
Formyl Peptide Receptor 2-Dependent cPLA2 and 5-LOX Activation Requires a Functional NADPH Oxidase
 , , , and
, , , and 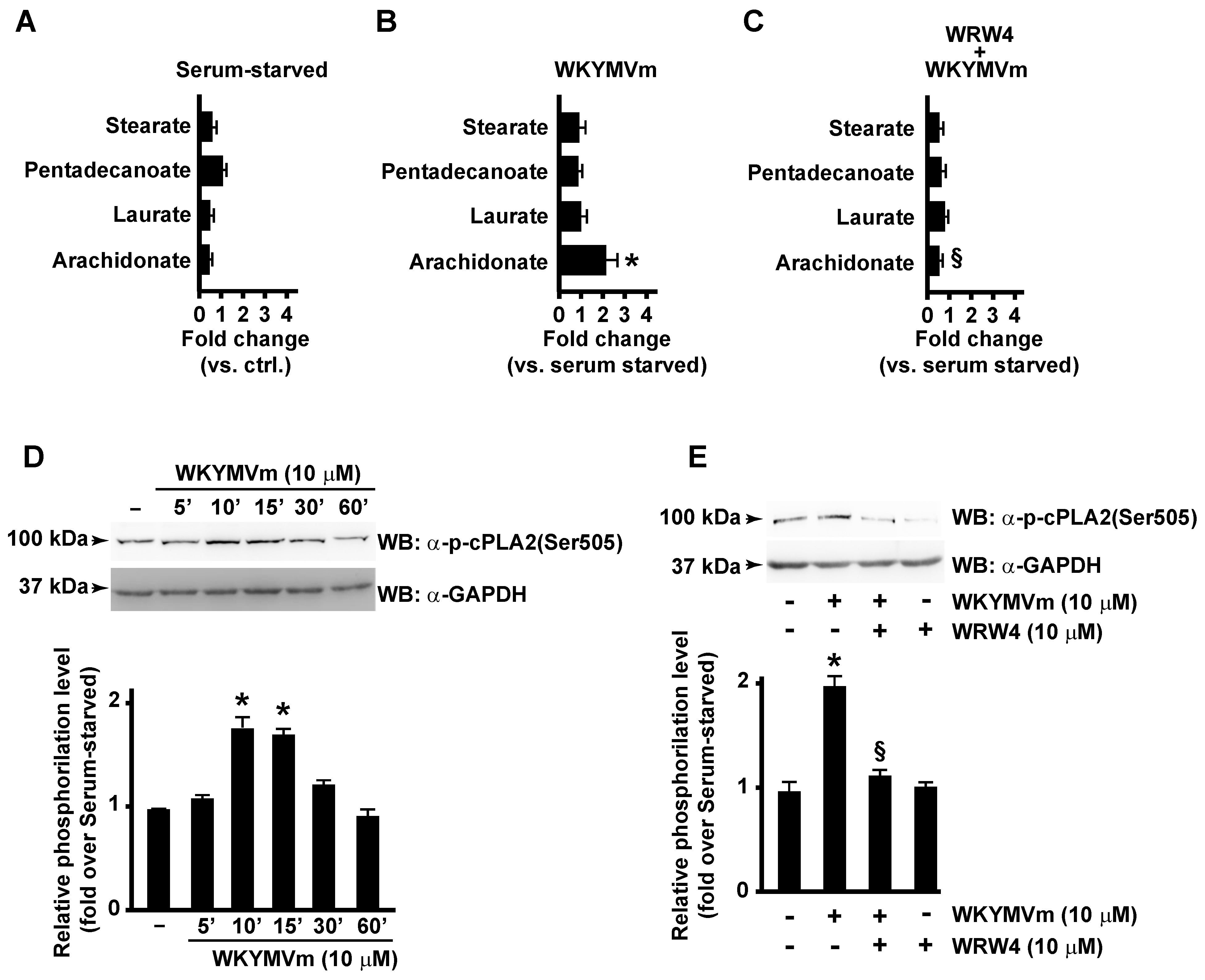{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. p22phoxCrispr/Cas9 Double-Nickase CaLu-6 Cells
2.3. Metabolomic Analysis by LC-MS
2.4. Protein Extraction and Western Blot
2.5. Statistical Analysis
3. Results and Discussion
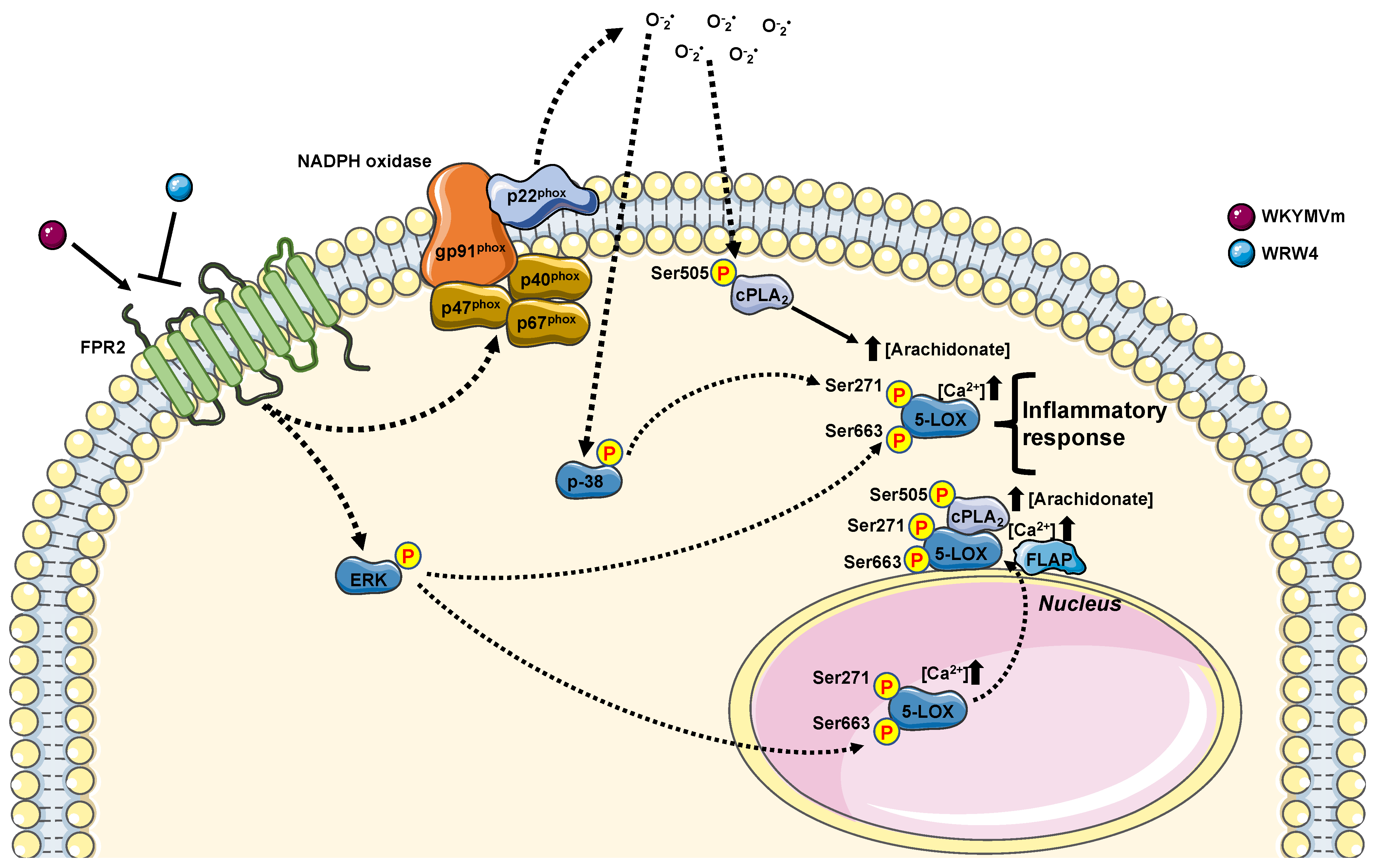
3.1. FPR2 Stimulation Induces Arachidonic Acid Release by Activating cPLA2
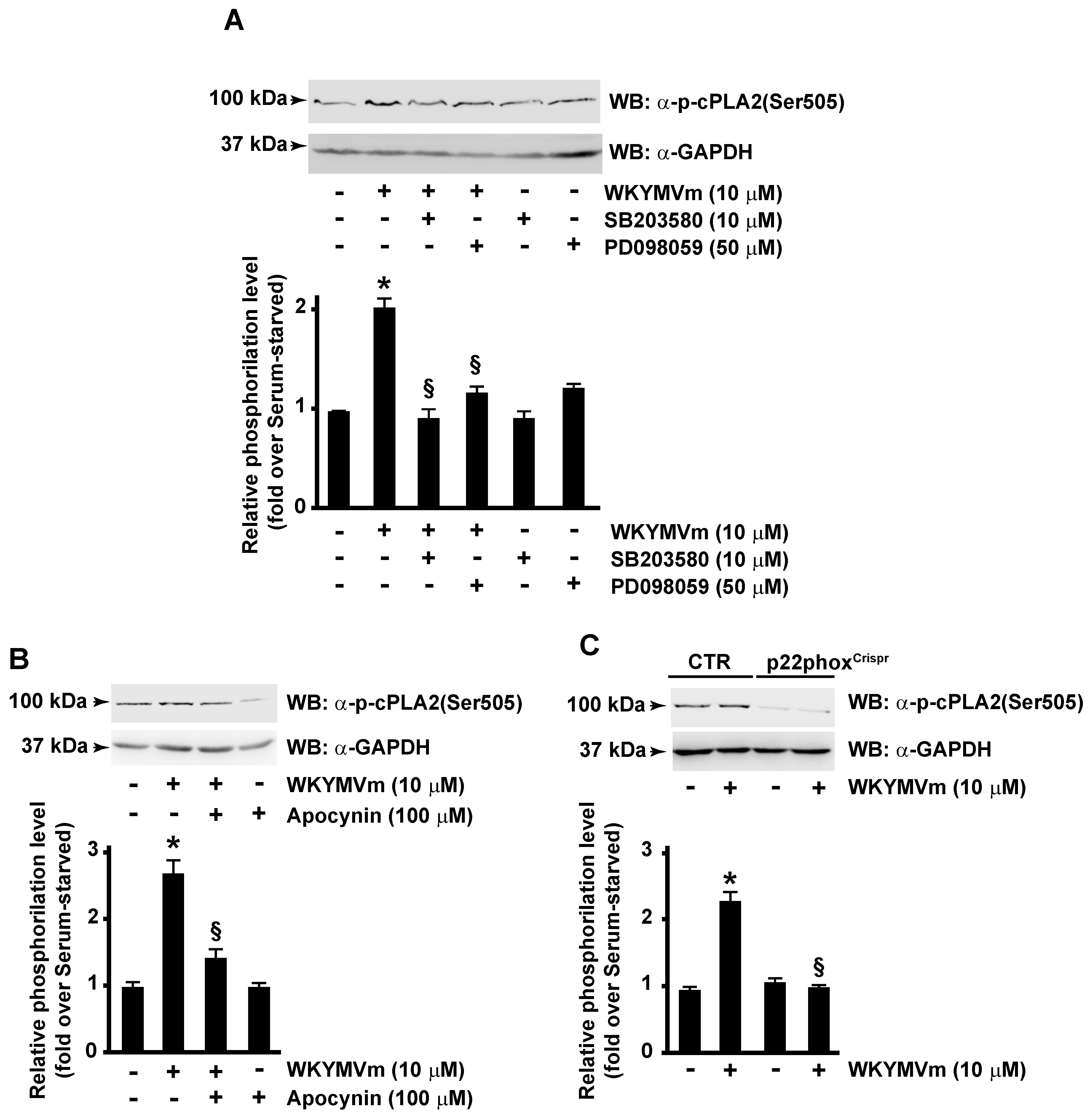
3.2. FPR2-Mediated cPLA2 Ser505 Phosphorylation Depends on NOX Activation
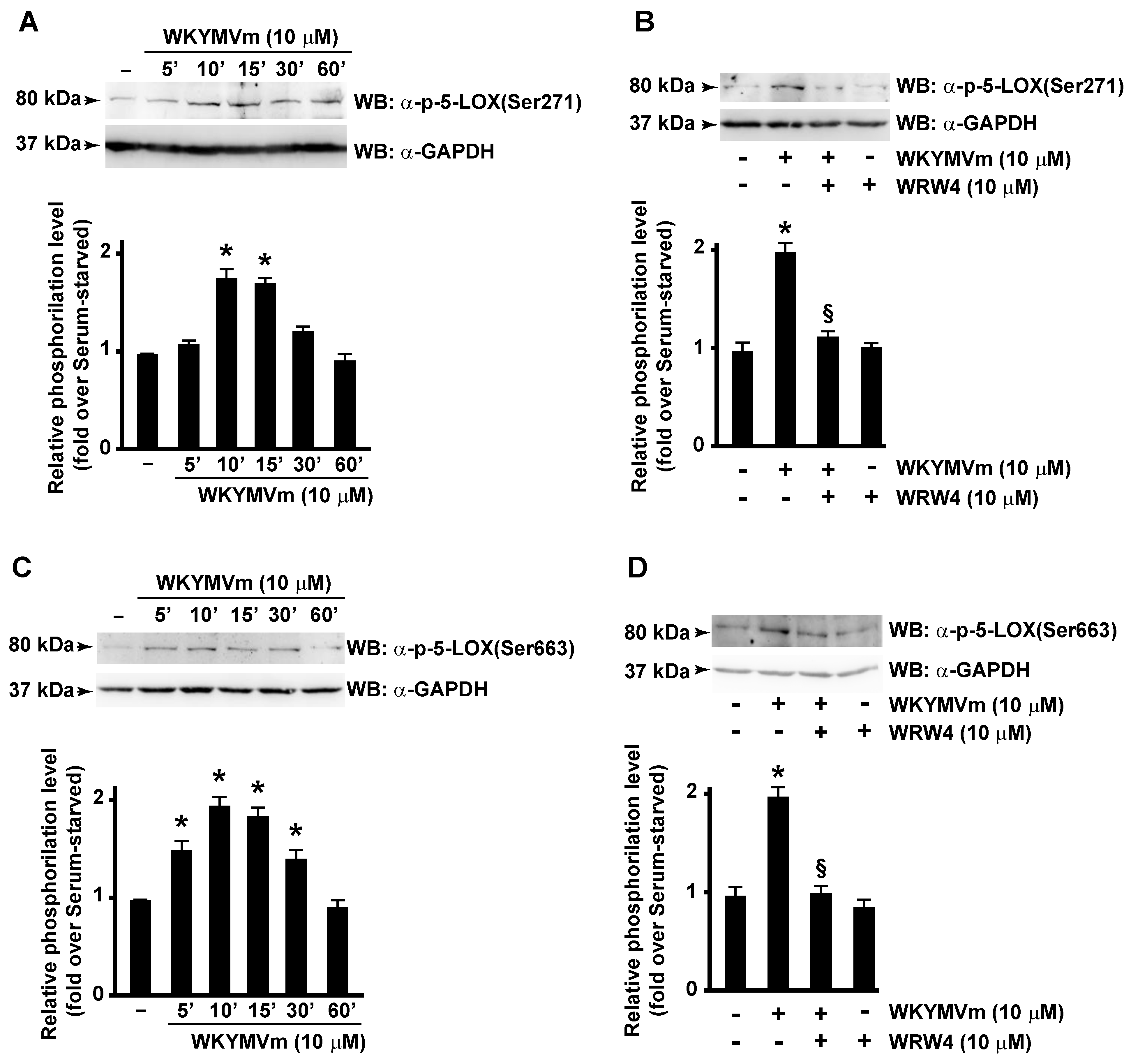
3.3. FPR2 Stimulation Induces 5-LOX Activation
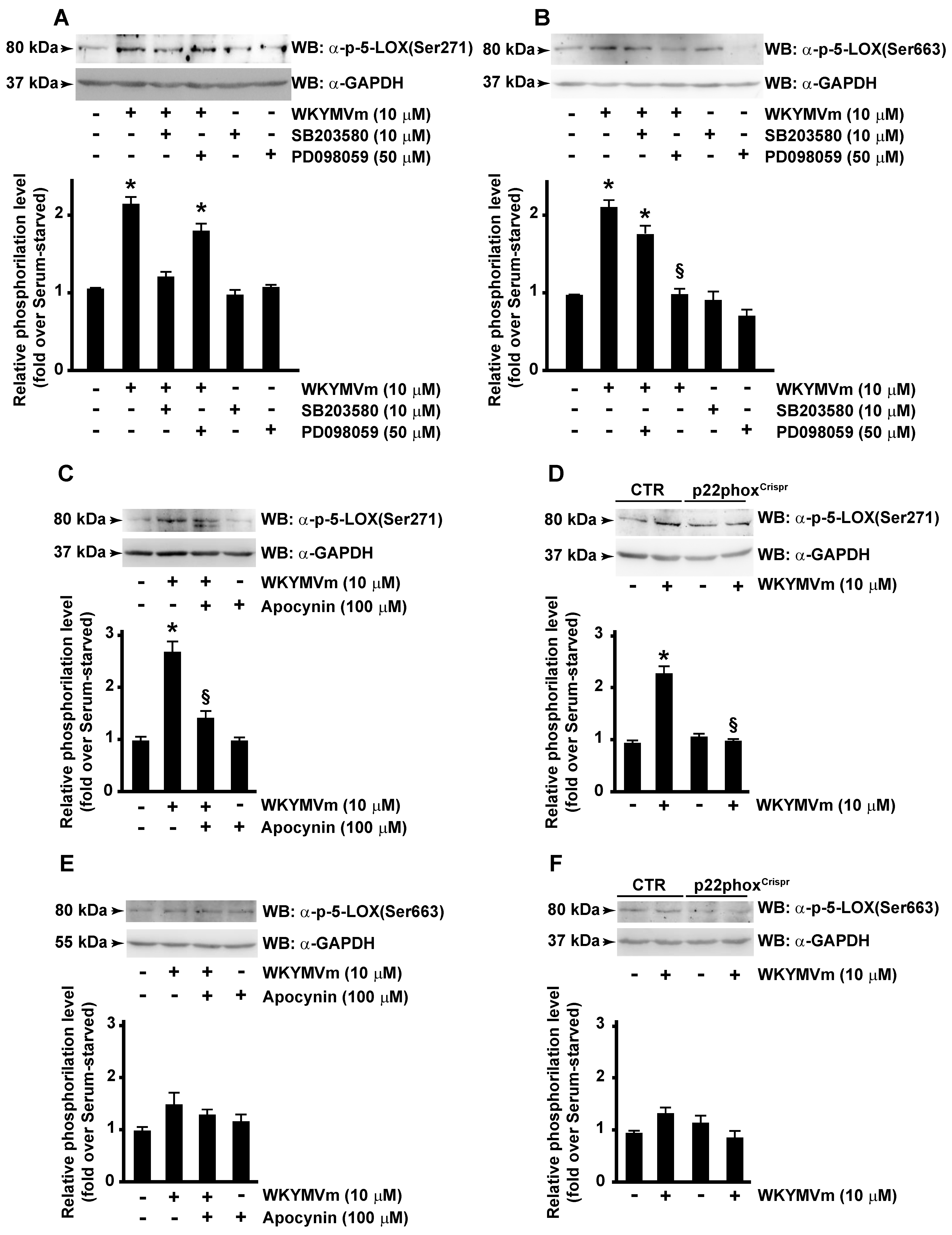
3.4. p38MAPK, ERKs and NOX Are Required for FPR2-Dependent 5-LOX Phosphorylation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Vecchi, L.; Araujo, T.G.; Azevedo, F.; Mota, S.T.S.; Avila, V.M.R.; Ribeiro, M.A.; Goulart, L.R. Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells 2021, 10, 1472. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Bollinger, J.M.; Gelb, M.; Mauro, T.M. sPLA2 and the epidermal barrier. Biochim. Biophys. Acta 2014, 1841, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 2006, 1761, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Tang, X.; Hou, Y.; Schwartz, T.W.; Haeggstrom, J.Z. Metabolite G-protein coupled receptor signaling: Potential regulation of eicosanoids. Biochem. Pharmacol. 2022, 204, 115208. [Google Scholar] [CrossRef]
- Lin, L.L.; Wartmann, M.; Lin, A.Y.; Knopf, J.L.; Seth, A.; Davis, R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell 1993, 72, 269–278. [Google Scholar] [CrossRef]
- Nito, C.; Kamada, H.; Endo, H.; Niizuma, K.; Myer, D.J.; Chan, P.H. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2008, 28, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Gijon, M.A.; Spencer, D.M.; Siddiqi, A.R.; Bonventre, J.V.; Leslie, C.C. Cytosolic phospholipase A2 is required for macrophage arachidonic acid release by agonists that Do and Do not mobilize calcium. Novel role of mitogen-activated protein kinase pathways in cytosolic phospholipase A2 regulation. J. Biol. Chem. 2000, 275, 20146–20156. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, M.C.; Parisi, M.; Esposito, G.; Fabbrocini, G.; Ammendola, R.; Cattaneo, F. Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling. Int. J. Mol. Sci. 2020, 21, 3818. [Google Scholar] [CrossRef]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. FPRL1-mediated induction of superoxide in LL-37-stimulated IMR90 human fibroblast. Arch. Biochem. Biophys. 2009, 481, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Parisi, M.; Fioretti, T.; Sarnataro, D.; Esposito, G.; Ammendola, R. Nuclear localization of Formyl-Peptide Receptor 2 in human cancer cells. Arch. Biochem. Biophys. 2016, 603, 10–19. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxidative Med. Cell. Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef]
- Jang, I.H.; Heo, S.C.; Kwon, Y.W.; Choi, E.J.; Kim, J.H. Role of formyl peptide receptor 2 in homing of endothelial progenitor cells and therapeutic angiogenesis. Adv. Biol. Regul. 2015, 57, 162–172. [Google Scholar] [CrossRef]
- Cattaneo, F.; Russo, R.; Castaldo, M.; Chambery, A.; Zollo, C.; Esposito, G.; Pedone, P.V.; Ammendola, R. Phosphoproteomic analysis sheds light on intracellular signaling cascades triggered by Formyl-Peptide Receptor 2. Sci. Rep. 2019, 9, 17894. [Google Scholar] [CrossRef]
- Li, Y.; Ye, D. Molecular biology for formyl peptide receptors in human diseases. J. Mol. Med. 2013, 91, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Prevete, N.; Liotti, F.; Marone, G.; Melillo, R.M.; de Paulis, A. Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth. Pharmacol. Res. 2015, 102, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Parisi, M.; Lucariello, A.; De Luca, A.; De Rosa, N.; Mazzarella, G.; Bianco, A.; Ammendola, R. Expression of Formyl-peptide Receptors in Human Lung Carcinoma. Anticancer. Res. 2015, 35, 2769–2774. [Google Scholar] [PubMed]
