
Cardioprotective Effects of Dexmedetomidine in an Oxidative-Stress In Vitro Model of Neonatal Rat Cardiomyocytes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture: Cell Line H9c2
2.2. Cell Culture: Primary Neonatal Rat Cardiomyocytes (NRCM)
2.3. Cellular Model and Defining Oxygen Level
2.4. Oxygen Exposure and Drug Administration
2.5. RNA Extraction and qPCR
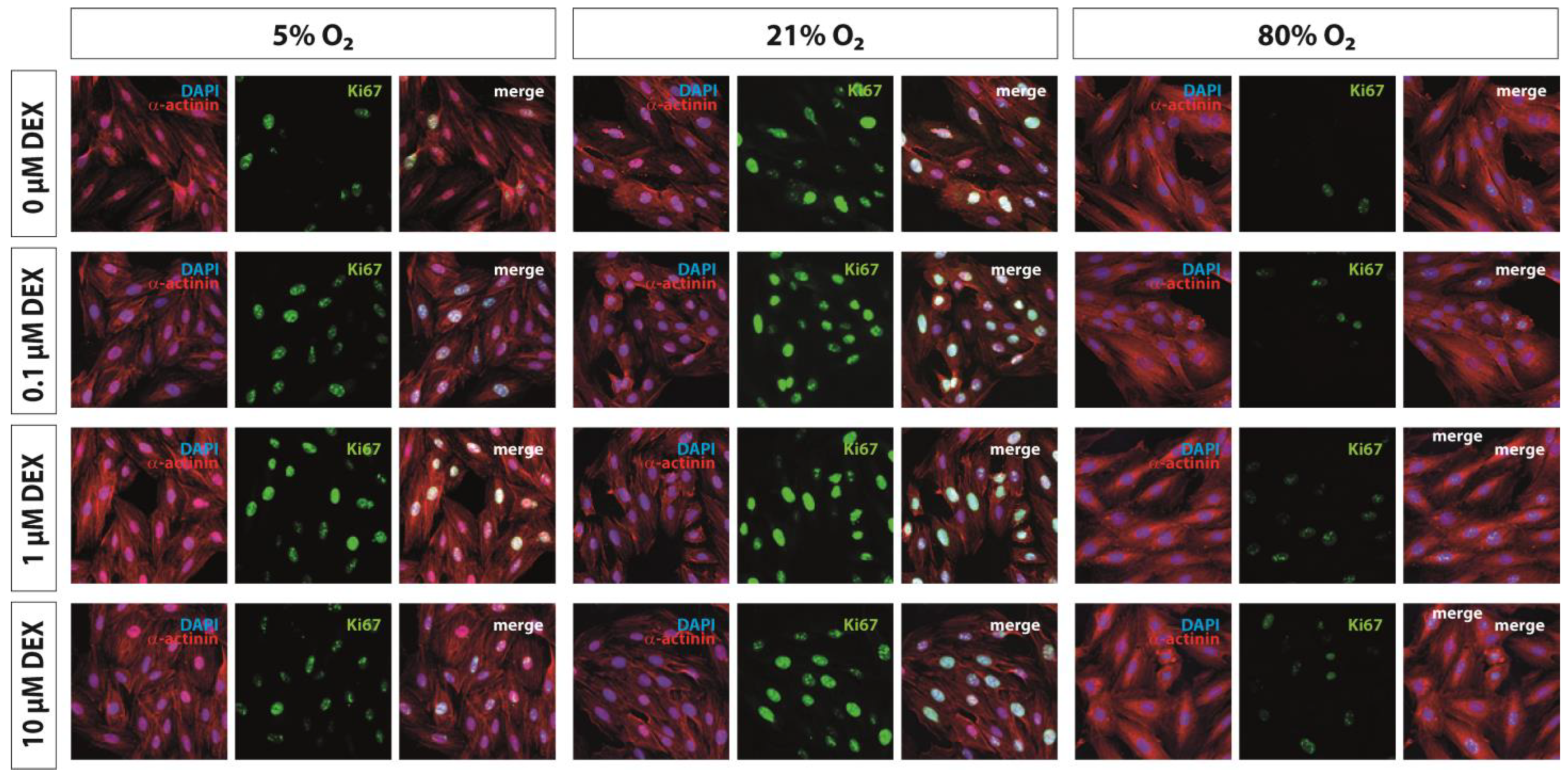
2.6. Immunohistochemistry
2.7. LDH-Assay and CCK8
2.8. Statistical Analyses
3. Results
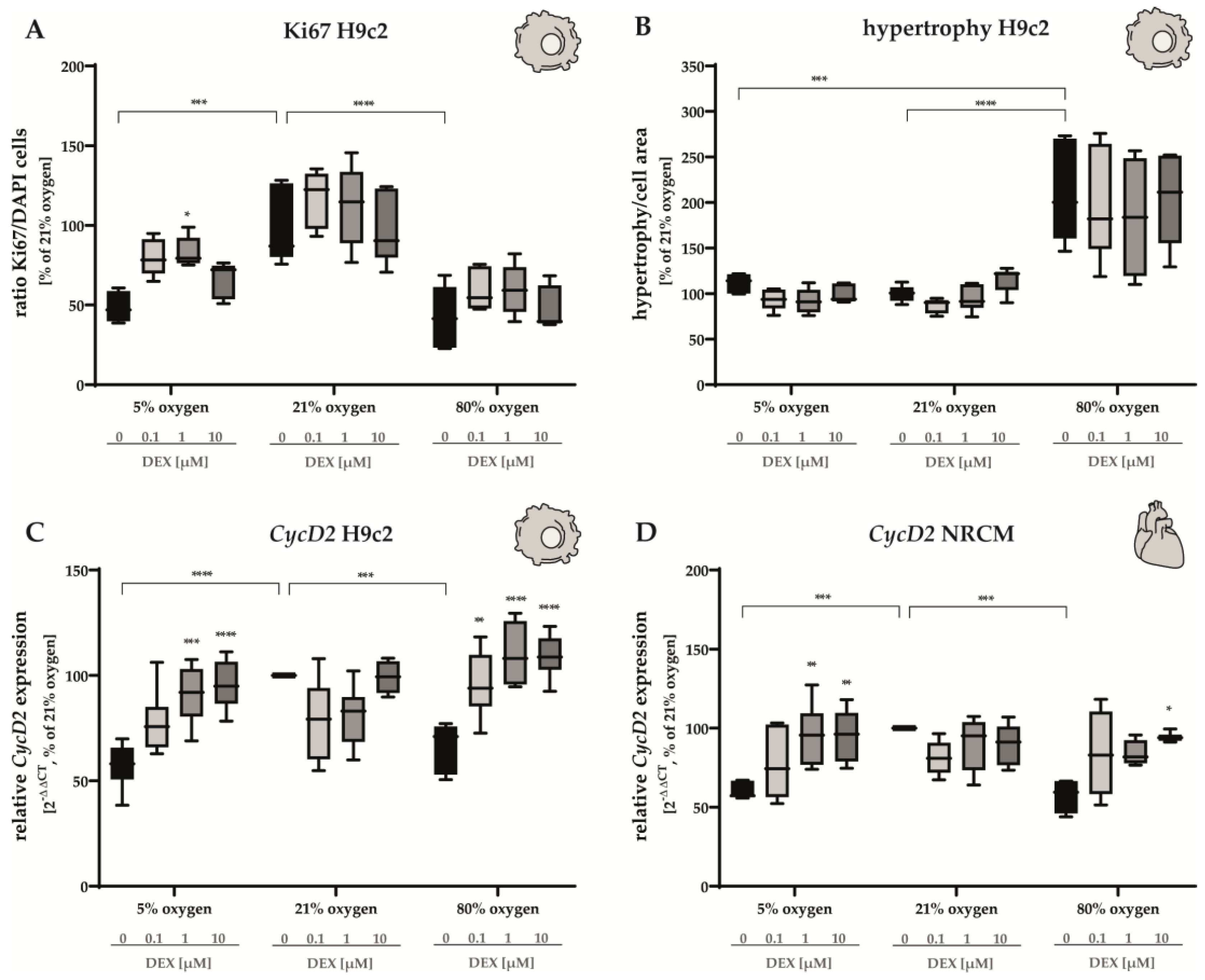
3.1. Proliferation Capacity and Hypertrophy
3.2. Cell-Death-Associated Factors
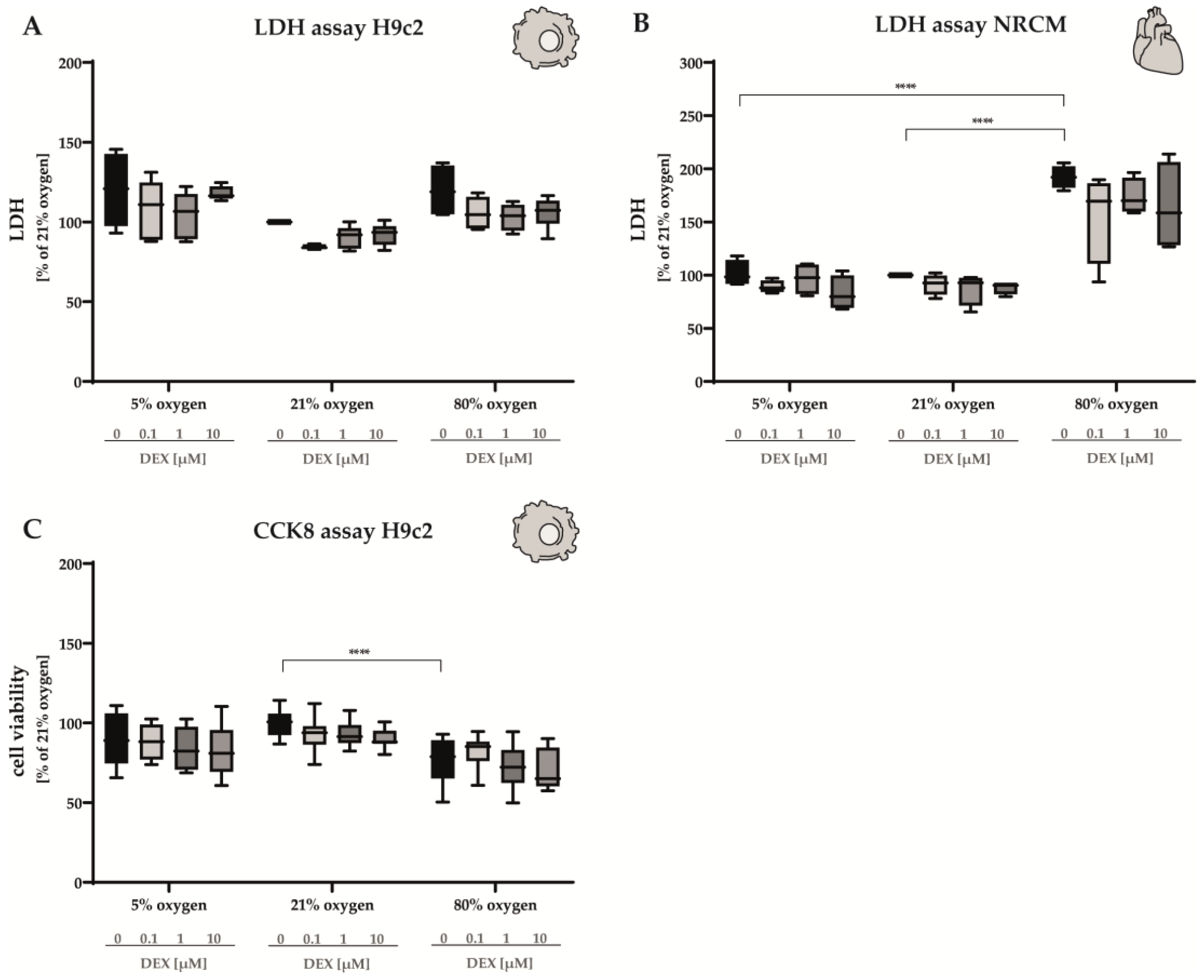
3.2.1. LDH Release and Cell Viability
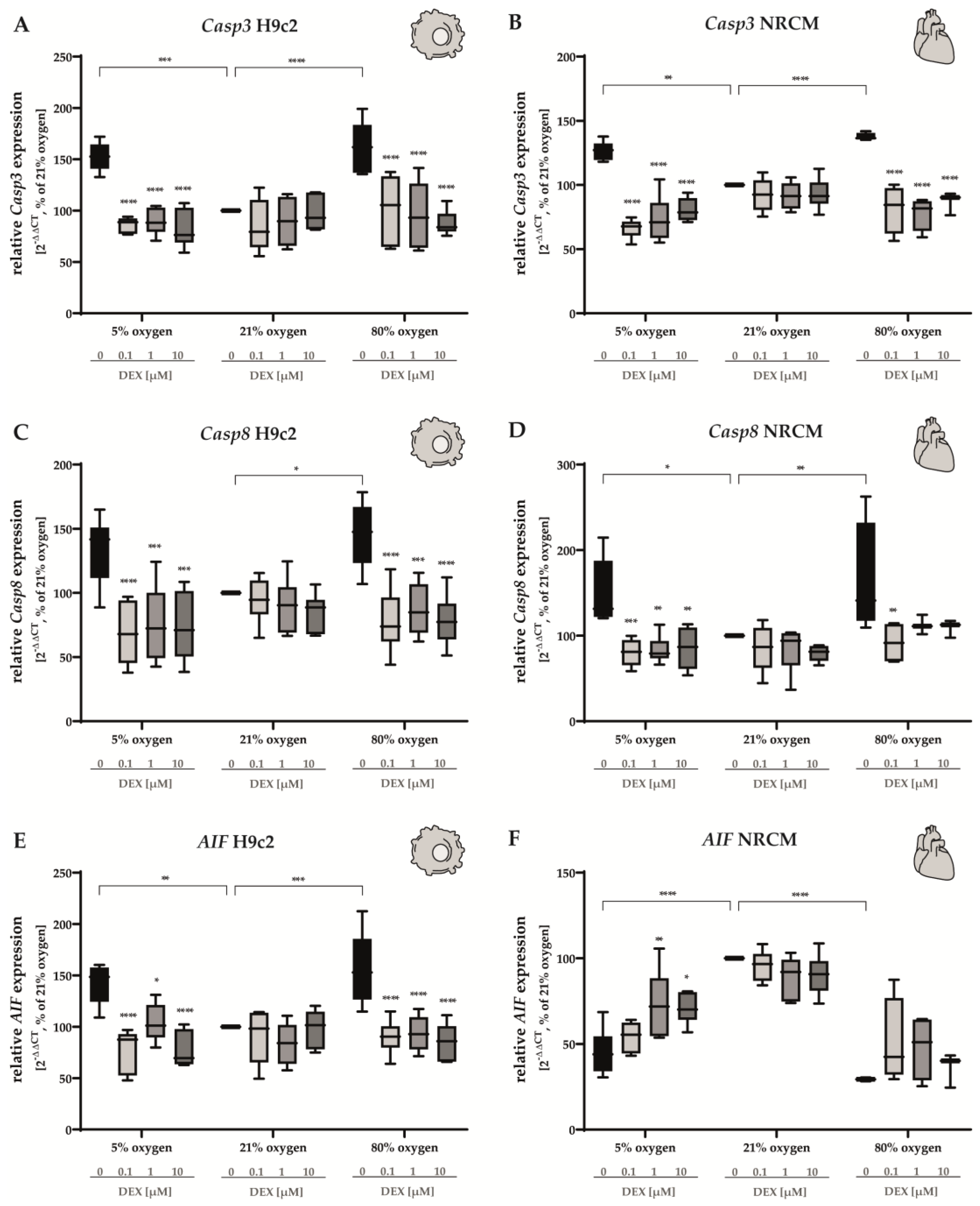
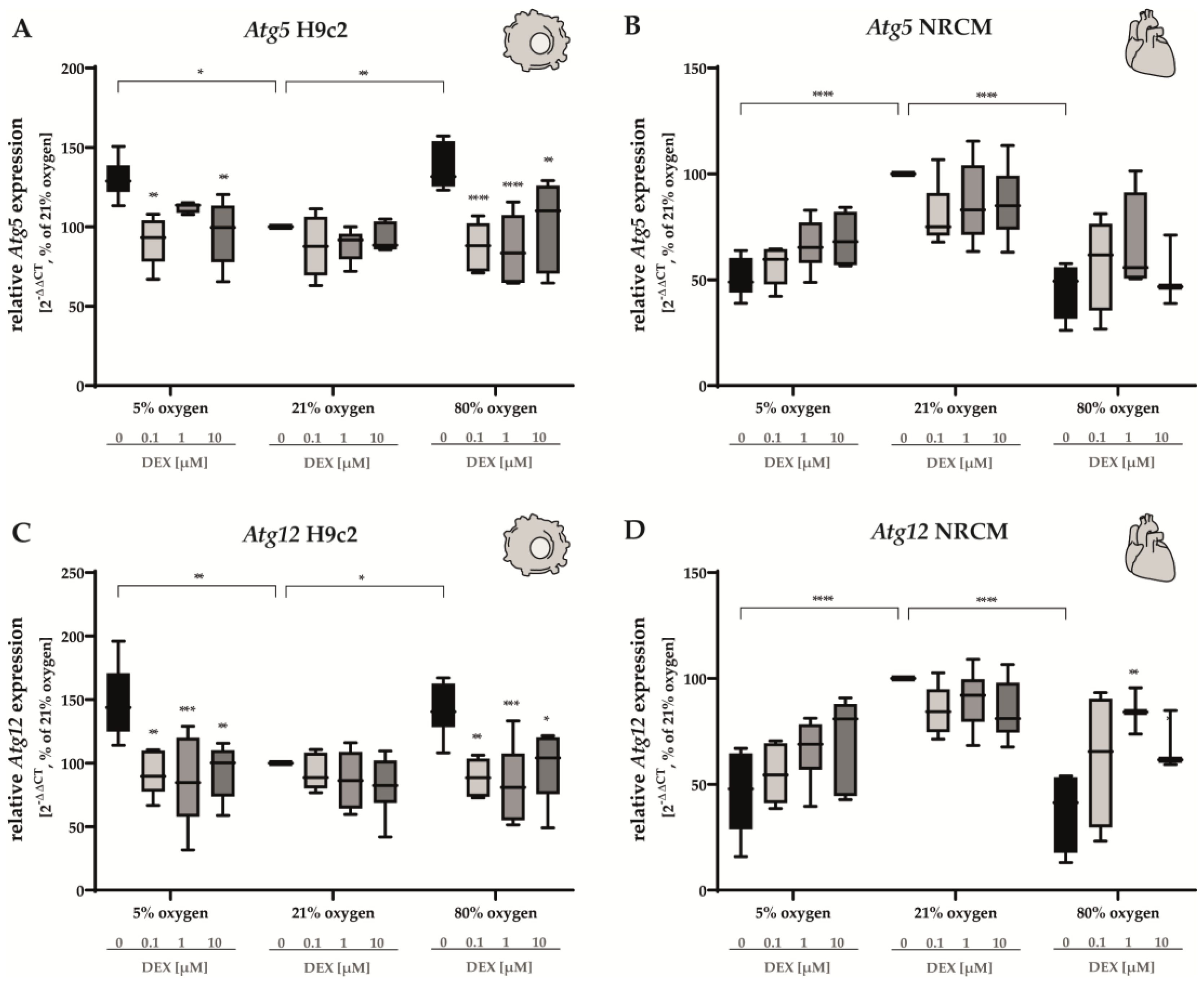
3.2.2. Apoptosis and Autophagy
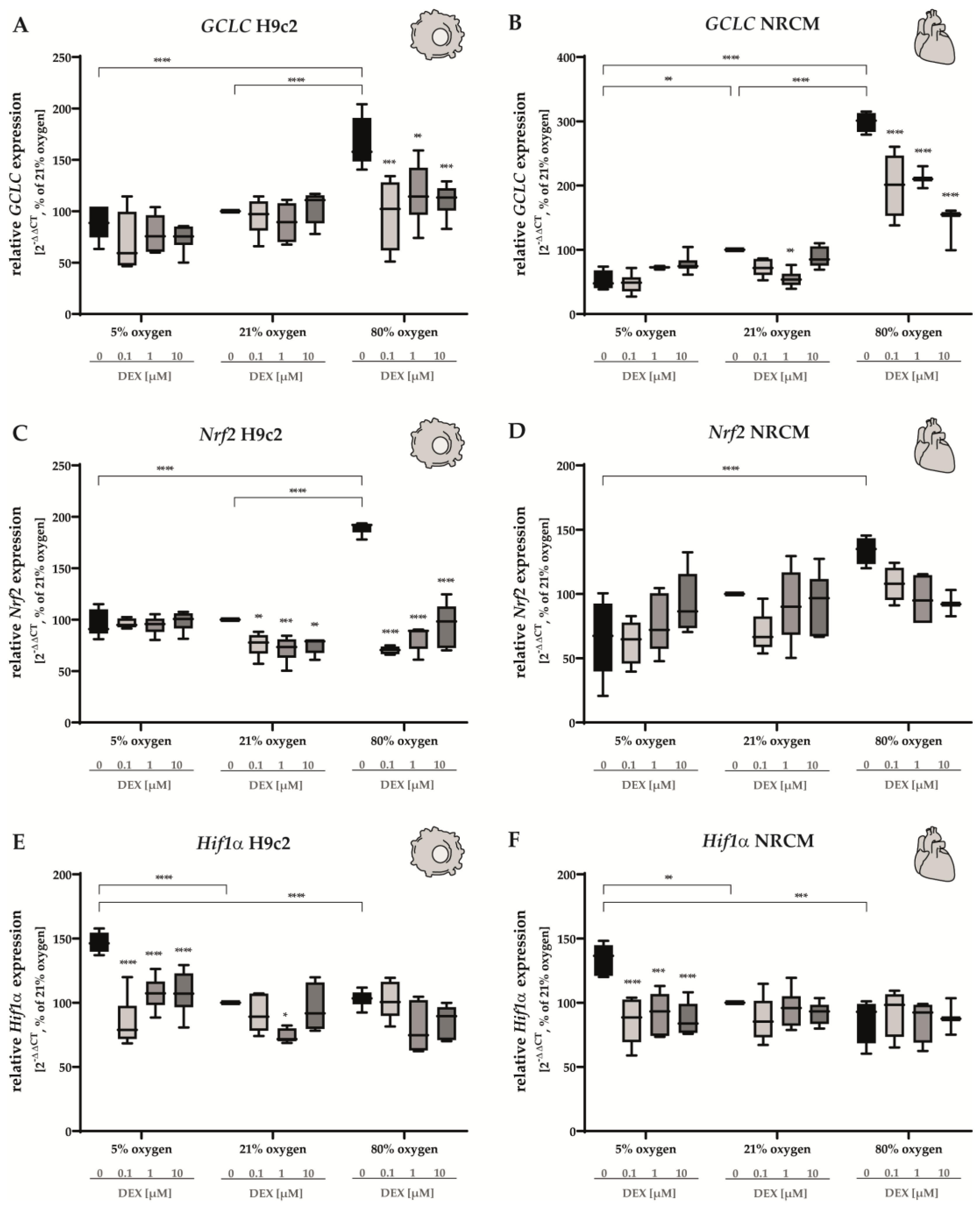
3.3. Oxidative Stress Response
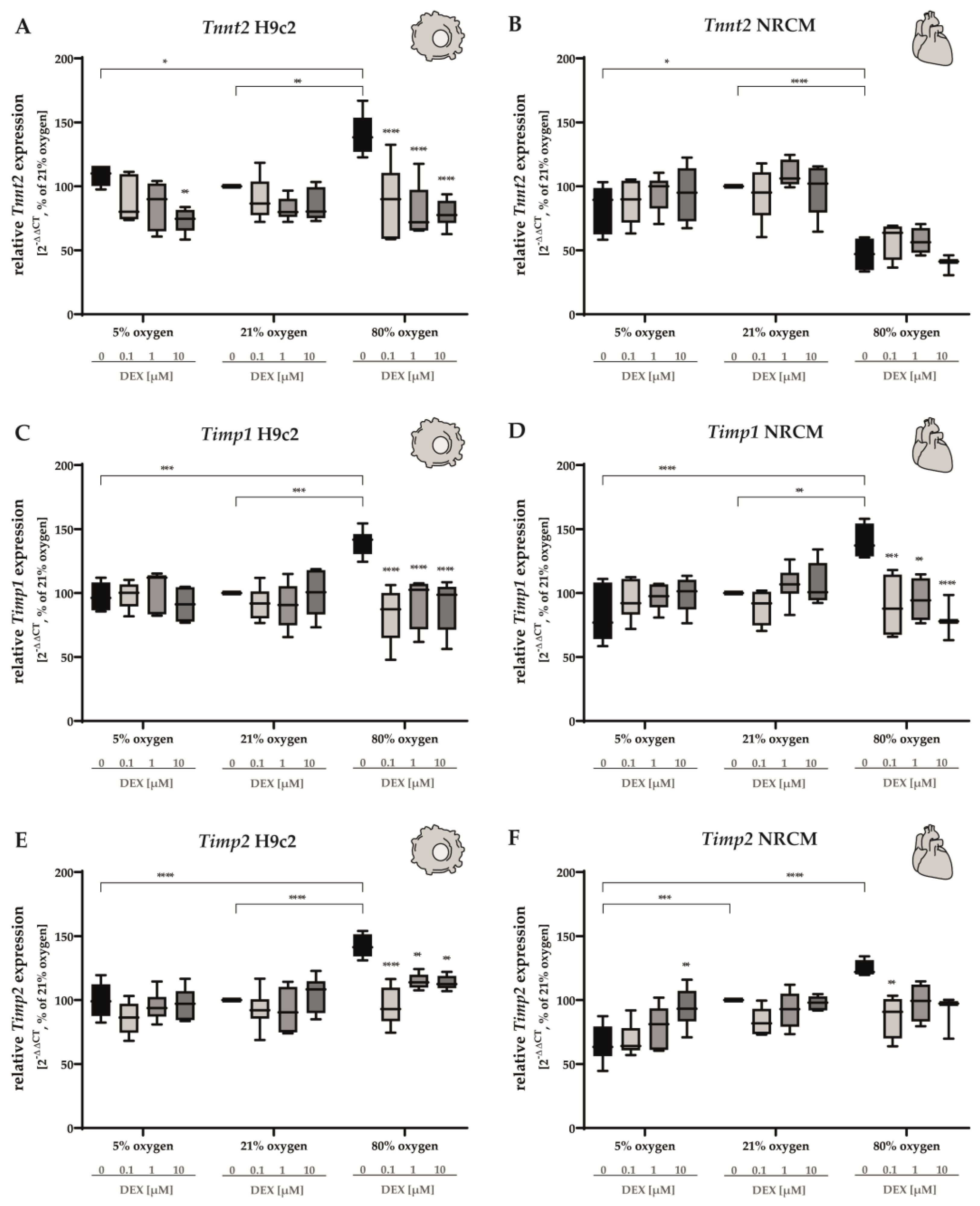
3.4. Cardiomyocyte Tissue Structure
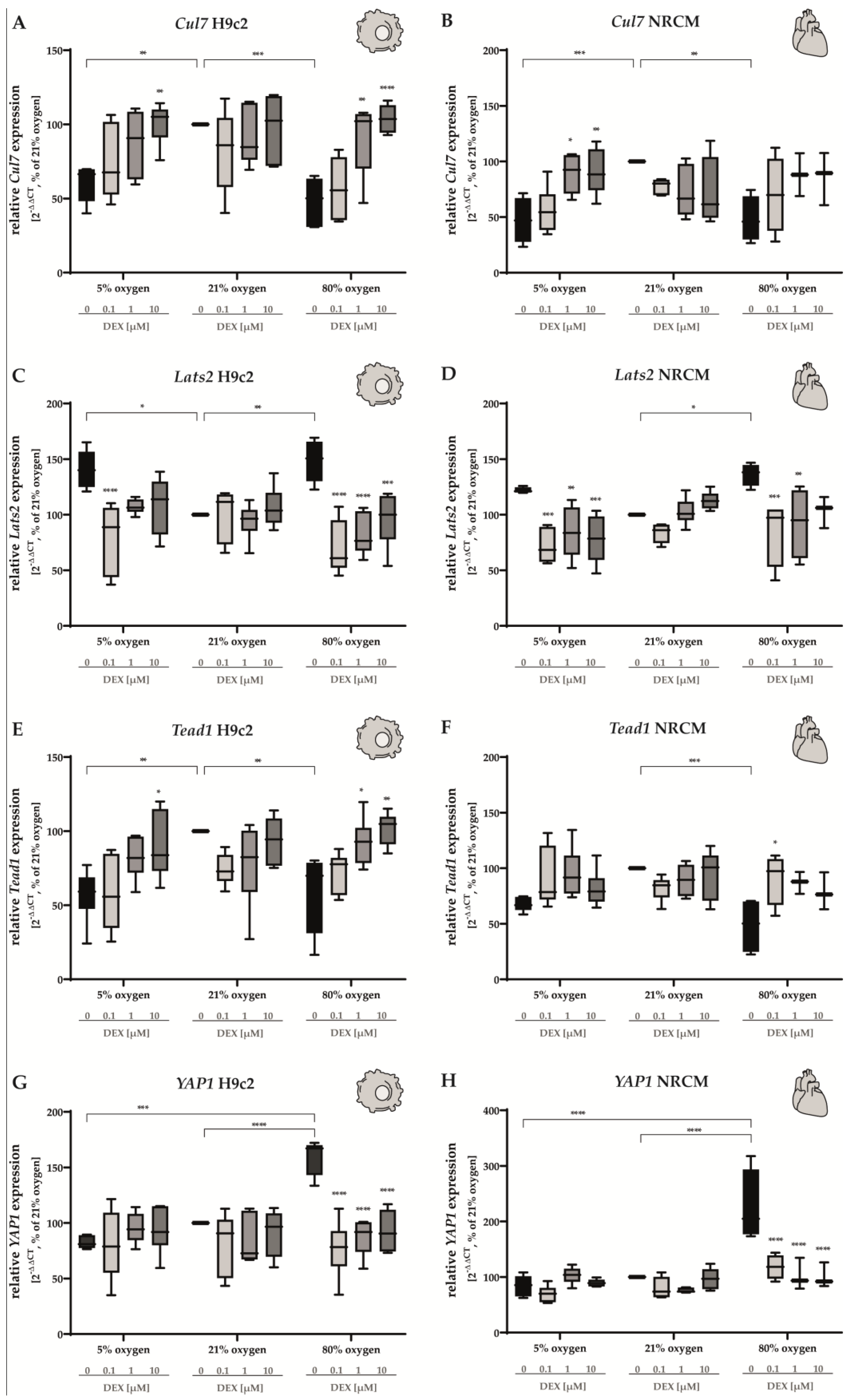
3.5. Hippo Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, G.; Liu, J.; Liu, M. Global, Regional, and National Incidence and Mortality of Neonatal Preterm Birth, 1990–2019. JAMA Pediatr. 2022, 176, 787–796. [Google Scholar] [CrossRef]
- Spoto, G.; Amore, G.; Vetri, L.; Quatrosi, G.; Cafeo, A.; Gitto, E.; Nicotera, A.G.; Di Rosa, G. Cerebellum and Prematurity: A Complex Interplay Between Disruptive and Dysmaturational Events. Front. Syst. Neurosci. 2021, 15, 655164. [Google Scholar] [CrossRef] [PubMed]
- Morsing, E.; Lundgren, P.; Hård, A.L.; Rakow, A.; Hellström-Westas, L.; Jacobson, L.; Johnson, M.; Nilsson, S.; Smith, L.E.H.; Sävman, K.; et al. Neurodevelopmental disorders and somatic diagnoses in a national cohort of children born before 24 weeks of gestation. Acta Paediatr. 2022, 111, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Pascal, A.; Govaert, P.; Oostra, A.; Naulaers, G.; Ortibus, E.; Van den Broeck, C. Neurodevelopmental outcome in very preterm and very-low-birthweight infants born over the past decade: A meta-analytic review. Dev. Med. Child Neurol. 2018, 60, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Laverty, C.; Surtees, A.; O’Sullivan, R.; Sutherland, D.; Jones, C.; Richards, C. The prevalence and profile of autism in individuals born preterm: A systematic review and meta-analysis. J. Neurodev. Disord. 2021, 13, 41. [Google Scholar] [CrossRef]
- Markopoulou, P.; Papanikolaou, E.; Analytis, A.; Zoumakis, E.; Siahanidou, T. Preterm Birth as a Risk Factor for Metabolic Syndrome and Cardiovascular Disease in Adult Life: A Systematic Review and Meta-Analysis. J. Pediatr. 2019, 210, 69–80.e5. [Google Scholar] [CrossRef]
- Norman, M. Preterm Birth—An Emerging Risk Factor for Adult Hypertension? Semin. Perinatol. 2010, 34, 183–187. [Google Scholar] [CrossRef]
- Hovi, P.; Andersson, S.; Eriksson, J.G.; Järvenpää, A.L.; Strang-Karlsson, S.; Mäkitie, O.; Kajantie, E. Glucose regulation in young adults with very low birth weight. N. Engl. J. Med. 2007, 356, 2053–2063. [Google Scholar] [CrossRef]
