
PD-1 Alleviates Cisplatin-Induced Muscle Atrophy by Regulating Inflammation and Oxidative Stress

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cisplatin-Induced Skeletal Muscle Atrophy
2.3. RNA Extraction and Gene Expression Analysis
2.4. Histological Analysis
2.5. Western Blot Analysis
2.6. Statistical Analysis
3. Results
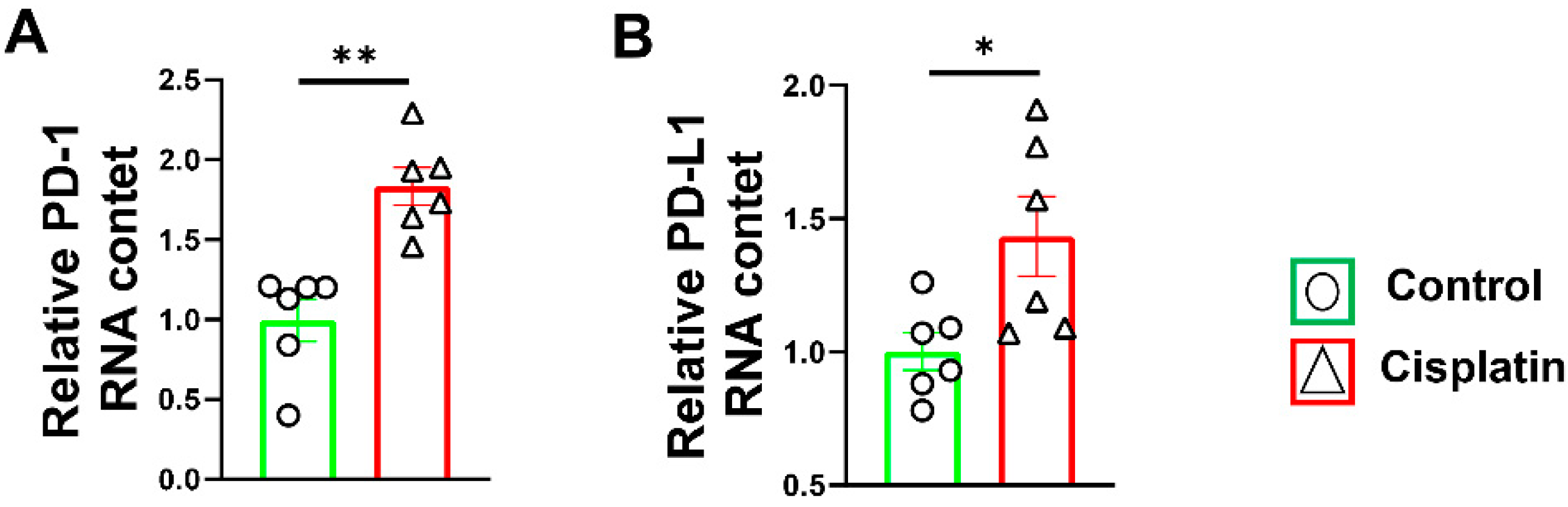
3.1. Increased PD-1/PD-L1 Expression after Cisplatin Treatment in Wild-Type Mice
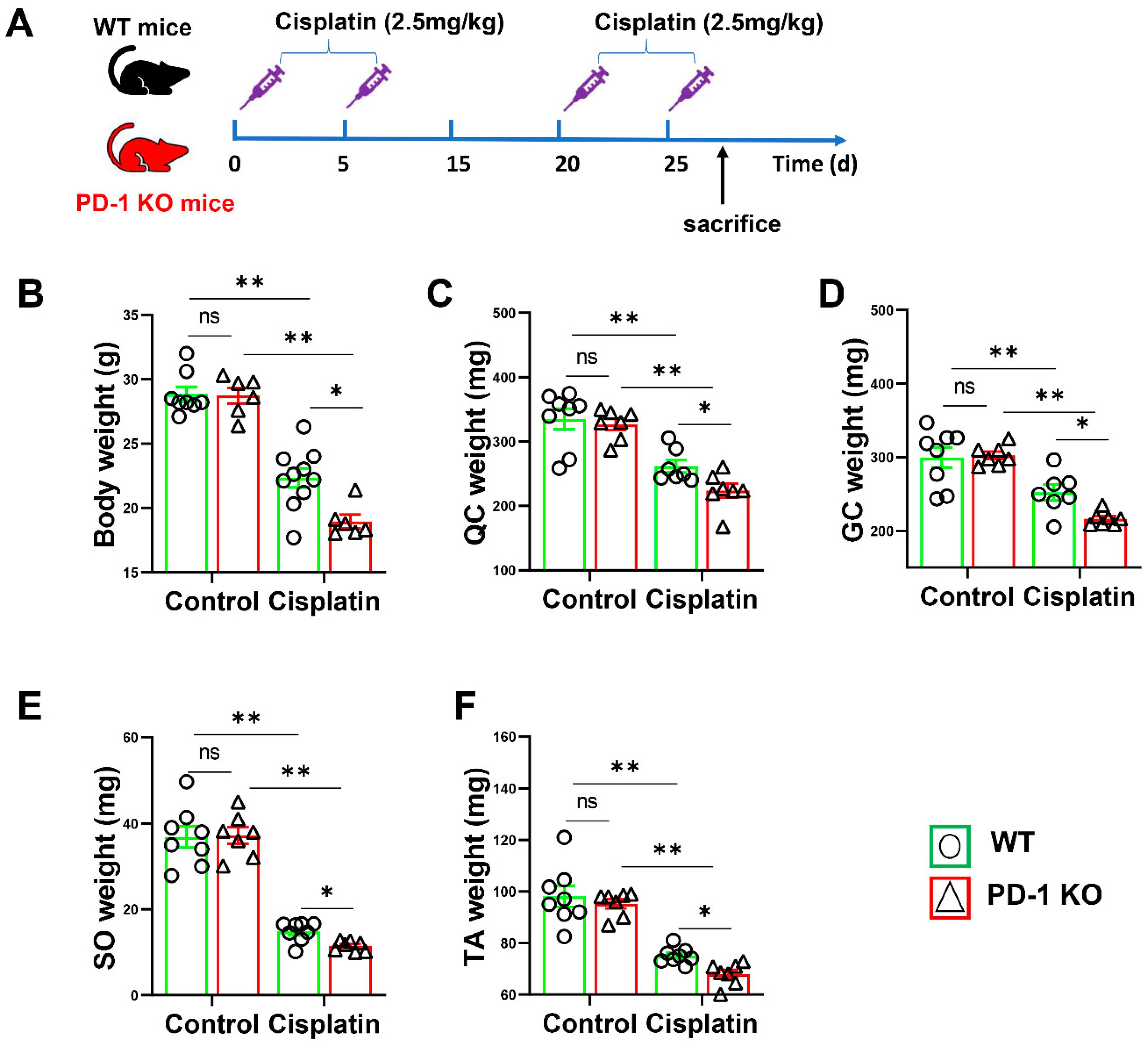
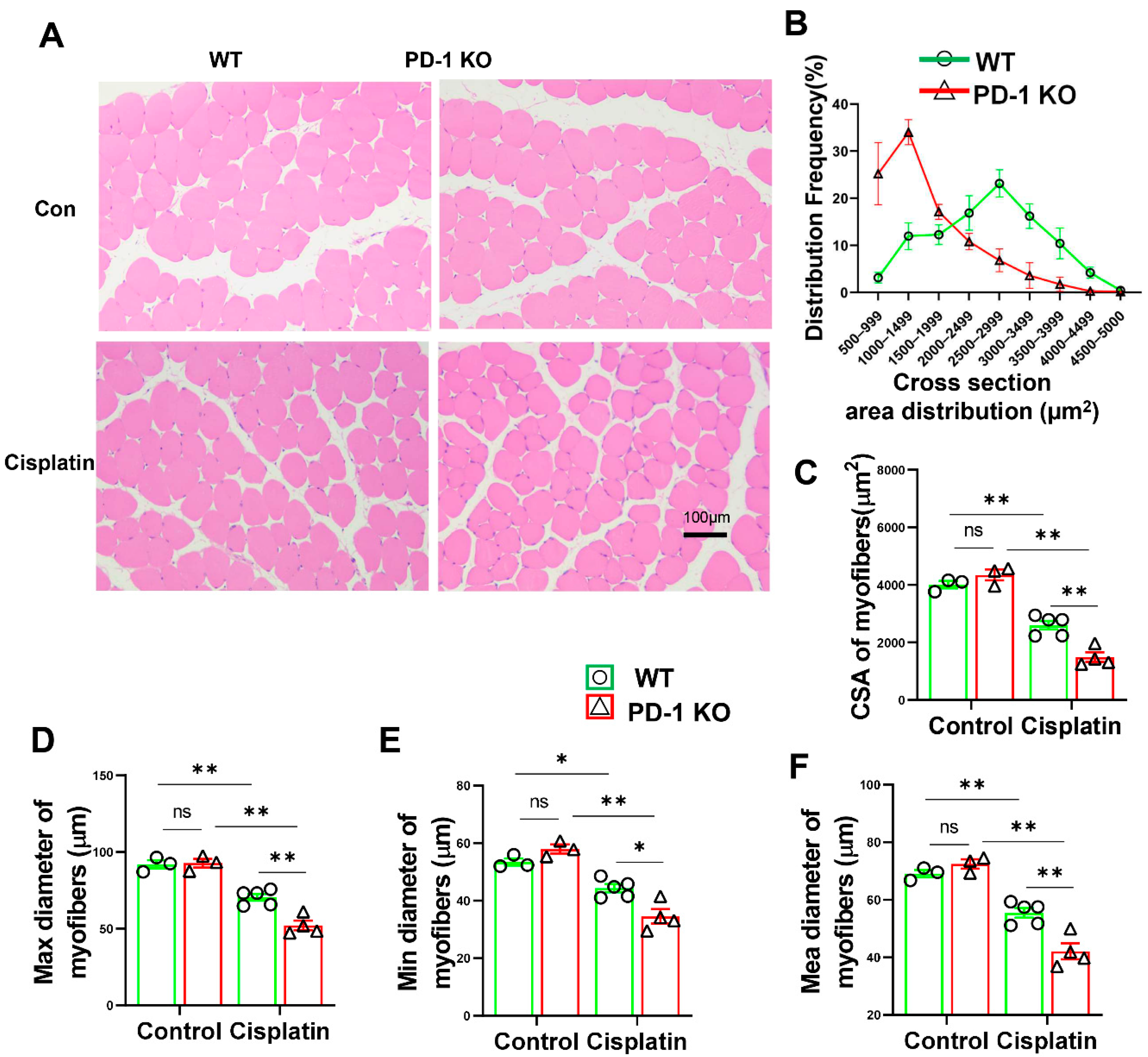
3.2. PD-1−/− Contributed to Skeletal Muscle Atrophy after Cisplatin Treatment
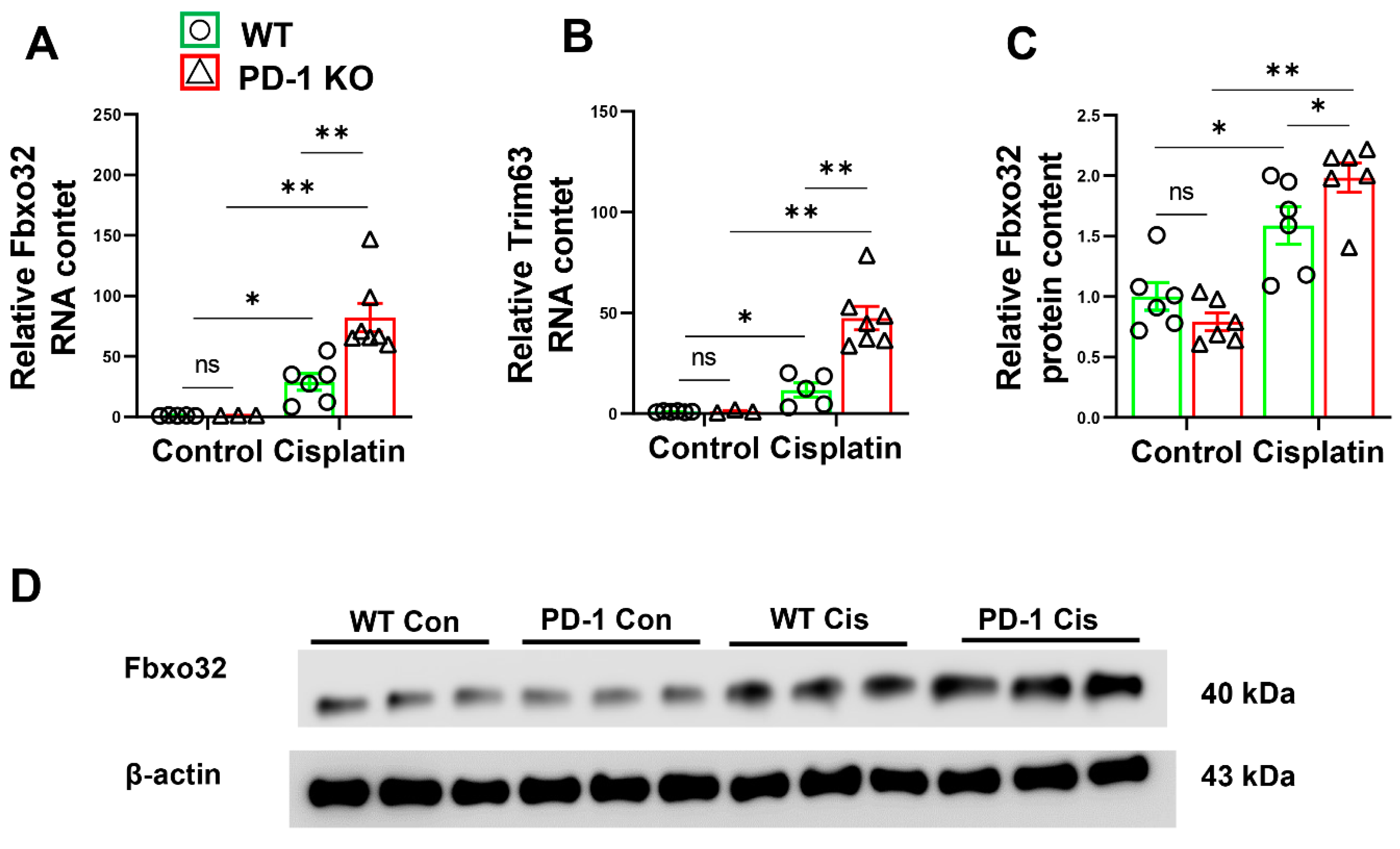
3.3. PD-1−/− Enhanced the Expression of E3 Ubiquitin-Protein Ligases after Cisplatin Treatment
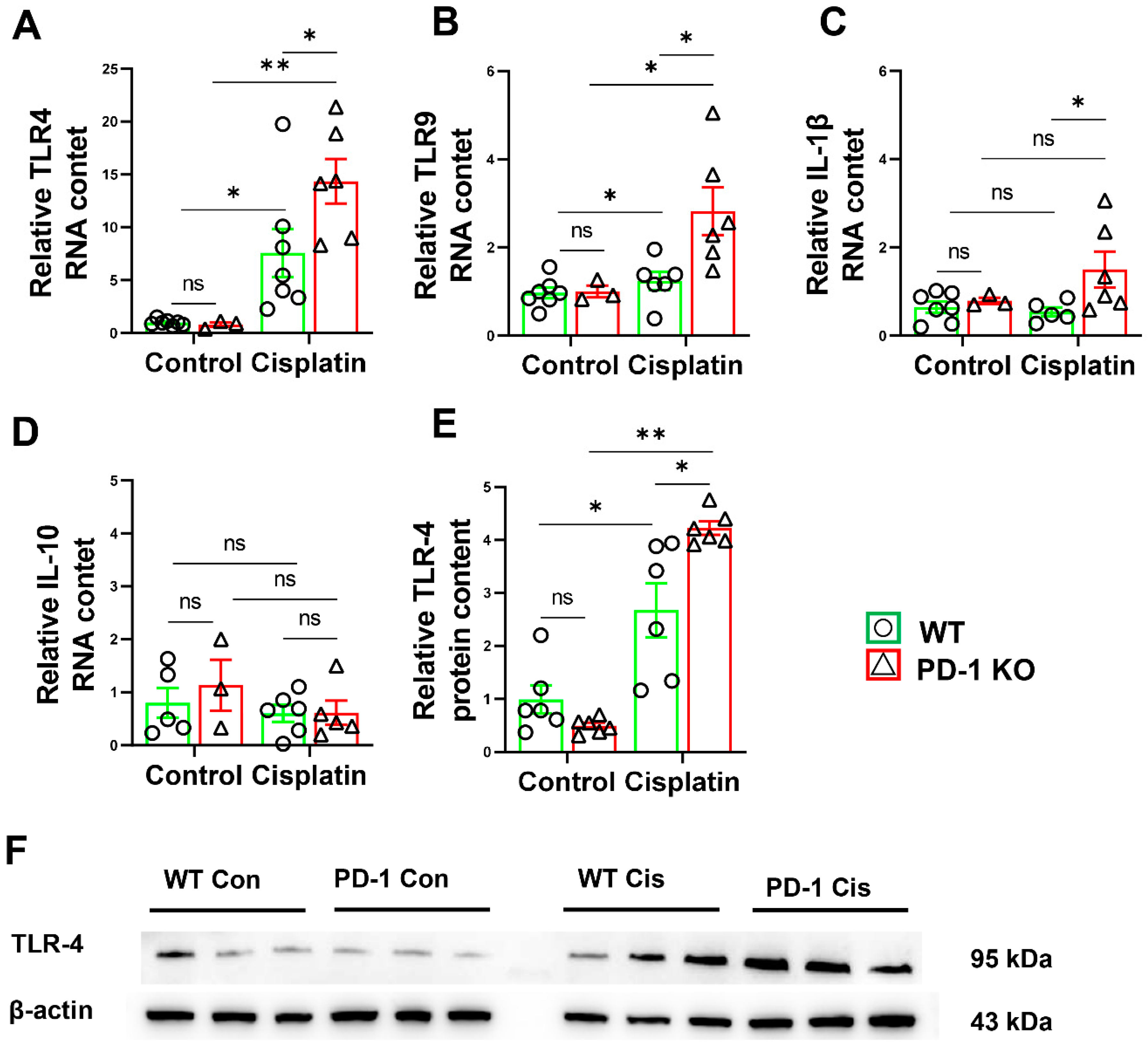
3.4. PD-1−/− Exacerbated Muscle Inflammation in Mice after Cisplatin Treatment
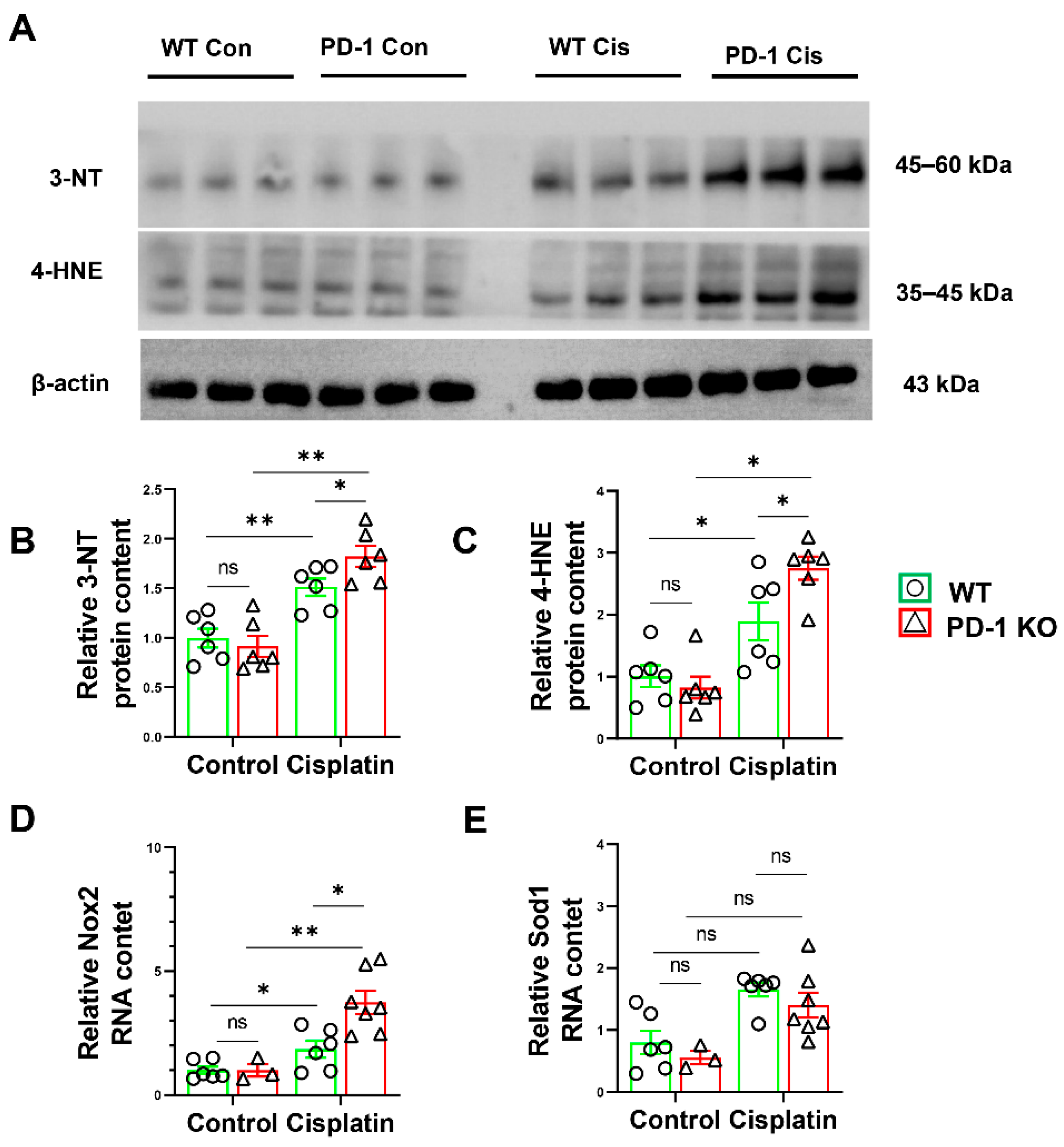
3.5. PD-1−/− Exacerbated Muscle Oxidative Stress in Mice after Cisplatin Treatment
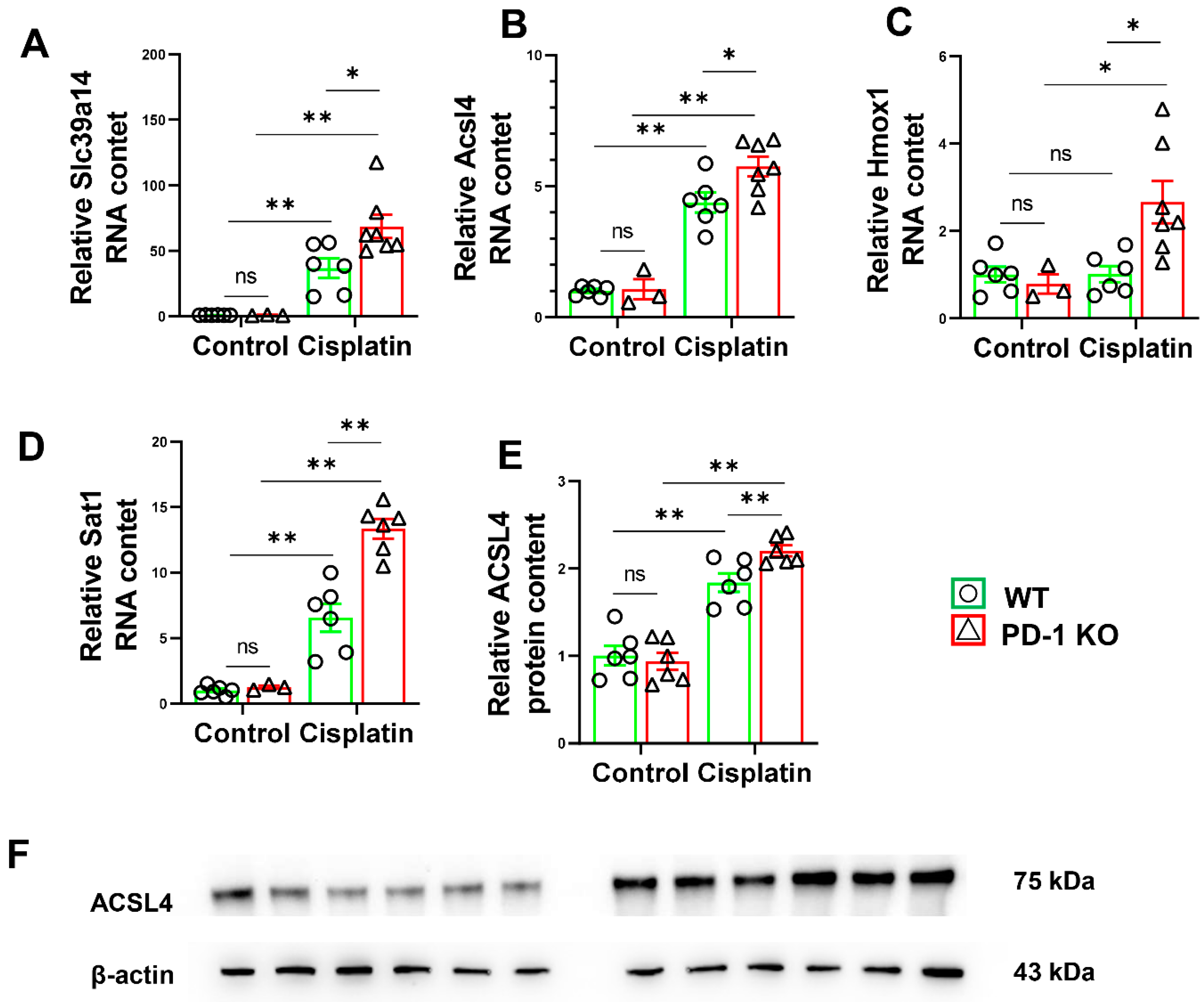
3.6. PD-1−/− Exacerbated the Expression of Ferroptosis-Related Genes in Mice after Cisplatin Treatment
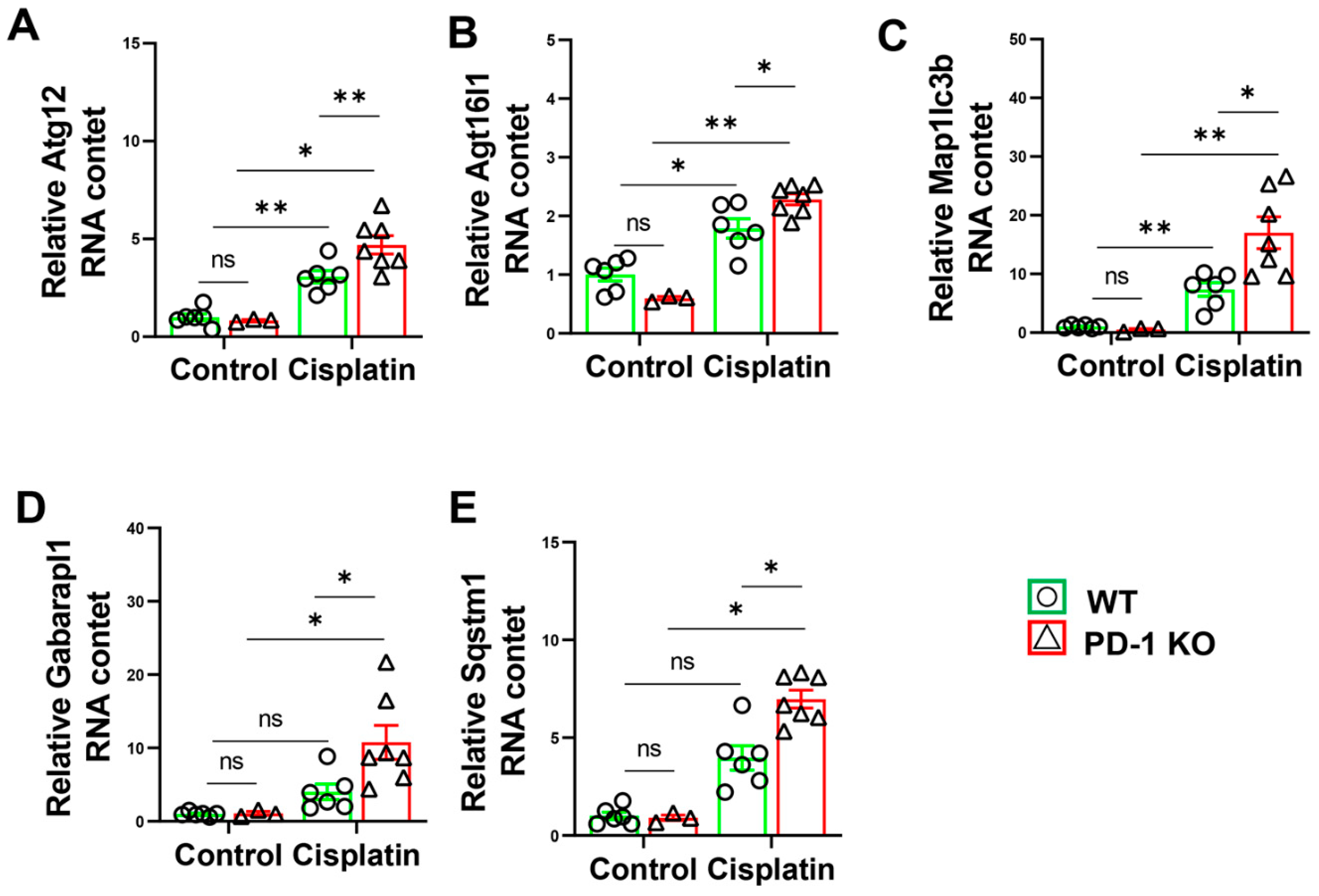
3.7. PD-1−/− Exacerbated the Expression of Autophagy-Related Genes in Mice after Cisplatin Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-A.; Splenser, A.; Guillory, B.; Luo, J.; Mendiratta, M.; Belinova, B.; Halder, T.; Zhang, G.; Li, Y.