- Liang, W.; Chen, K.; Gong, W.; Yoshimura, T.; Le, Y.; Wang, Y.; Wang, J.M. The Contribution of Chemoattractant GPCRs, Formylpeptide Receptors, to Inflammation and Cancer. Front. Endocrinol. 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Cattaneo, F.; Lippiello, P.; Cristiano, C.; Zurlo, F.; Castaldo, M.; Irace, C.; Borsello, T.; Santamaria, R.; Ammendola, R.; et al. Motor coordination and synaptic plasticity deficits are associated with increased cerebellar activity of NADPH oxidase, CAMKII, and PKC at preplaque stage in the TgCRND8 mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 68, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Busch, L.; Vieten, S.; Brodel, S.; Endres, K.; Bufe, B. Emerging contributions of formyl peptide receptors to neurodegenerative diseases. Biol. Chem. 2022, 403, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Caso, V.M.; Manzo, V.; Pecchillo Cimmino, T.; Conti, V.; Caso, P.; Esposito, G.; Russo, V.; Filippelli, A.; Ammendola, R.; Cattaneo, F. Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression. Life 2021, 11, 243. [Google Scholar] [CrossRef]
- Lupisella, J.A.; Shirude, P.S.; Wurtz, N.R.; Garcia, R.A. Formyl peptide receptor 2 and heart disease. Semin. Immunol. 2022, 59, 101602. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar] [CrossRef] [PubMed]
- Raabe, C.A.; Groper, J.; Rescher, U. Biased perspectives on formyl peptide receptors. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 305–316. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, L.; Guo, J.; Sun, D.; Wang, Y.; Liu, W.; Xu, H.E.; Zhang, C. Molecular recognition of formylpeptides and diverse agonists by the formylpeptide receptors FPR1 and FPR2. Nat. Commun. 2022, 13, 1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, G.; Chen, X.; Xue, X.; Guo, Q.; Liu, M.; Zhao, J. Formyl peptide receptors promotes neural differentiation in mouse neural stem cells by ROS generation and regulation of PI3K-AKT signaling. Sci. Rep. 2017, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Mottola, G.; Chatterjee, A.; Wu, B.; Chen, M.; Conte, M.S. Aspirin-triggered resolvin D1 attenuates PDGF-induced vascular smooth muscle cell migration via the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway. PLoS ONE 2017, 12, e0174936. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar] [CrossRef]