- Luu, T.M.; Rehman Mian, M.O.; Nuyt, A.M. Long-Term Impact of Preterm Birth: Neurodevelopmental and Physical Health Outcomes. Clin. Perinatol. 2017, 44, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Ryckman, K.K. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab. Syndr. Obes. Targets Ther. 2015, 8, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Risnes, K.; Bilsteen, J.F.; Brown, P.; Pulakka, A.; Andersen, A.N.; Opdahl, S.; Kajantie, E.; Sandin, S. Mortality Among Young Adults Born Preterm and Early Term in 4 Nordic Nations. JAMA Netw. Open 2021, 4, e2032779. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.H.S. Cardiovascular Morbidities in Adults Born Preterm: Getting to the Heart of the Matter! Children 2022, 9, 1843. [Google Scholar] [CrossRef]
- Vrselja, A.; Pillow, J.J.; Black, M.J. Effect of Preterm Birth on Cardiac and Cardiomyocyte Growth and the Consequences of Antenatal and Postnatal Glucocorticoid Treatment. J. Clin. Med. 2021, 10, 3896. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, A.J. Acute and chronic cardiac adaptations in adults born preterm. Exp. Physiol. 2022, 107, 405–409. [Google Scholar] [CrossRef]
- Chuai, Y.; Jiang, W.; Xu, X.; Wang, A.; Yao, Y.; Chen, L. Maternal oxygen exposure may not change umbilical cord venous partial pressure of oxygen: Non-random, paired venous and arterial samples from a randomised controlled trial. BMC Pregnancy Childbirth 2020, 20, 510. [Google Scholar] [CrossRef] [PubMed]
- Torres-Cuevas, I.; Parra-Llorca, A.; Sanchez-Illana, A.; Nunez-Ramiro, A.; Kuligowski, J.; Chafer-Pericas, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674–681. [Google Scholar] [CrossRef]
- Perez, M.; Robbins, M.E.; Revhaug, C.; Saugstad, O.D. Oxygen radical disease in the newborn, revisited: Oxidative stress and disease in the newborn period. Free Radic. Biol. Med. 2019, 142, 61–72. [Google Scholar] [CrossRef]
- Abdel Ghany, E.A.; Alsharany, W.; Ali, A.A.; Younass, E.R.; Hussein, J.S. Anti-oxidant profiles and markers of oxidative stress in preterm neonates. Paediatr. Int. Child Health 2015, 36, 134–140. [Google Scholar] [CrossRef]
- Perrone, S.; Santacroce, A.; Longini, M.; Proietti, F.; Bazzini, F.; Buonocore, G. The Free Radical Diseases of Prematurity: From Cellular Mechanisms to Bedside. Oxidative Med. Cell. Longev. 2018, 2018, 7483062. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. What is Oxidative Stress? In Oxidative Stress and Vascular Disease; Keaney, J.F., Ed.; Springer: Boston, MA, USA, 2000; pp. 1–8. [Google Scholar] [CrossRef]
- Alva, R.; Mirza, M.; Baiton, A.; Lazuran, L.; Samokysh, L.; Bobinski, A.; Cowan, C.; Jaimon, A.; Obioru, D.; Al Makhoul, T.; et al. Oxygen toxicity: Cellular mechanisms in normobaric hyperoxia. Cell Biol. Toxicol. 2023, 39, 111–143. [Google Scholar] [CrossRef]
- Bouayed, J.; Bohn, T. Exogenous antioxidants—Double-edged swords in cellular redox state: Health beneficial effects at physiologic doses versus deleterious effects at high doses. Oxidative Med. Cell. Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, T.A.; Ahmad, I.M.; Zimmerman, M.C. Oxidative Stress and Preterm Birth: An Integrative Review. Biol. Res. Nurs. 2018, 20, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Maroli, G.; Braun, T. The long and winding road of cardiomyocyte maturation. Cardiovasc. Res. 2021, 117, 712–726. [Google Scholar] [CrossRef]
