
Different Effects of SSRIs, Bupropion, and Trazodone on Mitochondrial Functions and Monoamine Oxidase Isoform Activity

Abstract
:1. Introduction
2. Materials and Methods
2.1. Media and Chemicals
2.2. Isolation of Pig Brain Mitochondria
2.3. Mitochondrial Enzymes Activities
2.3.1. Activity of Citrate Synthase
2.3.2. Activity of Malate Dehydrogenase
2.3.3. Activity of Complex I (NADH Dehydrogenase)
2.3.4. Activity of Complex II+III (Succinate Cytochrome c Oxidoreductase)
2.3.5. Activity of Complex IV (Cytochrome c Oxidase)
2.4. ATP Content and Kinetics
2.4.1. Total Complex I- and Complex II-Linked ATP Content
2.4.2. Complex I- and Complex II-Linked ATP Kinetics
2.5. Mitochondrial Respiration
2.6. Activity of Monoamine Oxidase
2.7. Data Analysis and Statistics
3. Results
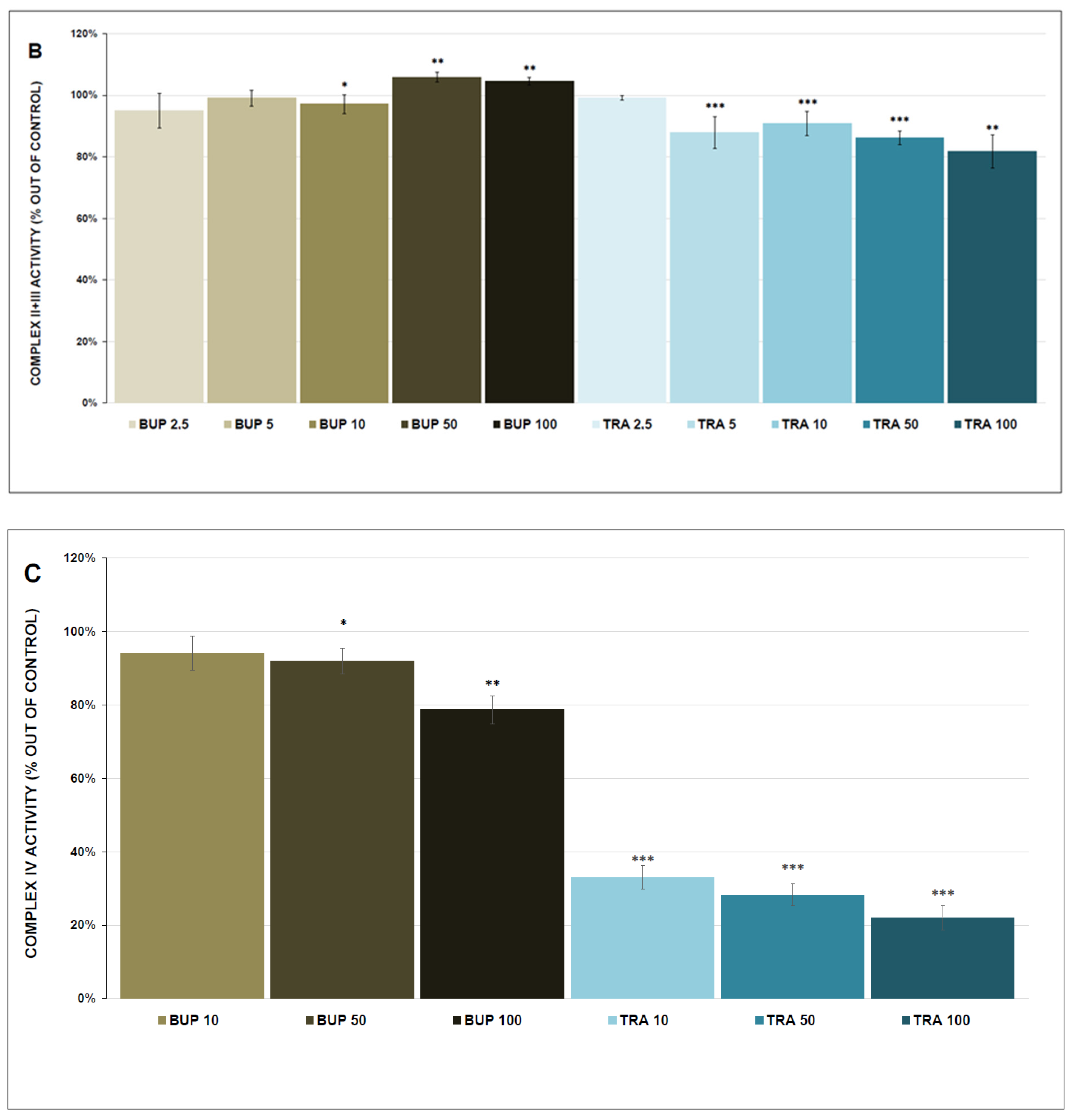
3.1. Activity of Mitochondrial Enzymes
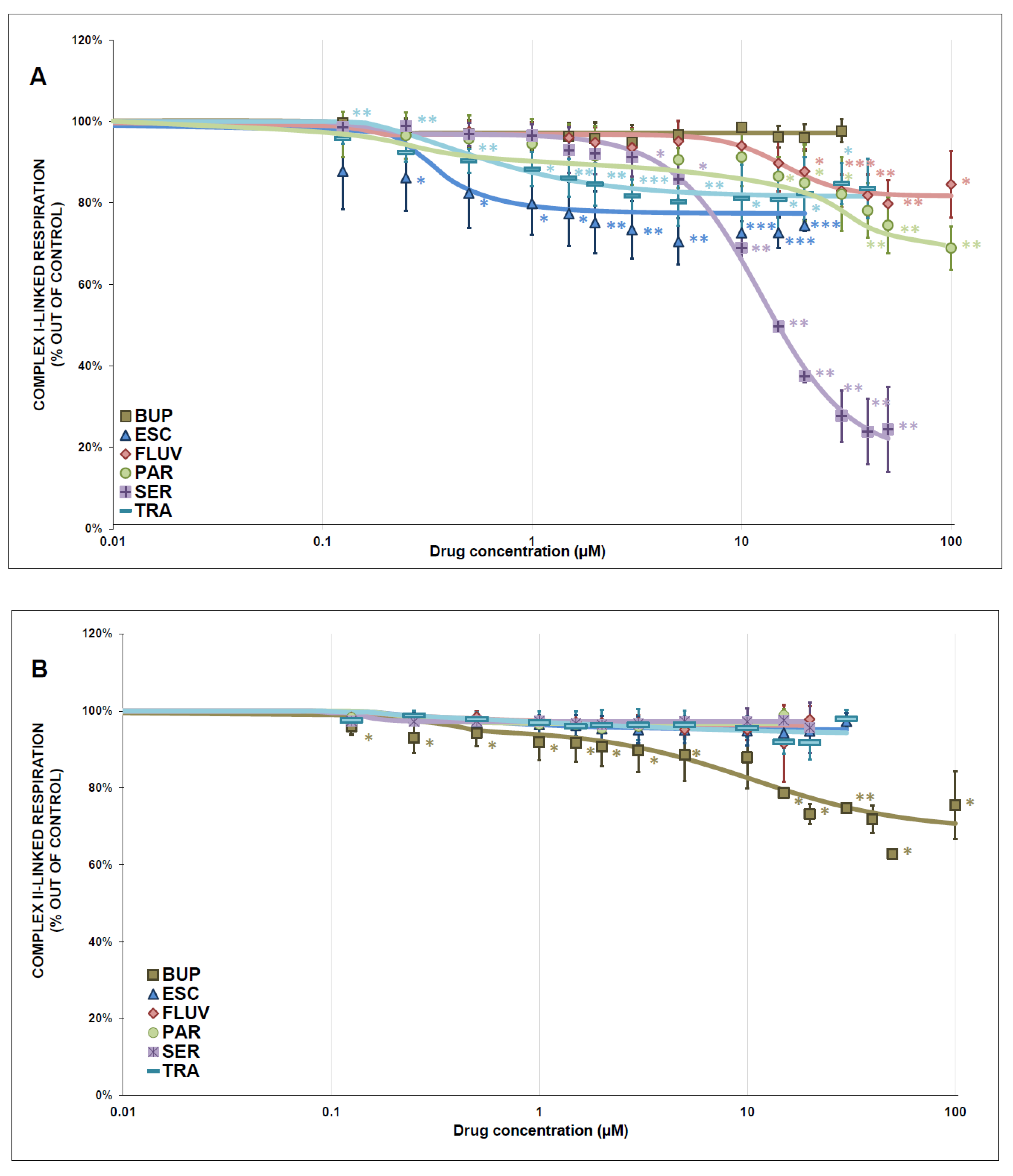
3.2. Mitochondrial Respiration
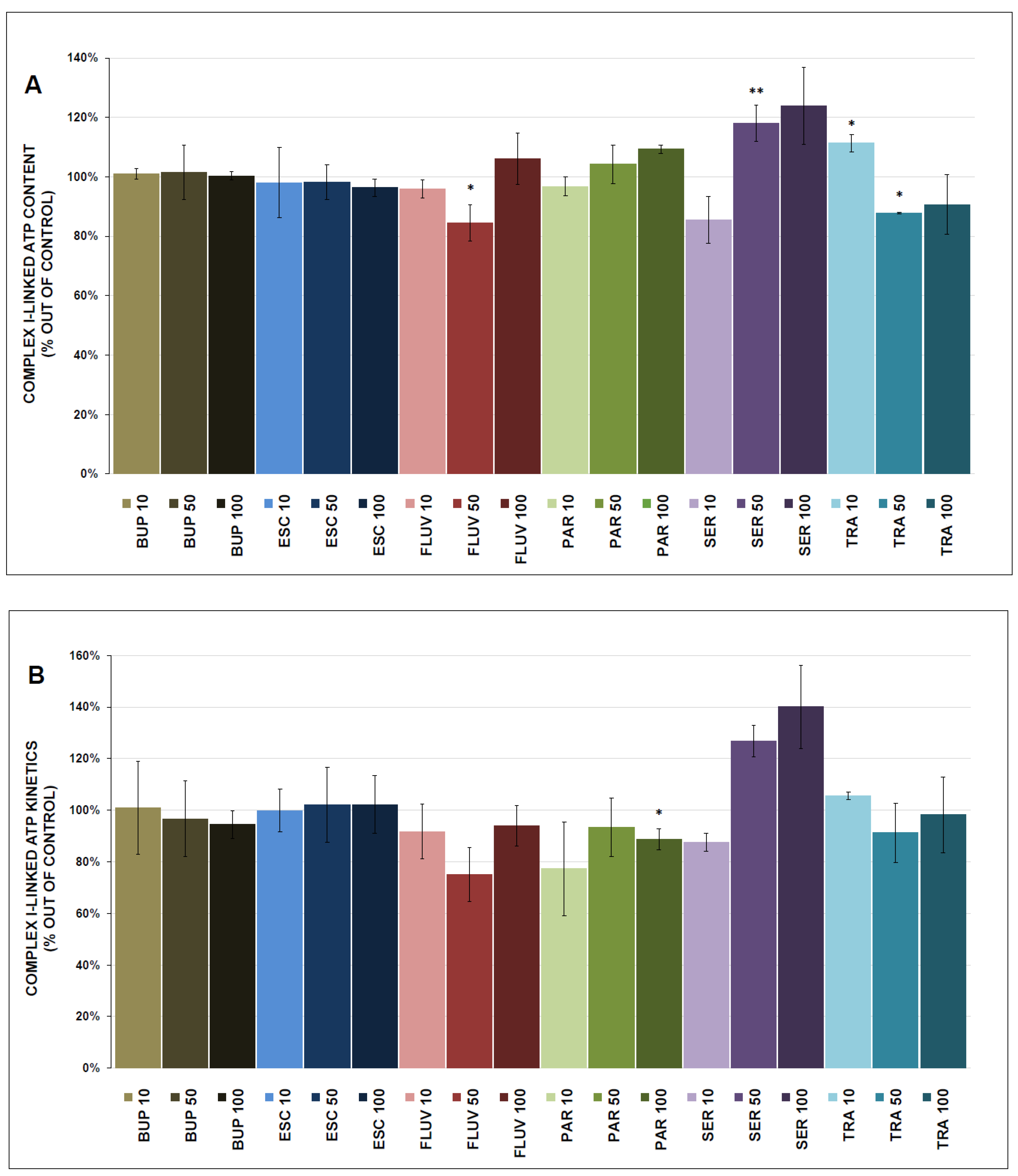
3.3. ATP Content and Kinetics
3.4. MAO Activity
3.5. Correlations
4. Discussion
4.1. Mitochondrial Enzyme Activity and Respiration
4.2. ATP Production
4.3. MAO Inhibition
4.4. Correlations
4.5. Study Limitations
4.6. Possible Clinical Impact
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BUP | bupropion |
| CS | citrate synthase |
| ESC | escitalopram |
| ETC | electron transport chain |
| FLUV | fluvoxamine |
| IC50 | half-maximal inhibitory concentration |
| MAOI | monoamine oxidase inhibitor |
| MAO | monoamine oxidase |
| MDD | major depressive disorder |
| MDH | malate dehydrogenase |
| NADH | Reduced nicotinamide adenine dinucleotide |
| OXPHOS | oxidative phosphorylation |
| PAR | paroxetine |
| ROS | reactive oxygen species |
| SD | standard deviation |
| SEM | standard error of the mean |
| SER | sertraline |
| SSRI | selective serotonin reuptake inhibitor |
| TRA | trazodone |
References
- Caruso, G.; Benatti, C.; Blom, J.M.C.; Caraci, F.; Tascedda, F. The Many Faces of Mitochondrial Dysfunction in Depression: From Pathology to Treatment. Front. Pharmacol. 2019, 10, 995. [Google Scholar] [CrossRef] [Green Version]
- Bansal, Y.; Kuhad, A. Mitochondrial Dysfunction in Depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Ľupták, M.; Hroudová, J. Important role of mitochondria and the effect of mood stabilizers on mitochondrial function. Physiol. Res. 2019, 68 (Suppl. 1), S3–S15. [Google Scholar] [CrossRef]
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef]
- Czarny, P.; Wigner, P.; Galecki, P.; Sliwinski, T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 80 Pt C, 309–321. [Google Scholar] [CrossRef]
- Cohen, G.; Kesler, N. Monoamine oxidase and mitochondrial respiration. J. Neurochem. 1999, 73, 2310–2315. [Google Scholar] [CrossRef] [Green Version]
- Gamaro, G.D.; Streck, E.L.; Matté, C.; Prediger, M.E.; Wyse, A.T.; Dalmaz, C. Reduction of hippocampal Na+, K+-ATPase activity in rats subjected to an experimental model of depression. Neurochem. Res. 2003, 28, 1339–1344. [Google Scholar] [CrossRef]
- Gong, Y.; Chai, Y.; Ding, J.H.; Sun, X.L.; Hu, G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 2011, 488, 76–80. [Google Scholar] [CrossRef]
- Rappeneau, V.; Wilmes, L.; Touma, C. Molecular correlates of mitochondrial dysfunctions in major depression: Evidence from clinical and rodent studies. Mol. Cell. Neurosci. 2020, 109, 103555. [Google Scholar] [CrossRef]