- Filina, Y.; Gabdoulkhakova, A.; Rizvanov, A.; Safronova, V. MAP kinases in regulation of NOX activity stimulated through two types of formyl peptide receptors in murine bone marrow granulocytes. Cell Signal. 2022, 90, 110205. [Google Scholar] [CrossRef]
- Ammendola, R.; Parisi, M.; Esposito, G.; Cattaneo, F. Pro-Resolving FPR2 Agonists Regulate NADPH Oxidase-Dependent Phosphorylation of HSP27, OSR1, and MARCKS and Activation of the Respective Upstream Kinases. Antioxidants 2021, 10, 134. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef]
- Castaldo, M.; Zollo, C.; Esposito, G.; Ammendola, R.; Cattaneo, F. NOX2-Dependent Reactive Oxygen Species Regulate Formyl-Peptide Receptor 1-Mediated TrkA Transactivation in SH-SY5Y Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2051235. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 2013, 587, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, T.P.; Pagano, E.; Stornaiuolo, M.; Esposito, G.; Ammendola, R.; Cattaneo, F. Formyl-peptide receptor 2 signalling triggers aerobic metabolism of glucose through Nox2-dependent modulation of pyruvate dehydrogenase activity. Open Biol. 2023, 13, 230336. [Google Scholar] [CrossRef] [PubMed]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef] [PubMed]
- Pecchillo Cimmino, T.; Pagano, E.; Stornaiuolo, M.; Esposito, G.; Ammendola, R.; Cattaneo, F. Formyl-Peptide Receptor 2 Signaling Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming of Lung Cancer Cells. Antioxidants 2022, 11, 1692. [Google Scholar] [CrossRef] [PubMed]
- Pavone, L.M.; Cattaneo, F.; Rea, S.; De Pasquale, V.; Spina, A.; Sauchelli, E.; Mastellone, V.; Ammendola, R. Intracellular signaling cascades triggered by the NK1 fragment of hepatocyte growth factor in human prostate epithelial cell line PNT1A. Cell Signal. 2011, 23, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Casas, J.; Balsinde, J.; Balboa, M.A. Phosphorylation of cPLA2α at Ser505 Is Necessary for Its Translocation to PtdInsP2-Enriched Membranes. Molecules 2022, 27, 2347. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Shindou, H.; Shimizu, T. Cytosolic phospholipase A2 and lysophospholipid acyltransferases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V.; Subramanian, P.; Vora, M.; Cho, W.; Chalfant, C.E. Ceramide-1-phosphate binds group IVA cytosolic phospholipase a2 via a novel site in the C2 domain. J. Biol. Chem. 2007, 282, 20467–20474. [Google Scholar] [CrossRef] [PubMed]
- Perez-Chacon, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim. Biophys. Acta 2009, 1791, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dong, Z.; Liu, H.; Xia, Y.F.; Liu, X.M.; Luo, B.B.; Wang, W.K.; Li, B.; Gao, F.; Zhang, C.; et al. Serum amyloid A stimulates lipoprotein-associated phospholipase A2 expression in vitro and in vivo. Atherosclerosis 2013, 228, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Lyngstadaas, A.V.; Olsen, M.V.; Bair, J.A.; Hodges, R.R.; Utheim, T.P.; Serhan, C.N.; Dartt, D.A. Pro-Resolving Mediator Annexin A1 Regulates Intracellular Ca2+ and Mucin Secretion in Cultured Goblet Cells Suggesting a New Use in Inflammatory Conjunctival Diseases. Front. Immunol. 2021, 12, 618653. [Google Scholar] [CrossRef]
- Bae, Y.S.; Park, J.C.; He, R.; Ye, R.D.; Kwak, J.Y.; Suh, P.G.; Ho Ryu, S. Differential signaling of formyl peptide receptor-like 1 by Trp-Lys-Tyr-Met-Val-Met-CONH2 or lipoxin A4 in human neutrophils. Mol. Pharmacol. 2003, 64, 721–730. [Google Scholar] [CrossRef]
- Zhu, D.; Lai, Y.; Shelat, P.B.; Hu, C.; Sun, G.Y.; Lee, J.C. Phospholipases A2 mediate amyloid-β peptide-induced mitochondrial dysfunction. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 11111–11119. [Google Scholar] [CrossRef]
- Pessach, I.; Leto, T.L.; Malech, H.L.; Levy, R. Essential requirement of cytosolic phospholipase A2 for stimulation of NADPH oxidase-associated diaphorase activity in granulocyte-like cells. J. Biol. Chem. 2001, 276, 33495–33503. [Google Scholar] [CrossRef]
- Cherny, V.V.; Henderson, L.M.; Xu, W.; Thomas, L.L.; DeCoursey, T.E. Activation of NADPH oxidase-related proton and electron currents in human eosinophils by arachidonic acid. J. Physiol. 2001, 535, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bey, E.A.; Wientjes, F.B.; Cathcart, M.K. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phosphorylation of p67phox and p47phox. J. Biol. Chem. 2002, 277, 25385–25392. [Google Scholar] [CrossRef] [PubMed]