-P.; Garcia, J.M. Ghrelin prevents tumour- and cisplatin-induced muscle wasting: Characterization of multiple mechanisms involved. J. Cachexia Sarcopenia Muscle 2015, 6, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Shi, X.; Liu, X.; Chen, Y.; Chen, P.; Xiao, W. Programmed death-1 promotes contused skeletal muscle regeneration by regulating Treg cells and macrophages. Lab. Investig. 2021, 101, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Jeong, H.; Lee, H.; Hong, M.; Park, S.-Y.; Bae, H. Magnolol Attenuates Cisplatin-Induced Muscle Wasting by M2c Macrophage Activation. Front. Immunol. 2020, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Bresciani, E.; Rizzi, L.; Cappellari, O.; de Luca, A.; Torsello, A.; Liantonio, A. Cisplatin-Induced Skeletal Muscle Dysfunction: Mechanisms and Counteracting Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 1242. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020, 38, 685–700.e688. [Google Scholar] [CrossRef]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef]
- Berger, K.N.; Pu, J.J. PD-1 pathway and its clinical application: A 20year journey after discovery of the complete human PD-1 gene. Gene 2018, 638, 20–25. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Sun, L.; Li, C.W.; Chung, E.M.; Yang, R.; Kim, Y.S.; Park, A.H.; Lai, Y.J.; Yang, Y.; Wang, Y.H.; Liu, J.; et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res. 2020, 80, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Gambichler, T.; Strutzmann, S.; Tannapfel, A.; Susok, L. Paraneoplastic acral vascular syndrome in