- Videbech, P. PET measurements of brain glucose metabolism and blood flow in major depressive disorder: A critical review. Acta Psychiatr. Scand. 2000, 101, 11–20. [Google Scholar] [CrossRef]
- Mayberg, H.S.; Lewis, P.J.; Regenold, W.; Wagner, H.N., Jr. Paralimbic hypoperfusion in unipolar depression. J. Nucl. Med. 1994, 35, 929–934. [Google Scholar]
- Newton, S.S.; Thome, J.; Wallace, T.L.; Shirayama, Y.; Schlesinger, L.; Sakai, N.; Chen, J.; Neve, R.; Nestler, E.J.; Duman, R.S. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J. Neurosci. 2002, 22, 10883–10890. [Google Scholar] [CrossRef] [Green Version]
- Baxter, L.R., Jr.; Schwartz, J.M.; Phelps, M.E.; Mazziotta, J.C.; Guze, B.H.; Selin, C.E.; Gerner, R.H.; Sumida, R.M. Reduction of prefrontal cortex glucose metabolism common to three types of depression. Arch. Gen. Psychiatry 1989, 46, 243–250. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Karry, R. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS ONE 2008, 3, e3676. [Google Scholar] [CrossRef] [Green Version]
- Moretti, A.; Gorini, A.; Villa, R.F. Affective disorders, antidepressant drugs and brain metabolism. Mol. Psychiatry 2003, 8, 773–785. [Google Scholar] [CrossRef]
- Hroudová, J.; Fišar, Z.; Kitzlerová, E.; Zvěřová, M.; Raboch, J. Mitochondrial respiration in blood platelets of depressive patients. Mitochondrion 2013, 13, 795–800. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Böck, C.; Salinas-Manrique, J.; Kolassa, S.; Calzia, E.; Dietrich, D.E.; Kolassa, I.T. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl. Psychiatry 2014, 4, e397. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef]
- Trumpff, C.; Marsland, A.L.; Basualto-Alarcón, C.; Martin, J.L.; Carroll, J.E.; Sturm, G.; Vincent, A.E.; Mosharov, E.V.; Gu, Z.; Kaufman, B.A.; et al. Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology 2019, 106, 268–276. [Google Scholar] [CrossRef]
- Emmerzaal, T.L.; Nijkamp, G.; Veldic, M.; Rahman, S.; Andreazza, A.C.; Morava, E.; Rodenburg, R.J.; Kozicz, T. Effect of neuropsychiatric medications on mitochondrial function: For better or for worse. Neurosci. Biobehav. Rev. 2021, 127, 555–571. [Google Scholar] [CrossRef]
- Fernström, J.; Mellon, S.H.; McGill, M.A.; Picard, M.; Reus, V.I.; Hough, C.M.; Lin, J.; Epel, E.S.; Wolkowitz, O.M.; Lindqvist, D. Blood-based mitochondrial respiratory chain function in major depression. Transl. Psychiatry 2021, 11, 593. [Google Scholar] [CrossRef]
- Edmondson, D.E. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr. Pharm. Des. 2014, 20, 155–160. [Google Scholar] [CrossRef]
- Fišar, Z. Drugs related to monoamine oxidase activity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 69, 112–124. [Google Scholar] [CrossRef]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase a levels in the brain: An explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.A.; Schrag, A. Impact of newer pharmacological treatments on quality of life in patients with Parkinson’s disease. CNS Drugs 2008, 22, 563–586. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z.; Ghosh, K.K. Progress in drug development for Alzheimer’s disease: An overview in relation to mitochondrial energy metabolism. Eur. J. Med. Chem. 2016, 121, 774–784. [Google Scholar] [CrossRef]
