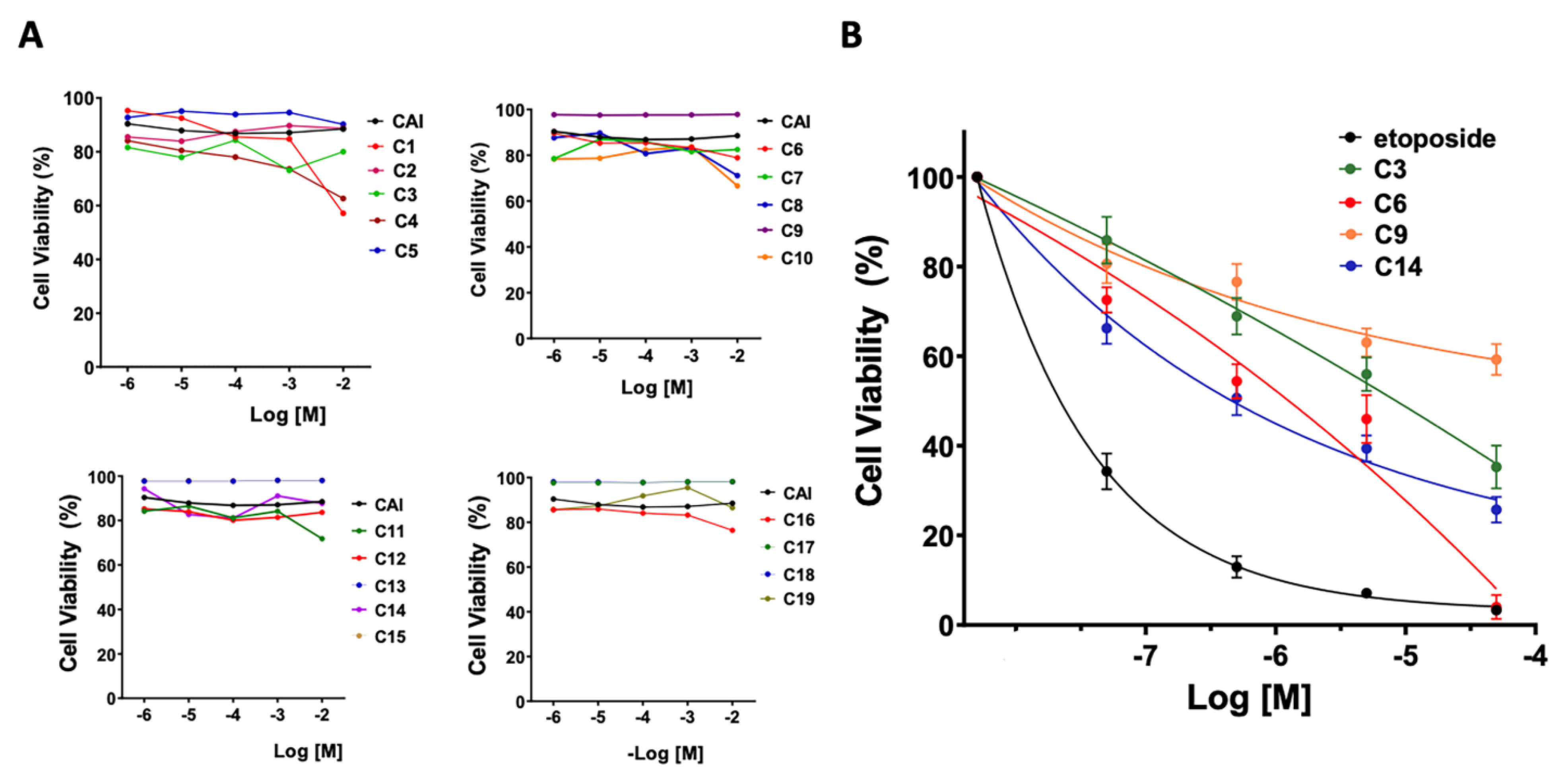
3.3. Cytotoxicity
3.4. Activity of NADPH Oxidase by Chemiluminescence
3.5. NADPH Oxidase Activity by Reduction of Cytochrome C
3.6. ROS Production Assay by the Conversion of 2′,7′-Dichlorofluorescein Diacetate (DCF-DA) in Cardiomyocytes Isolated from Mdx Mice
3.7. Translocation of the p47phox Cytosolic Subunit by PMA Stimulation
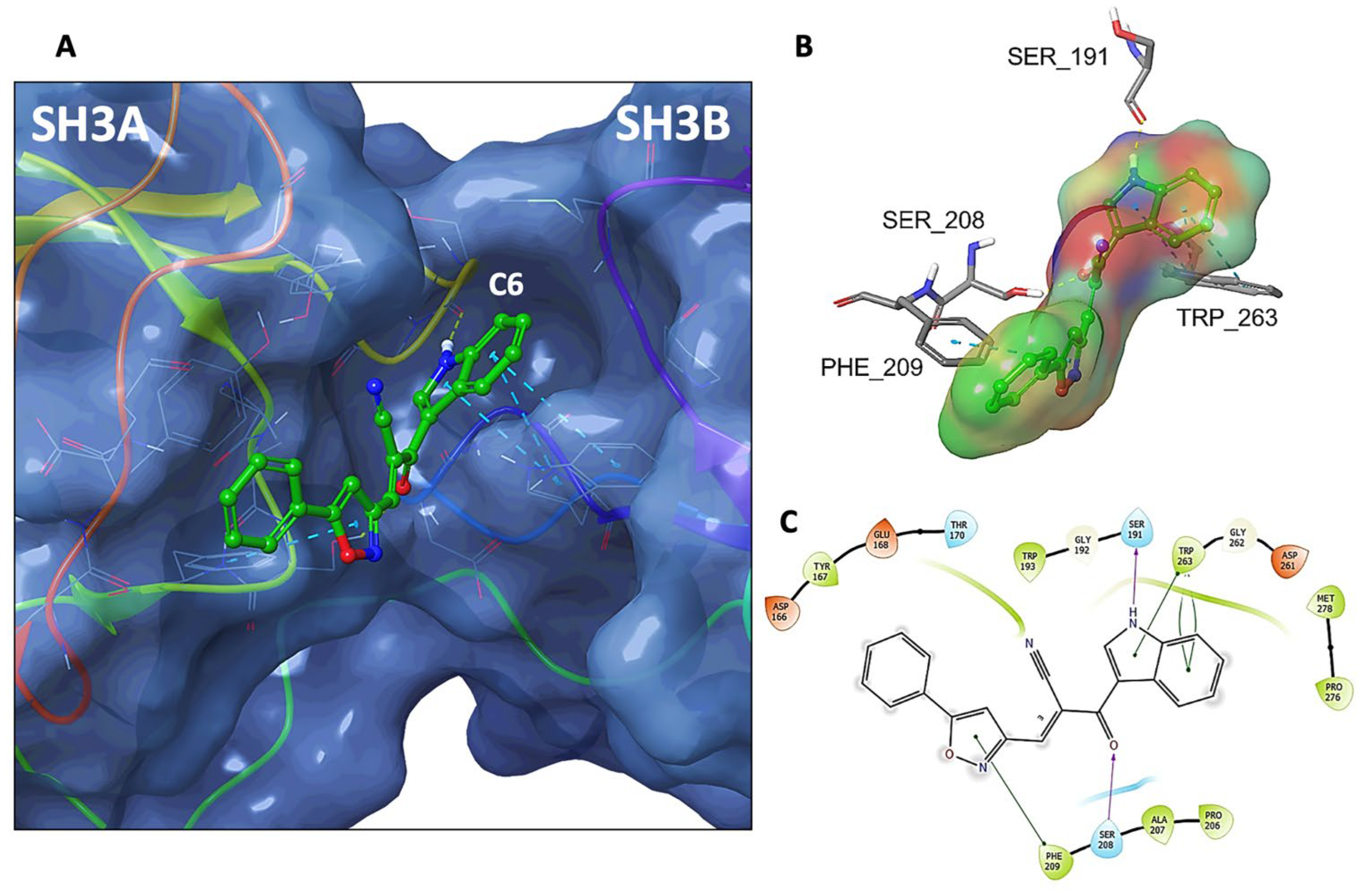
3.8. Molecular Docking of C6 and C14 with p47phox
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Noreng, S.; Ota, N.; Sun, Y.; Ho, H.; Johnson, M.; Arthur, C.P.; Schneider, K.; Lehoux, I.; Davies, C.W.; Mortara, K.; et al. Structure of the core human NADPH oxidase NOX2. Nat. Commun. 2022, 13, 6079. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Quinn, M.T.; Lambeth, J.D. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol. Biol. 2007, 7, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Liu, R.; Song, K.; Wu, J.X.; Geng, X.P.; Zheng, L.; Gao, X.; Peng, H.; Chen, L. Structure of human phagocyte NADPH oxidase in the resting state. Elife 2022, 11, e83743. [Google Scholar] [CrossRef]
- Groemping, Y.; Lapouge, K.; Smerdon, S.J.; Rittinger, K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 2003, 113, 343–355. [Google Scholar] [CrossRef]
- Touyz, R.M.; Yao, G.; Schiffrin, E.L. c-Src induces phosphorylation and translocation of p47phox: Role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Marcoux, J.; Man, P.; Petit-Haertlein, I.; Vives, C.; Forest, E.; Fieschi, F. p47phox molecular activation for assembly of the neutrophil NADPH oxidase complex. J. Biol. Chem. 2010, 285, 28980–28990. [Google Scholar] [CrossRef] [Green Version]
- Ogura, K.; Nobuhisa, I.; Yuzawa, S.; Takeya, R.; Torikai, S.; Saikawa, K.; Sumimoto, H.; Inagaki, F. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J. Biol. Chem. 2006, 281, 3660–3668. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.Y.; Miralda, I.; Armstrong, C.L.; Uriarte, S.M.; Bagaitkar, J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol. Oral Microbiol. 2019, 34, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Tse, H.M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol. 2021, 48, 102159. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; San Martin, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Loffredo, L.; Carnevale, R.; Sanguigni, V.; Martino, E.; Catasca, E.; Zanoni, C.; Pignatelli, P.; Violi, F. Oxidative stress is associated with arterial dysfunction and enhanced intima-media thickness in children with hypercholesterolemia: The potential role of nicotinamide-adenine dinucleotide phosphate oxidase. Pediatrics 2008, 122, e648–e655. [Google Scholar] [CrossRef]