a patient with metastatic melanoma under immune checkpoint blockade. BMC Cancer 2017, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chavez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suarez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Liu, X.; Weng, X.; Xiao, W.; Xu, X.; Chen, Y.; Chen, P. Pharmacological and Genetic Inhibition of PD-1 Demonstrate an Important Role of PD-1 in Ischemia-Induced Skeletal Muscle Inflammation, Oxidative Stress, and Angiogenesis. Front. Immunol. 2021, 12, 586429. [Google Scholar] [CrossRef]
- Matas-García, A.; Milisenda, J.C.; Selva-O’Callaghan, A.; Prieto-González, S.; Padrosa, J.; Cabrera, C.; Reguart, N.; Castrejón, N.; Solé, M.; Ros, J.; et al. Emerging PD-1 and PD-1L inhibitors-associated myopathy with a characteristic histopathological pattern. Autoimmun. Rev. 2020, 19, 102455. [Google Scholar] [CrossRef]
- Liu, X.; Zeng, Z.; Zhao, L.; Chen, P.; Xiao, W. Impaired Skeletal Muscle Regeneration Induced by Macrophage Depletion Could Be Partly Ameliorated by MGF Injection. Front. Physiol. 2019, 10, 601. [Google Scholar] [CrossRef]
- Warren, S.A.; Briggs, L.E.; Zeng, H.; Chuang, J.; Chang, E.I.; Terada, R.; Li, M.; Swanson, M.S.; Lecker, S.H.; Willis, M.S.; et al. Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation 2012, 126, 2575–2588. [Google Scholar] [CrossRef]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef]