- Ruan, J.L.; Tulloch, N.L.; Saiget, M.; Paige, S.L.; Razumova, M.V.; Regnier, M.; Tung, K.C.; Keller, G.; Pabon, L.; Reinecke, H.; et al. Mechanical Stress Promotes Maturation of Human Myocardium From Pluripotent Stem Cell-Derived Progenitors. Stem Cells 2015, 33, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Neary, M.T.; Ng, K.E.; Ludtmann, M.H.; Hall, A.R.; Piotrowska, I.; Ong, S.B.; Hausenloy, D.J.; Mohun, T.J.; Abramov, A.Y.; Breckenridge, R.A. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell. Cardiol. 2014, 74, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensley, J.G.; Moore, L.; De Matteo, R.; Harding, R.; Black, M.J. Impact of preterm birth on the developing myocardium of the neonate. Pediatr. Res. 2018, 83, 880–888. [Google Scholar] [CrossRef]
- Bensley, J.G.; Stacy, V.K.; De Matteo, R.; Harding, R.; Black, M.J. Cardiac remodelling as a result of pre-term birth: Implications for future cardiovascular disease. Eur. Heart J. 2010, 31, 2058–2066. [Google Scholar] [CrossRef]
- Bubb, K.J.; Cock, M.L.; Black, M.J.; Dodic, M.; Boon, W.M.; Parkington, H.C.; Harding, R.; Tare, M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J. Physiol. 2007, 578, 871–881. [Google Scholar] [CrossRef]
- O’Mara, K.; Gal, P.; Wimmer, J.; Ransom, J.L.; Carlos, R.Q.; Dimaguila, M.A.; Davanzo, C.C.; Smith, M. Dexmedetomidine versus standard therapy with fentanyl for sedation in mechanically ventilated premature neonates. J. Pediatr. Pharmacol. Ther. 2012, 17, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Chrysostomou, C.; Schulman, S.R.; Herrera Castellanos, M.; Cofer, B.E.; Mitra, S.; da Rocha, M.G.; Wisemandle, W.A.; Gramlich, L. A phase II/III, multicenter, safety, efficacy, and pharmacokinetic study of dexmedetomidine in preterm and term neonates. J. Pediatr. 2014, 164, 276–282.e3. [Google Scholar] [CrossRef] [Green Version]
- Dersch-Mills, D.A.; Banasch, H.L.; Yusuf, K.; Howlett, A. Dexmedetomidine Use in a Tertiary Care NICU: A Descriptive Study. Ann. Pharmacother. 2018, 53, 464–470. [Google Scholar] [CrossRef]
- Bao, N.; Tang, B. Organ-Protective Effects and the Underlying Mechanism of Dexmedetomidine. Mediat. Inflamm. 2020, 2020, 6136105. [Google Scholar] [CrossRef]
- Endesfelder, S.; Makki, H.; von Haefen, C.; Spies, C.D.; Buhrer, C.; Sifringer, M. Neuroprotective effects of dexmedetomidine against hyperoxia-induced injury in the developing rat brain. PLoS ONE 2017, 12, e0171498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kho, W.; von Haefen, C.; Paeschke, N.; Nasser, F.; Endesfelder, S.; Sifringer, M.; González-López, A.; Lanzke, N.; Spies, C.D. Dexmedetomidine Restores Autophagic Flux, Modulates Associated microRNAs and the Cholinergic Anti-inflammatory Pathway upon LPS-Treatment in Rats. J. Neuroimmune Pharmacol. 2022, 17, 261–276. [Google Scholar] [CrossRef]
- Paeschke, N.; von Haefen, C.; Endesfelder, S.; Sifringer, M.; Spies, C.D. Dexmedetomidine Prevents Lipopolysaccharide-Induced MicroRNA Expression in the Adult Rat Brain. Int. J. Mol. Sci. 2017, 18, 1830. [Google Scholar] [CrossRef] [Green Version]