- Bhawna; Kumar, A.; Bhatia, M.; Kapoor, A.; Kumar, P.; Kumar, S. Monoamine oxidase inhibitors: A concise review with special emphasis on structure activity relationship studies. Eur. J. Med. Chem. 2022, 242, 114655. [Google Scholar] [CrossRef]
- Manzoor, S.; Hoda, N. A comprehensive review of monoamine oxidase inhibitors as Anti-Alzheimer’s disease agents: A review. Eur. J. Med. Chem. 2020, 206, 112787. [Google Scholar] [CrossRef]
- Bodkin, J.A.; Dunlop, B.W. Moving on With Monoamine Oxidase Inhibitors. Focus (Am. Psychiatr. Publ.) 2021, 19, 50–52. [Google Scholar] [CrossRef]
- Mandrioli, R.; Mercolini, L.; Saracino, M.A.; Raggi, M.A. Selective serotonin reuptake inhibitors (SSRIs): Therapeutic drug monitoring and pharmacological interactions. Curr. Med. Chem. 2012, 19, 1846–1863. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P.; McCormack, P.L. Escitalopram: A review of its use in the management of major depressive disorder in adults. CNS Drugs 2010, 24, 769–796. [Google Scholar] [CrossRef]
- Protti, M.; Mandrioli, R.; Marasca, C.; Cavalli, A.; Serretti, A.; Mercolini, L. New-generation, non-SSRI antidepressants: Drug-drug interactions and therapeutic drug monitoring. Part 2: NaSSAs, NRIs, SNDRIs, MASSAs, NDRIs, and others. Med. Res. Rev. 2020, 40, 1794–1832. [Google Scholar] [CrossRef]
- Khouzam, H.R. A review of trazodone use in psychiatric and medical conditions. Postgrad. Med. 2017, 129, 140–148. [Google Scholar] [CrossRef]
- Ľupták, M.; Fišar, Z.; Hroudová, J. Agomelatine, Ketamine and Vortioxetine Attenuate Energy Cell Metabolism-In Vitro Study. Int. J. Mol. Sci. 2022, 23, 13824. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J. Pig Brain Mitochondria as a Biological Model for Study of Mitochondrial Respiration. Folia Biol. 2016, 62, 15–25. [Google Scholar]
- Srere, P.A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 3–11. [Google Scholar]
- Barrie Kitto, G. [19] Intra- and extramitochondrial malate dehydrogenases from chicken and tuna heart: [EC 1.1.1.37 l-Malate: NAD oxidoreductase]. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 106–116. [Google Scholar]
- Folbergrová, J.; Jesina, P.; Haugvicová, R.; Lisý, V.; Houstek, J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 2010, 56, 394–403. [Google Scholar] [CrossRef]
- Trounce, I.A.; Kim, Y.L.; Jun, A.S.; Wallace, D.C. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996, 264, 484–509. [Google Scholar] [CrossRef]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gérard, B.; Rötig, A.; Saudubray, J.M.; Munnich, A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef]
- Manfredi, G.; Spinazzola, A.; Checcarelli, N.; Naini, A. Assay of mitochondrial ATP synthesis in animal cells. Methods Cell Biol. 2001, 65, 133–145. [Google Scholar] [CrossRef]
- Tonkonogi, M.; Sahlin, K. Rate of oxidative phosphorylation in isolated mitochondria from human skeletal muscle: Effect of training status. Acta Physiol. Scand. 1997, 161, 345–353. [Google Scholar] [CrossRef]
- Hroudová, J.; Fišar, Z. In Vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol. Lett. 2012, 213, 345–352. [Google Scholar] [CrossRef]