- Loffredo, L.; Martino, F.; Zicari, A.M.; Carnevale, R.; Battaglia, S.; Martino, E.; Cammisotto, V.; Peruzzi, M.; De Castro, G.; Duse, M.; et al. Enhanced NOX-2 derived oxidative stress in offspring of patients with early myocardial infarction. Int. J. Cardiol. 2019, 293, 56–59. [Google Scholar] [CrossRef]
- Andres, C.M.C.; Perez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Perez-Lebena, E. The Role of Reactive Species on Innate Immunity. Vaccines 2022, 10, 1735. [Google Scholar] [CrossRef]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- ten Freyhaus, H.; Huntgeburth, M.; Wingler, K.; Schnitker, J.; Baumer, A.T.; Vantler, M.; Bekhite, M.M.; Wartenberg, M.; Sauer, H.; Rosenkranz, S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc. Res. 2006, 71, 331–341. [Google Scholar] [CrossRef]
- Hirano, K.; Chen, W.S.; Chueng, A.L.; Dunne, A.A.; Seredenina, T.; Filippova, A.; Ramachandran, S.; Bridges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, J.; Massari, M.; Marchese, S.; Ceccon, M.; Aalbers, F.S.; Corana, F.; Valente, S.; Mai, A.; Magnani, F.; Mattevi, A. A closer look into NADPH oxidase inhibitors: Validation and insight into their mechanism of action. Redox Biol. 2020, 32, 101466. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, P.; Arora, N.; Thanikachalam, P.V.; Monga, V. Recent advances in the synthetic and medicinal perspective of quinolones: A review. Bioorg. Chem. 2019, 92, 103291. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Matus, M.F.; Poblete, T.; Amigo, J.; Vallejos, G.; Astudillo, L. Isoxazoles: Synthesis, evaluation and bioinformatic design as acetylcholinesterase inhibitors. J. Pharm. Pharmacol. 2013, 65, 1796–1804. [Google Scholar] [CrossRef]
- Gawande, S.S.; Warangkar, S.C.; Bandgar, B.P.; Khobragade, C.N. Synthesis of new heterocyclic hybrids based on pyrazole and thiazolidinone scaffolds as potent inhibitors of tyrosinase. Bioorg. Med. Chem. 2013, 21, 2772–2777. [Google Scholar] [CrossRef]
- Becerra, D.; Abonia, R.; Castillo, J.C. Recent Applications of the Multicomponent Synthesis for Bioactive Pyrazole Derivatives. Molecules 2022, 27, 4723. [Google Scholar] [CrossRef]
- Tolomeu, H.V.; Fraga, C.A.M. Imidazole: Synthesis, Functionalization and Physicochemical Properties of a Privileged Structure in Medicinal Chemistry. Molecules 2023, 28, 838. [Google Scholar] [CrossRef]
- Ling, Y.; Hao, Z.Y.; Liang, D.; Zhang, C.L.; Liu, Y.F.; Wang, Y. The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design. Drug Des. Dev. Ther. 2021, 15, 4289–4338. [Google Scholar] [CrossRef]