- Sifringer, M.; von Haefen, C.; Krain, M.; Paeschke, N.; Bendix, I.; Buhrer, C.; Spies, C.D.; Endesfelder, S. Neuroprotective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain. Oxidative Med. Cell. Longev. 2015, 2015, 530371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.X.; Wang, T.H.; Wu, L.X.; Xue, F.S.; Zhang, G.H.; Yan, T. Role of Keap1-Nrf2/ARE signal transduction pathway in protection of dexmedetomidine preconditioning against myocardial ischemia/reperfusion injury. Biosci. Rep. 2022, 42, BSR20221306. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, J.; Ding, Y.; Chen, D.; Sun, H.; Yuan, F.; Li, S.; Li, X.; Yang, P.; Fu, L.; et al. Dexmedetomidine post-conditioning alleviates myocardial ischemia-reperfusion injury in rats by ferroptosis inhibition via SLC7A11/GPX4 axis activation. Hum. Cell 2022, 35, 836–848. [Google Scholar] [CrossRef]
- Yang, F.Y.; Zhang, L.; Zheng, Y.; Dong, H. Dexmedetomidine attenuates ischemia and reperfusion-induced cardiomyocyte injury through p53 and forkhead box O3a (FOXO3a)/p53-upregulated modulator of apoptosis (PUMA) signaling signaling. Bioengineered 2022, 13, 1377–1387. [Google Scholar] [CrossRef]
- Weng, X.; Shi, W.; Zhang, X.; Du, J. Dexmedetomidine attenuates H(2)O(2)-induced apoptosis of rat cardiomyocytes independently of antioxidant enzyme expression. Rev. Port. Cardiol. (Engl. Ed.) 2021, 40, 273–281. [Google Scholar] [CrossRef]
- Martinez, C.A.; Cistulli, P.A.; Cook, K.M. A Cell Culture Model that Mimics Physiological Tissue Oxygenation Using Oxygen-permeable Membranes. Bio-Protocol 2019, 9, e3371. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, G.; Sonesson, S.E.; Flo, K.; Räsänen, J.; Odibo, A. Hemodynamic aspects of normal human feto-placental (umbilical) circulation. Acta Obstet. Gynecol. Scand. 2016, 95, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leviton, A.; Allred, E.; Kuban, K.C.; Dammann, O.; O’Shea, T.M.; Hirtz, D.; Schreiber, M.D.; Paneth, N. Early blood gas abnormalities and the preterm brain. Am. J. Epidemiol. 2010, 172, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Heise, J.; Schmitz, T.; Bührer, C.; Endesfelder, S. Protective Effects of Early Caffeine Administration in Hyperoxia-Induced Neurotoxicity in the Juvenile Rat. Antioxidants 2023, 12, 295. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Novo, N.; Ferreira, P.; Medina, M. The apoptosis-inducing factor family: Moonlighting proteins in the crosstalk between mitochondria and nuclei. IUBMB Life 2021, 73, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, O. Autophagy in the Heart. Circ. J. 2019, 83, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Forman, H.J. Glutathione synthesis and its role in redox signaling. Semin. Cell Dev. Biol. 2012, 23, 722–728. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Streng, A.S.; Jacobs, L.H.; Schwenk, R.W.; Cardinaels, E.P.; Meex, S.J.; Glatz, J.F.; Wodzig, W.K.; van Dieijen-Visser, M.P. Cardiac troponin in ischemic cardiomyocytes: Intracellular decrease before onset of cell death. Exp. Mol. Pathol. 