- Sjövall, F.; Morota, S.; Hansson, M.J.; Friberg, H.; Gnaiger, E.; Elmér, E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit. Care 2010, 14, R214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisar, Z. Inhibition of monoamine oxidase activity by cannabinoids. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2010, 381, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Egashira, T.; Takayama, F.; Yamanaka, Y. The inhibition of monoamine oxidase activity by various antidepressants: Differences found in various mammalian species. Jpn. J. Pharmacol. 1999, 81, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Jou, S.H.; Chiu, N.Y.; Liu, C.S. Mitochondrial dysfunction and psychiatric disorders. Chang. Gung Med. J. 2009, 32, 370–379. [Google Scholar]
- Hroudová, J.; Fišar, Z. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin. Neurosci. 2011, 65, 130–141. [Google Scholar] [CrossRef]
- Cikánková, T.; Fišar, Z.; Hroudová, J. In Vitro effects of antidepressants and mood-stabilizing drugs on cell energy metabolism. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.L.; Rezin, G.T.; Ferreira, G.K.; Jeremias, I.C.; Cardoso, M.R.; Carvalho-Silva, M.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Differential effects of escitalopram administration on metabolic parameters of cortical and subcortical brain regions of Wistar rats. Acta Neuropsychiatr. 2012, 24, 147–154. [Google Scholar] [CrossRef]
- Ľupták, M.; Fišar, Z.; Hroudová, J. Effect of Novel Antipsychotics on Energy Metabolism—In Vitro Study in Pig Brain Mitochondria. Mol. Neurobiol. 2021, 58, 5548–5563. [Google Scholar] [CrossRef]
- Luethi, D.; Liechti, M.E.; Krähenbühl, S. Mechanisms of hepatocellular toxicity associated with new psychoactive synthetic cathinones. Toxicology 2017, 387, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Woynillowicz, A.K.; Raha, S.; Nicholson, C.J.; Holloway, A.C. The effect of smoking cessation pharmacotherapies on pancreatic beta cell function. Toxicol. Appl. Pharmacol. 2012, 265, 122–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Couch, L.; Higuchi, M.; Fang, J.L.; Guo, L. Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicol. Sci. 2012, 127, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Hynes, J.; Nadanaciva, S.; Swiss, R.; Carey, C.; Kirwan, S.; Will, Y. A high-throughput dual parameter assay for assessing drug-induced mitochondrial dysfunction provides additional predictivity over two established mitochondrial toxicity assays. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2013, 27, 560–569. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Bernal, A.; Aggeler, R.; Capaldi, R.; Will, Y. Target identification of drug induced mitochondrial toxicity using immunocapture based OXPHOS activity assays. Toxicol. Vitr. 2007, 21, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.K.; Cardoso, M.R.; Jeremias, I.C.; Gonçalves, C.L.; Freitas, K.V.; Antonini, R.; Scaini, G.; Rezin, G.T.; Quevedo, J.; Streck, E.L. Fluvoxamine alters the activity of energy metabolism enzymes in the brain. Braz. J. Psychiatry 2014, 36, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Psychotropic and neurological medication effects on mitochondrial complex I and IV in rodent models. Eur. Neuropsychopharmacol. 2019, 29, 986–1002. [Google Scholar] [CrossRef]