- Dai, X.J.; Xue, L.P.; Ji, S.K.; Zhou, Y.; Gao, Y.; Zheng, Y.C.; Liu, H.M.; Liu, H.M. Triazole-fused pyrimidines in target-based anticancer drug discovery. Eur. J. Med. Chem. 2023, 249, 115101. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [Green Version]
- Frederich, M.; Tits, M.; Angenot, L. Potential antimalarial activity of indole alkaloids. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Ishikawa, H.; Kurihara, M.; Kitajima, M.; Aimi, N.; Ponglux, D.; Koyama, F.; Matsumoto, K.; Moriyama, T.; Yamamoto, L.T.; et al. Studies on the synthesis and opioid agonistic activities of mitragynine-related indole alkaloids: Discovery of opioid agonists structurally different from other opioid ligands. J. Med. Chem. 2002, 45, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, A.; Shi, M.; Ciszewski, G.; Park, K.; Ellingboe, J.W.; Orlowski, M.; Feld, B.; Howe, A.Y. Design and synthesis of 2,3,4,9-tetrahydro-1H-carbazole and 1,2,3,4-tetrahydro-cyclopenta[b]indole derivatives as non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA polymerase. Bioorg. Med. Chem. Lett. 2006, 16, 2532–2534. [Google Scholar] [CrossRef] [PubMed]
- Bovens, S.; Schulze Elfringhoff, A.; Kaptur, M.; Reinhardt, D.; Schafers, M.; Lehr, M. 1-(5-Carboxyindol-1-yl)propan-2-one inhibitors of human cytosolic phospholipase A2alpha: Effect of substituents in position 3 of the indole scaffold on inhibitory potency, metabolic stability, solubility, and bioavailability. J. Med. Chem. 2010, 53, 8298–8308. [Google Scholar] [CrossRef]
- Abdelhamid, A.O.; Gomha, S.M.; Abdelriheem, N.A.; Kandeel, S.M. Synthesis of New 3-Heteroarylindoles as Potential Anticancer Agents. Molecules 2016, 21, 929. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, J.; Cobo, D.; Insuasty, B.; Abonia, R.; Nogueras, M.; Cobo, J.; Vasquez, Y.; Gupta, M.; Derita, M.; Zacchino, S. Synthesis and evaluation of novel E-2-(2-thienyl)- and Z-2-(3-thienyl)-3-arylacrylonitriles as antifungal and anticancer agents. Arch. Pharm. 2007, 340, 603–606. [Google Scholar] [CrossRef]
- Brancale, A.; Silvestri, R. Indole, a core nucleus for potent inhibitors of tubulin polymerization. Med. Res. Rev. 2007, 27, 209–238. [Google Scholar] [CrossRef]
- Asati, V.; Bhupal, R.; Bhattacharya, S.; Kaur, K.; Gupta, G.D.; Pathak, A.; Mahapatra, D.K. Recent Updates on Indole Derivatives as Kinase Inhibitors in the Treatment of Cancer. Anticancer Agents Med. Chem. 2023, 23, 404–416. [Google Scholar] [CrossRef]