- Hong, M.; Han, I.H.; Choi, I.; Cha, N.; Kim, W.; Kim, S.K.; Bae, H. Magnoliae Cortex Alleviates Muscle Wasting by Modulating M2 Macrophages in a Cisplatin-Induced Sarcopenia Mouse Model. Int. J. Mol. Sci. 2021, 22, 3188. [Google Scholar] [CrossRef]
- Sirago, G.; Conte, E.; Fracasso, F.; Cormio, A.; Fehrentz, J.-A.; Martinez, J.; Musicco, C.; Camerino, G.M.; Fonzino, A.; Rizzi, L.; et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia. Sci. Rep. 2017, 7, 13017. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, B.; Shen, D.; Chen, J.; Yu, Z.; Chen, C. Ferroptosis in a sarcopenia model of senescence accelerated mouse prone 8 (SAMP8). Int. J. Biol. Sci. 2021, 17, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Dolly, A.; Dumas, J.F.; Servais, S. Cancer cachexia and skeletal muscle atrophy in clinical studies: What do we really know? J. Cachexia Sarcopenia Muscle 2020, 11, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Ikeno, Y.; Tsukimura, Y.; Inomata, M.; Suzuki, Y.; Kon, R.; Ikarashi, N.; Chiba, Y.; Yamada, T.; Kamei, J. Upregulation of ubiquitinated proteins and their degradation pathway in muscle atrophy induced by cisplatin in mice. Toxicol. Appl. Pharmacol. 2020, 403, 115165. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Berry, G.J.; Goronzy, J.J. The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J. Leukoc. Biol. 2018, 103, 565–575. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, S.; Wu, H.; Xie, H.; Zhang, T.; Deng, Z. Blocking the PD-1/PD-L1 axis enhanced cisplatin chemotherapy in osteosarcoma in vitro and in vivo. Environ. Health Prev. Med. 2019, 24, 79. [Google Scholar] [CrossRef]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Segal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Tredaniel, J.; et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Taillandier, D.; Polge, C. Skeletal muscle atrogenes: From rodent models to human pathologies. Biochimie 2019, 166, 251–269. [Google Scholar] [CrossRef]
- Zhang, H.; Chi, M.; Wang, Y.; Chen, L.; Sun, X.; Wan, L.; Yang, Q.; Guo, C. Naringenin alleviates cisplatin induced muscle atrophy by regulating RIPK1/AMPK/NF-κB pathway. J. Funct. Foods 2021, 86, 104714. [Google Scholar] [CrossRef]
- Leite, A.B.; Lima, H.N.; Flores, C.O.; Oliveira, C.A.; Cunha, L.E.C.; Neves, J.L.; Correia, T.M.L.; de Melo, F.F.; Oliveira, M.V.; de Magalhaes, A.C.M.; et al. High-intensity interval training is more effective than continuous training to reduce inflammation markers in female rats with cisplatin nephrotoxicity. Life Sci. 2021, 266, 118880. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B. Progressive Skeletal Muscle Atrophy in Muscular Dystrophies: A Role for Toll-like Receptor-Signaling in Disease Pathogenesis. Int. J. Mol. Sci. 2020, 21, 4440. [Google Scholar] [CrossRef]
- Rausch, V.; Sala, V.; Penna, F.; Porporato, P.E.; Ghigo, A. Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis 2021, 10, 1. [Google Scholar] [CrossRef]
- Ren, X.; Akiyoshi, K.; Vandenbark, A.A.; Hurn, P.D.; Offner, H. Programmed death-1 pathway limits central nervous system inflammation and neurologic deficits in murine experimental stroke. Stroke 2011, 42, 2578–2583. [Google Scholar] [CrossRef]