2014, 96, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavò, L.; Perrone, S.; Viola, V.; Marseglia, L.; Di Rosa, G.; Gitto, E. Oxidative Stress and Respiratory Diseases in Preterm Newborns. Int. J. Mol. Sci. 2021, 22, 12504. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.L.; Ang, X.; Yap, K.Y.; Ng, J.J.; Goh, E.C.H.; Khoo, B.B.J.; Richards, A.M.; Drum, C.L. Novel Oxidative Stress Biomarkers with Risk Prognosis Values in Heart Failure. Biomedicines 2023, 11, 917. [Google Scholar] [CrossRef]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [Green Version]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Tong, W.; Xiong, F.; Li, Y.; Zhang, L. Hypoxia inhibits cardiomyocyte proliferation in fetal rat hearts via upregulating TIMP-4. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R613–R620. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Xue, Q.; Li, Y.; Zhang, L. Maternal hypoxia alters matrix metalloproteinase expression patterns and causes cardiac remodeling in fetal and neonatal rats. Am. J. Physiology. Heart Circ. Physiol. 2011, 301, H2113–H2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teringova, E.; Tousek, P. Apoptosis in ischemic heart disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [Green Version]
- Moe, G.W.; Marín-García, J. Role of cell death in the progression of heart failure. Heart Fail. Rev. 2016, 21, 157–167. [Google Scholar] [CrossRef]
- Bai, Y.T.; Xiao, F.J.; Wang, H.; Ge, R.L.; Wang, L.S. Hypoxia protects H9c2 cells against Ferroptosis through SENP1-mediated protein DeSUMOylation. Int. J. Med. Sci. 2021, 18, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Finkel, T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic. Biol. Med. 2017, 109, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, X.; Yang, T.; Wang, L.; Feng, L.; Wang, Z.; Xu, J.; Jing, W.; Wang, T.; Su, H.; et al. The role of autophagy in cardiovascular disease: Cross-interference of signaling pathways and underlying therapeutic targets. Front. Cardiovasc. Med. 2023, 10, 1088575. [Google Scholar] [CrossRef]
- Manu, T.M.; Anand, T.; Pandareesh, M.D.; Kumar, P.B.; Khanum, F. Terminalia arjuna extract and arjunic acid mitigate cobalt chloride-induced hypoxia stress-mediated apoptosis in H9c2 cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 1107–1119. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, D.; Hu, H.; Zhang, P.; Xie, R.; Cui, W. HIF-1α/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef] [PubMed]
- Puls, R.; von Haefen, C.; Bührer, C.; Endesfelder, S. Dexmedetomidine Protects Cerebellar Neurons against Hyperoxia-Induced Oxidative Stress and Apoptosis in the Juvenile Rat. Int. J. Mol. Sci. 2023, 24, 7804. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, H.; Zhang, H.; Sui, Y.; Zhang, Z.; Zou, Y.; Li, K.; Zhao, Y.; Xie, J.; Zhang, L. The neuroprotective effect of dexmedetomidine and its mechanism. Front. Pharmacol. 2022, 13, 965661. [Google Scholar] [CrossRef]