- de la Torre, P.; Saavedra, L.A.; Caballero, J.; Quiroga, J.; Alzate-Morales, J.H.; Cabrera, M.G.; Trilleras, J. A novel class of selective acetylcholinesterase inhibitors: Synthesis and evaluation of (E)-2-(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 2012, 17, 12072–12085. [Google Scholar] [CrossRef] [Green Version]
- Treuer, A.V.; De-La-Torre, P.; Gutiérrez, M.I. Synthesis of New (E)-2-(1H-Indole-3-ylcarbonyl)-3-heteroaryl-acrylonitriles via Microwave-Assisted Knoevenagel Condensation. J. Chem. 2017, 2017, 8418930. [Google Scholar] [CrossRef] [Green Version]
- De-la-Torre, P.; Osorio, E.; Alzate-Morales, J.H.; Caballero, J.; Trilleras, J.; Astudillo-Saavedra, L.; Brito, I.; Cárdenas, A.; Quiroga, J.; Gutiérrez, M. Ultrasound-assisted phase-transfer catalysis method in an aqueous medium to promote the Knoevenagel reaction: Advantages over the conventional and microwave-assisted solvent-free/catalyst-free method. Ultrason. Sonochem. 2014, 21, 1666–1674. [Google Scholar] [CrossRef]
- Unsal Tan, O.; Zengin, M. Insights into the chemistry and therapeutic potential of acrylonitrile derivatives. Arch. Pharm. 2022, 355, e2100383. [Google Scholar] [CrossRef]
- Solangi, M.; Khan, K.M.; Saleem, F.; Hameed, S.; Iqbal, J.; Shafique, Z.; Qureshi, U.; Ul-Haq, Z.; Taha, M.; Perveen, S. Indole acrylonitriles as potential anti-hyperglycemic agents: Synthesis, alpha-glucosidase inhibitory activity and molecular docking studies. Bioorg. Med. Chem. 2020, 28, 115605. [Google Scholar] [CrossRef] [PubMed]
- Penthala, N.R.; Sonar, V.N.; Horn, J.; Leggas, M.; Yadlapalli, J.S.; Crooks, P.A. Synthesis and evaluation of a series of benzothiophene acrylonitrile analogs as anticancer agents. Medchemcomm 2013, 4, 1073–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethencourt-Estrella, C.J.; Delgado-Hernandez, S.; Lopez-Arencibia, A.; San Nicolas-Hernandez, D.; Sifaoui, I.; Tejedor, D.; Garcia-Tellado, F.; Lorenzo-Morales, J.; Pinero, J.E. Acrylonitrile Derivatives against Trypanosoma cruzi: In Vitro Activity and Programmed Cell Death Study. Pharmaceuticals 2021, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Saczewski, F.; Reszka, P.; Gdaniec, M.; Grunert, R.; Bednarski, P.J. Synthesis, X-ray crystal structures, stabilities, and in vitro cytotoxic activities of new heteroarylacrylonitriles. J. Med. Chem. 2004, 47, 3438–3449. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Ismael, M.; Del Carpio, C.A.; Tsuboi, H.; Koyama, M.; Endou, A.; Kubo, M.; Broclawik, E.; Miyamoto, A. Three-dimensional quantitative structure-activity relationship (3 D-QSAR) and docking studies on (benzothiazole-2-yl) acetonitrile derivatives as c-Jun N-terminal kinase-3 (JNK3) inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5917–5925. [Google Scholar] [CrossRef]