- Bu, D.X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Cao, Y.; Lu, J.; Li, J.; Liu, L.; Wang, H.; Su, F.; Zheng, Q. Programmed cell death-1 deficiency results in atrial remodeling in C57BL/6 mice. Int. J. Mol. Med. 2013, 31, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; Lomicronpez-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Shakri, A.R.; Zhong, T.J.; Ma, W.; Coker, C.; Kim, S.; Calluori, S.; Scholze, H.; Szabolcs, M.; Caffrey, T.; Grandgenett, P.M.; et al. Upregulation of ZIP14 and Altered Zinc Homeostasis in Muscles in Pancreatic Cancer Cachexia. Cancers 2019, 12, 3. [Google Scholar] [CrossRef]
- Sandri, M. Autophagy in skeletal muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef]
- Sakuma, K.; Kinoshita, M.; Ito, Y.; Aizawa, M.; Aoi, W.; Yamaguchi, A. p62/SQSTM1 but not LC3 is accumulated in sarcopenic muscle of mice. J. Cachexia Sarcopenia Muscle 2016, 7, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Goljanek-Whysall, K.; Soriano-Arroquia, A.; McCormick, R.; Chinda, C.; McDonagh, B. miR-181a regulates p62/SQSTM1, parkin, and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle aging. Aging Cell 2020, 19, e13140. [Google Scholar] [CrossRef] [PubMed]