- Puls, R.; von Haefen, C.; Bührer, C.; Endesfelder, S. Protective Effect of Dexmedetomidine against Hyperoxia-Damaged Cerebral Neurodevelopment in the Juvenile Rat. Antioxidants 2023, 12, 980. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, J.; Fang, J.; Li, X.; Huang, C.; Liang, W.; Wu, K. Naringin protects H9C2 cardiomyocytes from chemical hypoxia-induced injury by promoting the autophagic flux via the activation of the HIF-1α/BNIP3 signaling pathway. Int. J. Mol. Med. 2021, 47, 102. [Google Scholar] [CrossRef]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Chen, Q.M. Nrf2 for cardiac protection: Pharmacological options against oxidative stress. Trends Pharmacol. Sci. 2021, 42, 729–744. [Google Scholar] [CrossRef]
- Lu, Y.; An, L.; Taylor, M.R.G.; Chen, Q.M. Nrf2 signaling in heart failure: Expression of Nrf2, Keap1, antioxidant, and detoxification genes in dilated or ischemic cardiomyopathy. Physiol. Genom. 2022, 54, 115–127. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Cao, X.; Wang, W.; Cui, X.; Yang, X.; Wang, Y.; Shi, J. Nrf2 Promotes Inflammation in Early Myocardial Ischemia-Reperfusion via Recruitment and Activation of Macrophages. Front. Immunol. 2021, 12, 763760. [Google Scholar] [CrossRef]
- Vashi, R.; Patel, B.M. NRF2 in Cardiovascular Diseases: A Ray of Hope! J. Cardiovasc. Transl. Res. 2021, 14, 573–586. [Google Scholar] [CrossRef]
- Ott, C.; Jung, T.; Brix, S.; John, C.; Betz, I.R.; Foryst-Ludwig, A.; Deubel, S.; Kuebler, W.M.; Grune, T.; Kintscher, U.; et al. Hypertrophy-Reduced Autophagy Causes Cardiac Dysfunction by Directly Impacting Cardiomyocyte Contractility. Cells 2021, 10, 805. [Google Scholar] [CrossRef]
- Li, B.; Zhan, Y.; Liang, Q.; Xu, C.; Zhou, X.; Cai, H.; Zheng, Y.; Guo, Y.; Wang, L.; Qiu, W.; et al. Isogenic human pluripotent stem cell disease models reveal ABRA deficiency underlies cTnT mutation-induced familial dilated cardiomyopathy. Protein Cell 2022, 13, 65–71. [Google Scholar] [CrossRef]
- Ramani, M.; Bradley, W.E.; Dell’Italia, L.J.; Ambalavanan, N. Early exposure to hyperoxia or hypoxia adversely impacts cardiopulmonary development. Am. J. Respir. Cell Mol. Biol. 2015, 52, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Shivanna, B. Long-term pulmonary and cardiovascular morbidities of neonatal hyperoxia exposure in mice. Int. J. Biochem. Cell Biol. 2018, 94, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Panguluri, S.K.; Tur, J.; Fukumoto, J.; Deng, W.; Sneed, K.B.; Kolliputi, N.; Bennett, E.S.; Tipparaju, S.M. Hyperoxia-induced hypertrophy and ion channel remodeling in left ventricle. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1651–H1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapalamadugu, K.C.; Panguluri, S.K.; Bennett, E.S.; Kolliputi, N.; Tipparaju, S.M. High level of oxygen treatment causes cardiotoxicity with arrhythmias and redox modulation. Toxicol. Appl. Pharmacol. 2015, 282, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.D.; Yee, M.; Porter, G.A., Jr.; Ritzer, E.; McDavid, A.N.; Brookes, P.S.; Pryhuber, G.S.; O’Reilly, M.A. Neonatal hyperoxia inhibits proliferation and survival of atrial cardiomyocytes by suppressing fatty acid synthesis. JCI Insight 2021, 6, e140785. [Google Scholar] [CrossRef]