- Velasco, A.; González-Calvo, V.; Alvarez, F.J.; Dueñas, A.; García-Roldán, J.L. Effect of trazodone on oxidative metabolism of rat brain in vitro. Rev. Esp. Fisiol. 1985, 41, 201–205. [Google Scholar]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Nadanaciva, S.; Will, Y. Mitochondrial membrane potential measurement of H9c2 cells grown in high-glucose and galactose-containing media does not provide additional predictivity towards mitochondrial assessment. Toxicol. Vitr. 2011, 25, 580–587. [Google Scholar] [CrossRef]
- Swiss, R.; Niles, A.; Cali, J.J.; Nadanaciva, S.; Will, Y. Validation of a HTS-amenable assay to detect drug-induced mitochondrial toxicity in the absence and presence of cell death. Toxicol. Vitr. 2013, 27, 1789–1797. [Google Scholar] [CrossRef]
- Nabekura, T.; Ishikawa, S.; Tanase, M.; Okumura, T.; Kawasaki, T. Antidepressants induce toxicity in human placental BeWo cells. Curr. Res. Toxicol. 2022, 3, 100073. [Google Scholar] [CrossRef]
- Rodrigues, D.O.; Bristot, I.J.; Klamt, F.; Frizzo, M.E. Sertraline reduces glutamate uptake in human platelets. Neurotoxicology 2015, 51, 192–197. [Google Scholar] [CrossRef]
- Gerö, D.; Szoleczky, P.; Suzuki, K.; Módis, K.; Oláh, G.; Coletta, C.; Szabo, C. Cell-based screening identifies paroxetine as an inhibitor of diabetic endothelial dysfunction. Diabetes 2013, 62, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salin, K.; Auer, S.K.; Rey, B.; Selman, C.; Metcalfe, N.B. Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance. Proc. Biol. Sci. 2015, 282, 20151028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. The efficiency and plasticity of mitochondrial energy transduction. Biochem. Soc. Trans. 2005, 33 Pt 5, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finberg, J.P.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef] [Green Version]
- Bryant, S.G.; Guernsey, B.G.; Ingrim, N.B. Review of bupropion. Clin. Pharm. 1983, 2, 525–537. [Google Scholar] [PubMed]
- Gandolfi, O.; Barbaccia, M.L.; Chuang, D.M.; Costa, E. Daily bupropion injections for 3 weeks attenuate the NE stimulation of adenylate cyclase and the number of beta-adrenergic recognition sites in rat frontal cortex. Neuropharmacology 1983, 22, 927–929. [Google Scholar] [CrossRef]
- Ferris, R.M.; Cooper, B.R.; Maxwell, R.A. Studies of bupropion’s mechanism of antidepressant activity. J. Clin. Psychiatry 1983, 44 Pt 2, 74–78. [Google Scholar]
- Johnson, A.M. An overview of the animal pharmacology of paroxetine. Acta Psychiatr. Scand. Suppl. 1989, 350, 14–20. [Google Scholar] [CrossRef]
- Pivac, N.; Mück-Seler, D.; Sagud, M.; Jakovljević, M.; Mustapić, M.; Mihaljević-Peles, A. Long-term sertraline treatment and peripheral biochemical markers in female depressed patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Yang, Z.Y. Monoamine oxidase A inhibition by fluoxetine: An in vitro and in vivo study. Synapse 1999, 31, 285–289. [Google Scholar] [CrossRef]
- Mukherjee, J.; Yang, Z.Y. Evaluation of monoamine oxidase B inhibition by fluoxetine (Prozac): An In Vitro and In Vivo study. Eur. J. Pharmacol. 1997, 337, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Fisar, Z.; Hroudová, J.; Raboch, J. Inhibition of monoamine oxidase activity by antidepressants and mood stabilizers. Neuro Endocrinol. Lett. 2010, 31, 645–656. [Google Scholar]