- Carta, A.; Briguglio, I.; Piras, S.; Boatto, G.; La Colla, P.; Loddo, R.; Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Pipitone, R.M.; et al. 3-Aryl-2-[1H-benzotriazol-1-yl]acrylonitriles: A novel class of potent tubulin inhibitors. Eur. J. Med. Chem. 2011, 46, 4151–4167. [Google Scholar] [CrossRef]
- De-la-Torre, P.; Treuer, A.V.; Gutierrez, M.; Poblete, H.; Alzate-Morales, J.H.; Trilleras, J.; Astudillo-Saavedra, L.; Caballero, J. Synthesis and in silico analysis of the quantitative structure–activity relationship of heteroaryl–acrylonitriles as AChE inhibitors. J. Taiwan Inst. Chem. Eng. 2016, 59, 45–60. [Google Scholar] [CrossRef]
- Gonzalez, D.R.; Treuer, A.V.; Lamirault, G.; Mayo, V.; Cao, Y.; Dulce, R.A.; Hare, J.M. NADPH oxidase-2 inhibition restores contractility and intracellular calcium handling and reduces arrhythmogenicity in dystrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H710–H721. [Google Scholar] [CrossRef] [Green Version]
- Castillo, O.A.; Herrera, G.; Manriquez, C.; Rojas, A.F.; Gonzalez, D.R. Pharmacological Inhibition of S-Nitrosoglutathione Reductase Reduces Cardiac Damage Induced by Ischemia-Reperfusion. Antioxidants 2021, 10, 555. [Google Scholar] [CrossRef] [PubMed]
- Boly, R.; Franck, T.; Kohnen, S.; Lompo, M.; Guissou, I.P.; Dubois, J.; Serteyn, D.; Mouithys-Mickalad, A. Evaluation of Antiradical and Anti-Inflammatory Activities of Ethyl Acetate and Butanolic Subfractions of Agelanthus dodoneifolius (DC.) Polhill & Wiens (Loranthaceae) Using Equine Myeloperoxidase and Both PMA-Activated Neutrophils and HL-60 Cells. Evid. Based Complement. Altern. Med. 2015, 2015, 707524. [Google Scholar]
- Vielma, A.Z.; Boric, M.P.; Gonzalez, D.R. Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. Int. J. Mol. Sci. 2020, 21, 5415. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, S.; Sadoshima, J. Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion. Antioxidants 2022, 11, 1069. [Google Scholar] [CrossRef]
- Mohammad, A.; Babiker, F.; Al-Bader, M. Effects of Apocynin, a NADPH Oxidase Inhibitor, in the Protection of the Heart from Ischemia/Reperfusion Injury. Pharmaceuticals 2023, 16, 492. [Google Scholar] [CrossRef]
- Fan, L.M.; Liu, F.; Du, J.; Geng, L.; Li, J.M. Inhibition of endothelial Nox2 activation by LMH001 protects mice from angiotensin II-induced vascular oxidative stress, hypertension and aortic aneurysm. Redox Biol. 2022, 51, 102269. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef]