- Paolini, A.; Omairi, S.; Mitchell, R.; Vaughan, D.; Matsakas, A.; Vaiyapuri, S.; Ricketts, T.; Rubinsztein, D.C.; Patel, K. Attenuation of autophagy impacts on muscle fibre development, starvation induced stress and fibre regeneration following acute injury. Sci. Rep. 2018, 8, 9062. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Skipworth, R.J.; Gallagher, I.J.; Greig, C.A.; Guttridge, D.C.; Ross, J.A.; Fearon, K.C. Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J. Cachexia Sarcopenia Muscle 2015, 6, 53–61. [Google Scholar] [CrossRef]
- Paul, P.K.; Bhatnagar, S.; Mishra, V.; Srivastava, S.; Darnay, B.G.; Choi, Y.; Kumar, A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol. Cell. Biol. 2012, 32, 1248–1259. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Accession Number | Forward Primer | Reverse Primer |
|---|---|---|---|
| GAPDH | NM_008084 | ACTCCACTCACGGCAAATTC | TCTCCATGGTGGTGAAGACA |
| Trim63 | NM_001369245 | TGTCTGGAGGTCGTTTCCG | ATGCCGGTCCATGATCACTT |
| Fbxo32 | NM_026346 | TCACAGCTCACATCCCTGAG | AGACTTGCCGACTCTTTGGA |
| IL-1β | NM_008361 | TGACGTTCCCATTAGACAACTG | CCGTCTTTCATTACACAGGACA |
| IL-10 | NM_010548 | CAAGGAGCATTTGAATTCCC | GGCCTTGTAGACACCTTGGTC |
| TLR4 | NM_021297 | GCTTTCACCTCTGCCTTCAC | GAAACTGCCATGTTTGAGCA |
| TLR9 | NM_031178 | GAAAGCATCAACCACACCAA | ACAAGTCCACAAAGCGAAGG |
| Slc39a14 | NM_144808 | GCTGCTGCTATTTGGGTCTG | GACAAAGGGGACCAGAAAGC |
| Acsl4 | NM_019477 | CAATAGAGCAGAGTACCCTGAG | TAGAACCACTGGTGTACATGAC |
| Hmox1 | NM_010442 | GAGGTCAAGCACAGGGTGA | CAGGCCTCTGACGAAGTGA |
| Sat1 | NM_001291865 | CTGAAGGACATAGCATTGTTGG | TTCCATTCTGCTACCAAGAAGT |
| Atg12 | NM_026217 | GCCTCGGAACAGTTGTTTATTT | CAGTTTACCATCACTGCCAAAA |