- Baumann, P.; Hiemke, C.; Ulrich, S.; Eckermann, G.; Gaertner, I.; Gerlach, M.; Kuss, H.J.; Laux, G.; Müller-Oerlinghausen, B.; Rao, M.L.; et al. The AGNP-TDM Expert Group Consensus Guidelines: Therapeutic Drug Monitoring in Psychiatry. Pharmacopsychiatry 2004, 37, 243–265. [Google Scholar] [CrossRef]
- Laib, K.; Brünen, S.; Pfeifer, P.; Vincent, P.; Hiemke, C. 1023—Therapeutic drug-monitoring of bupropion for depression. Eur. Psychiatry 2013, 28, 1. [Google Scholar] [CrossRef]
- Szewczyk, A.; Wojtczak, L. Mitochondria as a pharmacological target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [Green Version]
- D’Sa, C.; Duman, R.S. Antidepressants and neuroplasticity. Bipolar Disord. 2002, 4, 183–194. [Google Scholar] [CrossRef]
- Stahl, S.M.; Pradko, J.F.; Haight, B.R.; Modell, J.G.; Rockett, C.B.; Learned-Coughlin, S. A Review of the Neuropharmacology of Bupropion, a Dual Norepinephrine and Dopamine Reuptake Inhibitor. Prim. Care Companion J. Clin. Psychiatry 2004, 6, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Bundgaard, C.; Jørgensen, M.; Larsen, F. Pharmacokinetic modelling of blood-brain barrier transport of escitalopram in rats. Biopharm. Drug Dispos. 2007, 28, 349–360. [Google Scholar] [CrossRef]
- Jacobsen, J.; Plenge, P.; Sachs, B.; Pehrson, A.; Cajina, M.; Du, Y.; Roberts, W.; Rudder, M.; Dalvi, P.; Robinson, T.; et al. The interaction of escitalopram and R-citalopram at the human serotonin transporter investigated in the mouse. Psychopharmacology 2014, 231, 4527–4540. [Google Scholar] [CrossRef] [Green Version]
- Bolo, N.R.; Hodé, Y.; Nédélec, J.F.; Lainé, E.; Wagner, G.; Macher, J.P. Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology 2000, 23, 428–438. [Google Scholar] [CrossRef]
- Nedahl, M.; Johansen, S.S.; Linnet, K. Reference Brain/Blood Concentrations of Citalopram, Duloxetine, Mirtazapine and Sertraline. J. Anal. Toxicol. 2018, 42, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.; Thompson, A.J.; Iqbal, F.; Zaidi, W.; Syed, N.I. The Antidepressant Sertraline Reduces Synaptic Transmission Efficacy and Synaptogenesis Between Identified Lymnaea Neurons. Front. Mar. Sci. 2020, 7, 603789. [Google Scholar] [CrossRef]
- Solek, P.; Mytych, J.; Tabecka-Lonczynska, A.; Sowa-Kucma, M.; Koziorowski, M. Toxic effect of antidepressants on male reproductive system cells: Evaluation of possible fertility reduction mechanism. J. Physiol. Pharmacol. 2021, 72, 367–379. [Google Scholar] [CrossRef]
- Chen, F.; Wegener, G.; Madsen, T.M.; Nyengaard, J.R. Mitochondrial plasticity of the hippocampus in a genetic rat model of depression after antidepressant treatment. Synapse 2013, 67, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Filipović, D.; Novak, B.; Xiao, J.; Yan, Y.; Bernardi, R.E.; Turck, C.W. Chronic fluoxetine treatment in socially-isolated rats modulates the prefrontal cortex synaptoproteome. J. Proteom. 2023, 282, 104925. [Google Scholar] [CrossRef]
- Ait Tayeb, A.E.K.; Poinsignon, V.; Chappell, K.; Bouligand, J.; Becquemont, L.; Verstuyft, C. Major Depressive Disorder and Oxidative Stress: A Review of Peripheral and Genetic Biomarkers According to Clinical Characteristics and Disease Stages. Antioxidants 2023, 12, 942. [Google Scholar] [CrossRef] [PubMed]