- Hafner, C.; Wu, J.; Tiboldi, A.; Hess, M.; Mitulovic, G.; Kaun, C.; Krychtiuk, K.A.; Wojta, J.; Ullrich, R.; Tretter, E.V.; et al. Hyperoxia Induces Inflammation and Cytotoxicity in Human Adult Cardiac Myocytes. Shock 2017, 47, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 2019, 7, 211. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhao, M. How Hippo Signaling Pathway Modulates Cardiovascular Development and Diseases. J. Immunol. Res. 2018, 2018, 3696914. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Wu, W.; Lin, X.; Shen, M.; Yang, Z.; Yu, S.; Luo, Y. Protective effects of dexmedetomidine in vital organ injury: Crucial roles of autophagy. Cell. Mol. Biol. Lett. 2022, 27, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Ma, W.; Li, J.; Littlejohn, R.; Zhou, H.; Kim, I.M.; Fulton, D.J.R.; Chen, W.; Weintraub, N.L.; Zhou, J.; et al. Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4101–E4110. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.-P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Zhang, M.; Hu, J.; Lin, J.; Feng, X.; Wang, S.; Wang, T.; Gao, E.; Wang, H.; Sun, D. Mst1 knockout enhances cardiomyocyte autophagic flux to alleviate angiotensin II-induced cardiac injury independent of angiotensin II receptors. J. Mol. Cell. Cardiol. 2018, 125, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Hassink, R.J.; Nakajima, H.; Nakajima, H.O.; Doevendans, P.A.; Field, L.J. Expression of a transgene encoding mutant p193/CUL7 preserves cardiac function and limits infarct expansion after myocardial infarction. Heart 2009, 95, 1159–1164. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Anger, M.; Scheufele, F.; Ramanujam, D.; Meyer, K.; Nakajima, H.; Field, L.J.; Engelhardt, S.; Sarikas, A. Genetic ablation of Cullin-RING E3 ubiquitin ligase 7 restrains pressure overload-induced myocardial fibrosis. PLoS ONE 2020, 15, e0244096. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, C.; Hong, H.; Liu, J.; Qiu, L.; Huang, Y.; Ye, L. Effects of hypoxia on cardiomyocyte proliferation and association with stage of development. Biomed. Pharmacother. 2019, 118, 109391. [Google Scholar] [CrossRef]
- von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onódi, Z.; Visnovitz, T.; Kiss, B.; Hambalkó, S.; Koncz, A.; Ágg, B.; Váradi, B.; Tóth, V.; Nagy, R.N.; Gergely, T.G.; et al. Systematic transcriptomic and phenotypic characterization of human and murine cardiac myocyte cell lines and primary cardiomyocytes reveals serious limitations and low resemblances to adult cardiac phenotype. J. Mol. Cell. Cardiol. 2022, 165, 19–30. [Google Scholar] [CrossRef]
- Bon-Mathier, A.-C.; Rignault-Clerc, S.; Bielmann, C.; Rosenblatt-Velin, N. Oxygen as a key regulator of cardiomyocyte proliferation: New results about cell culture conditions! Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118460. [Google Scholar] [CrossRef] [PubMed]
- Lembo, C.; Buonocore, G.; Perrone, S. Oxidative Stress in Preterm Newborns. Antioxidants 2021, 10, 1672. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide Sequence 5′−3′ | Accession No. | |
|---|---|---|
| AIF | ||
| forward | CACAAAGACACTGCAGTTCAGACA | NM_031356.1 |
| reverse | AGGTCCTGAGCAGAGACATAGAAAG | |
| probe | 6-FAM-AGAAGCATCTATTTCCAGCC-TAMRA | |
| Atg5 | ||
| forward | ACATCAGCATTGTGCCCCA | NM_001014250.1 |
| reverse | TGTCATGCTTCGGTGTCCTG | |