- Teuber, J.P.; Essandoh, K.; Hummel, S.L.; Madamanchi, N.R.; Brody, M.J. NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction. Antioxidants 2022, 11, 1822. [Google Scholar] [CrossRef]
- Elbatreek, M.H.; Mucke, H.; Schmidt, H. NOX Inhibitors: From Bench to Naxibs to Bedside. Handb. Exp. Pharmacol. 2021, 264, 145–168. [Google Scholar] [PubMed]
- Grauers Wiktorin, H.; Aydin, E.; Hellstrand, K.; Martner, A. NOX2-Derived Reactive Oxygen Species in Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 7095902. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.T.; Elbatreek, M.H.; Altenhofer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H. Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation. Free Radic. Biol. Med. 2020, 148, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Nenci, S.; Millana Fananas, E.; Ceccon, M.; Romero, E.; Fraaije, M.W.; Mattevi, A. Crystal structures and atomic model of NADPH oxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 6764–6769. [Google Scholar] [CrossRef]
- Wind, S.; Beuerlein, K.; Eucker, T.; Muller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.; Wingler, K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010, 161, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Solbak, S.M.O.; Zang, J.; Narayanan, D.; Hoj, L.J.; Bucciarelli, S.; Softley, C.; Meier, S.; Langkilde, A.E.; Gotfredsen, C.H.; Sattler, M.; et al. Developing Inhibitors of the p47phox-p22phox Protein-Protein Interaction by Fragment-Based Drug Discovery. J. Med. Chem. 2020, 63, 1156–1177. [Google Scholar] [CrossRef]
- Smith, S.M.; Min, J.; Ganesh, T.; Diebold, B.; Kawahara, T.; Zhu, Y.; McCoy, J.; Sun, A.; Snyder, J.P.; Fu, H.; et al. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem. Biol. 2012, 19, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Akbar, H.; Duan, X.; Piatt, R.; Saleem, S.; Davis, A.K.; Tandon, N.N.; Bergmeier, W.; Zheng, Y. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J. Thromb. Haemost. 2018, 16, 2083–2096. [Google Scholar] [CrossRef] [Green Version]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Zang, J.; Cambet, Y.; Jaquet, V.; Bach, A. Chemical synthesis of a reported p47phox/p22phox inhibitor and characterization of its instability and irreproducible activity. Front. Pharmacol. 2022, 13, 1075328. [Google Scholar] [CrossRef]
- Li, Y.; Cifuentes-Pagano, E.; DeVallance, E.R.; de Jesus, D.S.; Sahoo, S.; Meijles, D.N.; Koes, D.; Camacho, C.J.; Ross, M.; St Croix, C.; et al. NADPH oxidase 2 inhibitors CPP11G and CPP11H attenuate endothelial cell inflammation & vessel dysfunction and restore mouse hind-limb flow. Redox Biol. 2019, 22, 101143. [Google Scholar]