- Shao, A.; Lin, D.; Wang, L.; Tu, S.; Lenahan, C.; Zhang, J. Oxidative Stress at the Crossroads of Aging, Stroke and Depression. Aging Dis. 2020, 11, 1537–1566. [Google Scholar] [CrossRef]
- Vaváková, M.; Ďuračková, Z.; Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxidative Med. Cell. Longev. 2015, 2015, 898393. [Google Scholar] [CrossRef] [Green Version]
- Westenberg, H.G.; Sandner, C. Tolerability and safety of fluvoxamine and other antidepressants. Int. J. Clin. Pract. 2006, 60, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, C.; Reines, E.H.; Montgomery, S.A. A comparative review of escitalopram, paroxetine, and sertraline: Are they all alike? Int. Clin. Psychopharmacol. 2014, 29, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Nieuwstraten, C.E.; Dolovich, L.R. Bupropion versus selective serotonin-reuptake inhibitors for treatment of depression. Ann. Pharmacother. 2001, 35, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Allen, S.; Haque, M.N.; Angelescu, I.; Baumeister, D.; Tracy, D.K. Bupropion: A systematic review and meta-analysis of effectiveness as an antidepressant. Ther. Adv. Psychopharmacol. 2016, 6, 99–144. [Google Scholar] [CrossRef] [Green Version]
- Fagiolini, A.; Comandini, A.; Catena Dell’Osso, M.; Kasper, S. Rediscovering trazodone for the treatment of major depressive disorder. CNS Drugs 2012, 26, 1033–1049. [Google Scholar] [CrossRef] [Green Version]
- Prado, C.E.; Watt, S.; Crowe, S.F. A meta-analysis of the effects of antidepressants on cognitive functioning in depressed and non-depressed samples. Neuropsychol. Rev. 2018, 28, 32–72. [Google Scholar] [CrossRef]
- Bartels, C.; Wagner, M.; Wolfsgruber, S.; Ehrenreich, H.; Schneider, A. Impact of SSRI Therapy on Risk of Conversion from Mild Cognitive Impairment to Alzheimer’s Dementia in Individuals with Previous Depression. Am. J. Psychiatry 2018, 175, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, M.J.; Vaccarino, S.R.; McInerney, S.J. Procognitive Effects of Antidepressants and Other Therapeutic Agents in Major Depressive Disorder: A Systematic Review. J. Clin. Psychiatry 2020, 81, 19r13200. [Google Scholar] [CrossRef]
- Gonçalo, A.M.G.; Vieira-Coelho, M.A. The effects of trazodone on human cognition: A systematic review. Eur. J. Clin. Pharmacol. 2021, 77, 1623–1637. [Google Scholar] [CrossRef]
- Sommerlad, A.; Werbeloff, N.; Perera, G.; Smith, T.; Costello, H.; Mueller, C.; Kormilitzin, A.; Broadbent, M.; Nevado-Holgado, A.; Lovestone, S.; et al. Effect of trazodone on cognitive decline in people with dementia: Cohort study using UK routinely collected data. Int. J. Geriatr. Psychiatry 2021, 37, 1–12. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex I-Linked Respiration | ||||
|---|---|---|---|---|
| Drug | IC50 (µmol/L) | Hill Slope | Residual Activity (rel.u.) | Inhibition |
| escitalopram | 0.14 ± 0.08 | 1.39 ± 1.05 | 0.774 ± 0.015 | partial |
| fluvoxamine | 15.84 ± 2.57 | 3.09 ± 1.51 | 0.817 ± 0.017 | partial |
| paroxetine | 26.71 ± 6.73 | 1.27 ± 0.36 | 0.672 ± 0.029 | partial |
| sertraline | 12.38 ± 0.75 | 2.05 ± 0.23 | 0.179 ± 0.031 | partial |
| trazodone | 0.45 ± 0.19 | 1.10 ± 0.43 | 0.815 ± 0.015 | partial |
| complex II-linked respiration | ||||
| bupropion | 10.58 ± 4.49 | 1.17 ± 0.44 | 0.690 ± 0.045 | partial |
| MAO-A | ||||
|---|---|---|---|---|