- Shmelzer, Z.; Haddad, N.; Admon, E.; Pessach, I.; Leto, T.L.; Eitan-Hazan, Z.; Hershfinkel, M.; Levy, R. Unique targeting of cytosolic phospholipase A2 to plasma membranes mediated by the NADPH oxidase in phagocytes. J. Cell Biol. 2003, 162, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Horrocks, L.A.; Farooqui, A.A. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J. Neurochem. 2007, 103, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Lin, C.C.; Lee, I.T.; Lee, H.C.; Yang, C.M. Activation and induction of cytosolic phospholipase A2 by TNF-α mediated through Nox2, MAPKs, NF-κB, and p300 in human tracheal smooth muscle cells. J. Cell. Physiol. 2011, 226, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Roy, S.; Mandal, A.; Dey, K.; Chowdhury, A.; Shaikh, S.; Chakraborti, T. Role of PKCα-p38MAPK-Giα axis in NADPH oxidase derived O2·−-mediated activation of cPLA2 under U46619 stimulation in pulmonary artery smooth muscle cells. Arch. Biochem. Biophys. 2012, 523, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2013, 64, 409–421. [Google Scholar]
- Kefaloyianni, E.; Gaitanaki, C.; Beis, I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-κB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006, 18, 2238–2251. [Google Scholar] [CrossRef]
- Werz, O.; Burkert, E.; Fischer, L.; Szellas, D.; Dishart, D.; Samuelsson, B.; Radmark, O.; Steinhilber, D. 5-Lipoxygenase activation by MAPKAPK-2 and ERKs. Adv. Exp. Med. Biol. 2003, 525, 129–132. [Google Scholar] [CrossRef]
- Wan, M.; Tang, X.; Stsiapanava, A.; Haeggstrom, J.Z. Biosynthesis of leukotriene B4. Semin. Immunol. 2017, 33, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Srivastava, M.; Ahmad, N.; Sakamoto, K.; Bostwick, D.G.; Mukhtar, H. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer 2001, 91, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Hennig, R.; Grippo, P.; Ding, X.Z.; Rao, S.M.; Buchler, M.W.; Friess, H.; Talamonti, M.S.; Bell, R.H.; Adrian, T.E. 5-Lipoxygenase, a marker for early pancreatic intraepithelial neoplastic lesions. Cancer Res. 2005, 65, 6011–6016. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Skibbe, J.R.; Hu, C.; Dong, L.; Ferchen, K.; Su, R.; Li, C.; Huang, H.; Weng, H.; Huang, H.; et al. ALOX5 exhibits anti-tumor and drug-sensitizing effects in MLL-rearranged leukemia. Sci. Rep. 2017, 7, 1853. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, H.; Liu, Y.; Ou, Q.; Deng, S. Interference of ALOX5 alleviates inflammation and fibrosis in high glucose-induced renal mesangial cells. Exp. Ther. Med. 2023, 25, 34. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Brock, T.G. Intracellular compartmentalization of leukotriene biosynthesis. Am. J. Respir. Crit. Care Med. 2000, 161, S36–S40. [Google Scholar] [CrossRef]
- Radmark, O.P. The molecular biology and regulation of 5-lipoxygenase. Am. J. Respir. Crit. Care Med. 2000, 161, S11–S15. [Google Scholar] [CrossRef]
- Hammarberg, T.; Provost, P.; Persson, B.; Radmark, O. The N-terminal domain of 5-lipoxygenase binds calcium and mediates calcium stimulation of enzyme activity. J. Biol. Chem. 2000, 275, 38787–38793. [Google Scholar] [CrossRef]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta 2015, 1851, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Jones, S.M.; Phare, S.M.; Coffey, M.J.; Peters-Golden, M.; Brock, T.G. Protein kinase A inhibits leukotriene synthesis by phosphorylation of 5-lipoxygenase on serine 523. J. Biol. Chem. 2004, 279, 41512–41520. [Google Scholar] [CrossRef]
- Luo, M.; Jones, S.M.; Flamand, N.; Aronoff, D.M.; Peters-Golden, M.; Brock, T.G. Phosphorylation by protein kinase a inhibits nuclear import of 5-lipoxygenase. J. Biol. Chem. 2005, 280, 40609–40616. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Klemm, J.; Samuelsson, B.; Radmark, O. 5-lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc. Natl. Acad. Sci. USA 2000, 97, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Szellas, D.; Steinhilber, D.; Radmark, O. Arachidonic acid promotes phosphorylation of 5-lipoxygenase at Ser-271 by MAPK-activated protein kinase 2 (MK2). J. Biol. Chem. 2002, 277, 14793–14800. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Burkert, E.; Fischer, L.; Szellas, D.; Dishart, D.; Samuelsson, B.; Radmark, O.; Steinhilber, D. Extracellular signal-regulated kinases phosphorylate 5-lipoxygenase and stimulate 5-lipoxygenase product formation in leukocytes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1441–1443. [Google Scholar] [CrossRef] [PubMed]
- Hazan-Halevy, I.; Seger, R.; Levy, R. The requirement of both extracellular regulated kinase and p38 mitogen-activated protein kinase for stimulation of cytosolic phospholipase A2 activity by either FcgammaRIIA or FcgammaRIIIB in human neutrophils. A possible role for Pyk2 but not for the Grb2-Sos-Shc complex. J. Biol. Chem. 2000, 275, 12416–12423. [Google Scholar] [CrossRef]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase: Regulation of expression and enzyme activity. Trends Biochem. Sci. 2007, 32, 332–341. [Google Scholar] [CrossRef]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- Werz, O.; Burkert, E.; Samuelsson, B.; Radmark, O.; Steinhilber, D. Activation of 5-lipoxygenase by cell stress is calcium independent in human polymorphonuclear leukocytes. Blood 2002, 99, 1044–1052. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecchillo Cimmino, T.; Panico, I.; Scarano, S.; Stornaiuolo, M.; Esposito, G.; Ammendola, R.; Cattaneo, F. Formyl Peptide Receptor 2-Dependent cPLA2 and 5-LOX Activation Requires a Functional NADPH Oxidase. Antioxidants 2024, 13, 220. https://doi.org/10.3390/antiox13020220
Pecchillo Cimmino T, Panico I, Scarano S, Stornaiuolo M, Esposito G, Ammendola R, Cattaneo F. Formyl Peptide Receptor 2-Dependent cPLA2 and 5-LOX Activation Requires a Functional NADPH Oxidase. Antioxidants. 2024; 13(2):220. https://doi.org/10.3390/antiox13020220
Chicago/Turabian StylePecchillo Cimmino, Tiziana, Iolanda Panico, Simona Scarano, Mariano Stornaiuolo, Gabriella Esposito, Rosario Ammendola, and Fabio Cattaneo. 2024. "Formyl Peptide Receptor 2-Dependent cPLA2 and 5-LOX Activation Requires a Functional NADPH Oxidase" Antioxidants 13, no. 2: 220. https://doi.org/10.3390/antiox13020220