- Garsi, J.B.; Komjati, B.; Cullia, G.; Fejes, I.; Sipos, M.; Sipos, Z.; Fordos, E.; Markacz, P.; Balazs, B.; Lancelot, N.; et al. Targeting NOX2 via p47/phox-p22/phox Inhibition with Novel Triproline Mimetics. ACS Med. Chem. Lett. 2022, 13, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox Inhibitors & Therapies: Rational Design of Peptidic and Small Molecule Inhibitors. Curr. Pharm. Des. 2015, 21, 6023–6035. [Google Scholar] [PubMed] [Green Version]
- Sun, Q.A.; Hess, D.T.; Wang, B.; Miyagi, M.; Stamler, J.S. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870). Free Radic Biol. Med. 2012, 52, 1897–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.J.; Li, J.Y.; Chen, R.J.; Huang, L.T.; Lee, T.Y.; Lin, K.H. VAS2870 and VAS3947 attenuate platelet activation and thrombus formation via a NOX-independent pathway downstream of PKC. Sci. Rep. 2019, 9, 18852. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) |
|---|---|
| C1 | 21.5 ± 0.3 |
| C2 | 7.2 ± 0.3 |
| C3 | 3.0 ± 0.3 |
| C4 | 7.1 ± 0.4 |
| C5 | 6.2 ± 0.4 |
| C6 | 1.1 ± 0.2 |
| C7 | 68.1 ± 1.2 |
| C8 | 17.1 ± 0.2 |
| C9 | 4.7 ± 0.3 |
| C10 | 9.4 ± 0.3 |
| C11 | 19.9 ± 0.2 |
| C12 | 21.1 ± 0.2 |
| C13 | 16.3 ± 0.2 |
| C14 | 3.6 ± 0.2 |
| C15 | 30.4 ± 0.3 |
| C16 | 82.7 ± 0.2 |
| C17 | 19.7 ± 0.2 |
| C18 | 51.3 ± 0.2 |
| C19 | 69.3 ± 0.2 |
| Compound | XP GScore (kcal/mol) |
|---|---|
| C2 | −4.078 |
| C3 | −6.080 |
| C4 | −5.138 |
| C5 | −7.389 |
| C6 | −7.576 |
| C9 | −1.612 |
| C10 | −6.195 |
| C14 | −6.897 |
| VAS2870 | −5.482 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Treuer, A.V.; Faúndez, M.; Ebensperger, R.; Hovelmeyer, E.; Vergara-Jaque, A.; Perera-Sardiña, Y.; Gutierrez, M.; Fuentealba, R.; González, D.R. New NADPH Oxidase 2 Inhibitors Display Potent Activity against Oxidative Stress by Targeting p22phox-p47phox Interactions. Antioxidants 2023, 12, 1441. https://doi.org/10.3390/antiox12071441
Treuer AV, Faúndez M, Ebensperger R, Hovelmeyer E, Vergara-Jaque A, Perera-Sardiña Y, Gutierrez M, Fuentealba R, González DR. New NADPH Oxidase 2 Inhibitors Display Potent Activity against Oxidative Stress by Targeting p22phox-p47phox Interactions. Antioxidants. 2023; 12(7):1441. https://doi.org/10.3390/antiox12071441
Chicago/Turabian StyleTreuer, Adriana V., Mario Faúndez, Roberto Ebensperger, Erwin Hovelmeyer, Ariela Vergara-Jaque, Yunier Perera-Sardiña, Margarita Gutierrez, Roberto Fuentealba, and Daniel R. González. 2023. "New NADPH Oxidase 2 Inhibitors Display Potent Activity against Oxidative Stress by Targeting p22phox-p47phox Interactions" Antioxidants 12, no. 7: 1441. https://doi.org/10.3390/antiox12071441