| Drug | IC50 (µmol/L) | Hill Slope | Residual Activity (rel.u.) | Inhibition |
| bupropion | 20.15 ± 5.68 | 1.36 ± 0.33 | 0.235 ± 0.050 | partial |
| escitalopram | 8.78 ± 2.27 | 0.99 ± 0.29 | 0.005 ± 0.057 | full |
| fluvoxamine | 0.99 ± 0.23 | 0.91 ± 0.17 | 0.079 ± 0.041 | full |
| paroxetine | 0.47 ± 0.06 | 1.65 ± 0.22 | 0.007 ± 0.017 | full |
| sertraline | 4.91 ± 0.23 | 2.37 ± 0.39 | 0.074 ± 0.017 | partial |
| trazodone | 13.16 ± 2.55 | 1.23 ± 0.22 | 0.184 ± 0.042 | partial |
| MAO-B | ||||
| bupropion | 31.54 ± 3.32 | 1.68 ± 0.20 | 0.410 ± 0.018 | partial |
| escitalopram | 15.16 ± 2.23 | 1.13 ± 0.14 | 0.038 ± 0.032 | full |
| fluvoxamine | 39.04 ± 2.63 | 3.39 ± 1.35 | 0.306 ± 0.034 | partial |
| paroxetine | 2.19 ± 0.21 | 1.06 ± 0.10 | 0.015 ± 0.021 | full |
| sertraline | 10.95 ± 0.59 | 3.00 ± 0.68 | 0.372 ± 0.014 | partial |
| trazodone | 29.73 ± 5.77 | 2.75 ± 1.35 | 0.560 ± 0.079 | partial |
| Complex I Activity | Complex II + III Activity | Complex IV Activity | Complex I-Linked ATP Content | Complex I-Linked ATP Kinetics | Complex II-Linked ATP Content | Complex II-Linked ATP Kinetics | Complex I-Linked Respiration | Complex II-Linked Respiration | ||
|---|---|---|---|---|---|---|---|---|---|---|
| complex II + III activity | r | *** 0.58 | - | - | - | - | - | - | - | - |
| N | 153 | |||||||||
| complex IV activity | r | *** 0.75 | *** 0.66 | - | - | - | - | - | - | - |
| N | 64 | 64 | ||||||||
| complex I-linked ATP content | r | −0.16 | 0.03 | * −0.32 | ||||||
| N | 53 | 53 | 53 | - | - | - | - | - | - | |
| complex I-linked ATP kinetics | r | * −0.35 | 0.14 | −0.22 | −0.08 | |||||
| N | 46 | 46 | 46 | 44 | - | - | - | - | - | |
| complex II-linked ATP content | r | 0.12 | 0.19 | 0.00 | 0.06 | 0.11 | ||||
| N | 56 | 56 | 56 | 52 | 46 | - | - | - | - | |
| complex II-linked ATP kinetics | r | 0.04 | ** 0.42 | −0.03 | 0.12 | 0.19 | *** 0.50 | |||
| N | 45 | 45 | 45 | 45 | 43 | 45 | - | - | - | |
| complex I-linked respiration | r | *** 0.51 | *** 0.57 | *** 0.66 | −0.16 | −0.16 | −0.01 | 0.10 | ||
| N | 82 | 82 | 55 | 52 | 43 | 52 | 45 | - | - | |
| complex II-linked respiration | r | −0.05 | *** −0.61 | * −0.43 | 0.03 | −0.21 | −0.26 | *** −0.75 | −0.09 | |
| N | 54 | 54 | 27 | 24 | 21 | 25 | 21 | 294 | - | |
| MAO-A activity | r | *** 0.48 | 0.02 | 0.06 | 0.04 | 0.12 | 0.26 | 0.28 | * 0.23 | 0.12 |
| N | 71 | 71 | 52 | 50 | 42 | 51 | 44 | 100 | 74 | |
| MAO-B activity | r | *** 0.58 | −0.04 | * 0.30 | −0.12 | −0.20 | −0.06 | 0.06 | *** 0.37 | 0.15 |
| N | 87 | 87 | 63 | 53 | 46 | 56 | 45 | 104 | 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ľupták, M.; Fišar, Z.; Hroudová, J. Different Effects of SSRIs, Bupropion, and Trazodone on Mitochondrial Functions and Monoamine Oxidase Isoform Activity. Antioxidants 2023, 12, 1208. https://doi.org/10.3390/antiox12061208
Ľupták M, Fišar Z, Hroudová J. Different Effects of SSRIs, Bupropion, and Trazodone on Mitochondrial Functions and Monoamine Oxidase Isoform Activity. Antioxidants. 2023; 12(6):1208. https://doi.org/10.3390/antiox12061208
Chicago/Turabian StyleĽupták, Matej, Zdeněk Fišar, and Jana Hroudová. 2023. "Different Effects of SSRIs, Bupropion, and Trazodone on Mitochondrial Functions and Monoamine Oxidase Isoform Activity" Antioxidants 12, no. 6: 1208. https://doi.org/10.3390/antiox12061208