| probe | 6-FAM-CAGACTGAAGGCCGTGTCCTGCTCA-TAMRA | |
| Atg12 | ||
| forward | TCTGCCTAGCCTGGAACTCAG | NM_001038495.1 |
| reverse | TAGCCCTGTGTGCTCTGCTTT | |
| probe | 6-FAM-CCTGTCCGTGAAGCTCACCCAGC-TAMRA | |
| Casp3 | ||
| forward | ACAGTGGAACTGACGATGATATGG | NM_012922.2 |
| reverse | AATAGTAACCGGGTGCGGTAGA | |
| probe | 6-FAM-ATGCCAGAAGATACCAGTGG-TAMRA | |
| Casp8 | ||
| forward | GGACTATCCTGGCAGAAAAC | NM_022277.1 |
| reverse | TCACCTCATCCAAAACAGAAAC | |
| probe | 6-FAM-AGGATCGACGATTACGAACGATCAAGCACA-TAMRA | |
| Cul7 | ||
| forward | GATCCTTCTGTCACTGAGCC | XM_032900620.1 |
| reverse | TCCCAGCATTCAACTCCTCC | |
| probe | 6-FAM-CCGCTGCGCCCTGCTTGCACT-TAMRA | |
| CycD2 | ||
| forward | CGTACATGCGCAGGATGGT | NM_199501.1 |
| reverse | AATTCATGGCCAGAGGAAAGAC | |
| probe | 6-FAM-TGGATGCTAGAGGTCTGTGA-TAMRA | |
| GCLC | ||
| forward | GGAGGACAACATGAGGAAACG | NM_012815.2 |
| reverse | GCTCTGGCAGTGTGAATCCA | |
| probe | 6-FAM-TCAGGCTCTTTGCACGATAA-TAMRA | |
| Hif1α | ||
| forward | GCGCCTCTTCGACAAGCTT | NM_024359.2 |
| reverse | CTGCCGAAGTCCAGTGATATGA | |
| probe | 6-FAM-AGAGCCCGATGCCCTGACTCTGCT-TAMRA | |
| Lats2 | ||
| forward | ACTACCAGAAAGGGAACCAC | NM_001107267.1 |
| reverse | CAAAAAGAATCACACCGACAC | |
| probe | 6-FAM-CTGGTGACCTCTGGGACGACGTTTCCAA-TAMRA | |
| Nrf2 | ||
| forward | ACTCCCAGGTTGCCCACAT | NM_031789.2 |
| reverse | GCGACTCATGGTCATCTACAAATG | |
| probe | 6-FAM-CTTTGAAGACTGTATGCAGC-TAMRA | |
| TATAbp | ||
| forward | CACCGTGAATCTTGGCTGTAAAC | NM_001004198 |
| reverse | CGCAGTTGTTCGTGGCTCTC | |
| probe | 6-FAM-TCGTGCCAGAAATGCTGAATATAATCCCAA-TAMRA | |
| Tead1 | ||
| forward | AAACTGAGGACGGGAAAGAC | NM_001198589.2 |
| reverse | AGACGATCTGGGCTGATGAC | |
| probe | 6-FAM-ACAAGCATGGATCAGACTGCCAAGGACAA-TAMRA | |
| Timp1 | ||
| forward | CGGACCTGGTTATAAGGGCTAA | NM_053819.1 |
| reverse | CGTCGAATCCTTTGAGCATCT | |
| probe | 6-FAM-AGAAATCATCGAGACCACCT-TAMRA | |
| Timp2 | ||
| forward | GGCAACCCCATCAAGAGGAT | NM_021989.2 |
| reverse | GGGCCGTGTAGATAAATTCGAT | |
| probe | 6-FAM-AGATGTTCAAAGGACCTGAC-TAMRA | |
| Tnnt2 | ||
| forward | GCGAAGAAGAGGAAGACGAG | NM_012676.1 |
| reverse | CACCAAGTTGGGCATGAAG | |
| probe | 6-FAM-CAGTAGAGGACTCCAAACCCAAGCCCAGCA-TAMRA | |
| YAP1 | ||
| forward | TGCTGCTCAACATCTCAGAC | NM_001394328.1 |
| reverse | TGCTCCCATCCATCAGGAAG | |
| probe | 6-FAM-CCCGGAAGGCCATGCTCTCCCAACTGAA-TAMRA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borger, M.; von Haefen, C.; Bührer, C.; Endesfelder, S. Cardioprotective Effects of Dexmedetomidine in an Oxidative-Stress In Vitro Model of Neonatal Rat Cardiomyocytes. Antioxidants 2023, 12, 1206. https://doi.org/10.3390/antiox12061206
Borger M, von Haefen C, Bührer C, Endesfelder S. Cardioprotective Effects of Dexmedetomidine in an Oxidative-Stress In Vitro Model of Neonatal Rat Cardiomyocytes. Antioxidants. 2023; 12(6):1206. https://doi.org/10.3390/antiox12061206
Chicago/Turabian StyleBorger, Moritz, Clarissa von Haefen, Christoph Bührer, and Stefanie Endesfelder. 2023. "Cardioprotective Effects of Dexmedetomidine in an Oxidative-Stress In Vitro Model of Neonatal Rat Cardiomyocytes" Antioxidants 12, no. 6: 1206. https://doi.org/10.3390/antiox12061206