| Atg16l1 | NM_001205391 | TGCGTGGAATGATAGTCAACTA | TCAATCACCAACTGAGCTAACT |
| Map1lc3b | NM_026160 | CCACCAAGATCCCAGTGATTAT | TGATTATCTTGATGAGCTCGCT |
| Gabarapl1 | NM_020590 | TCCCTGATCTGGATAAGAGGAA | AAAGAAGAATAAGGCGTCCTCA |
| Sqstm1 | NM_011018 | GAACACAGCAAGCTCATCTTTC | AAAGTGTCCATGTTTCAGCTTC |
| PD-1 | NM_008798 | ATGACTTCCACATGAACATCCT | CTCCAGGATTCTCTCTGTTACC |
| PD-L1 | NM_021893 | TGAGCAAGTGATTCAGTTTGTG | CATTTCCCTTCAAAAGCTGGTC |
| NOX-2 | XM_011397013 | TGAATGCCAGAGTCGGGATT | CGAGTCACGGCCACATACA |
| SOD1 | NM_011434 | TATGGGGACAATACACAAGGCT | CGGGCCACCATGTTTCTTAGA |
| Primary Antibodies | Description |
|---|---|
| Fbxo32 | Rabbit polyclonal; 1:1000–1:2000; A3193; ABclonal, Wuhan, China |
| 4-hydroxynonenal | Rabbit polyclonal; 1:3000; ab46545; Cambridge, UK |
| 3-nitrotyrosine | Mouse monoclonal; 1:3000; ab61392; Cambridge, UK |
| TLR4 | Rabbit polyclonal; 1:500–1:1000; GB11519; Sercicebio, Wuhan, China |
| ACSL-4 | Mouse monoclonal; 1:100–1:1000; sc-365230; Santa Cruz; Dallas, TX, USA |
| Beta actin | Mouse monoclonal; 1:20000–1:100000; 66009-1-Ig; Proteintech; CHIX, USA |
| Secondary Antibodies | Description |
|---|---|
| Horseradish Peroxidase-conjugated Antibody | Goat Anti-Rabbit IgG; 1:3000; GB23204; Sercicebio, Wuhan, China |
| Horseradish Peroxidase-conjugated Antibody | Goat Anti-Mouse IgG; 1:3000; G1214; Sercicebio, Wuhan, China |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Xu, M.; Yu, Y.; Chen, Y.; Weng, X.; Zhu, L. PD-1 Alleviates Cisplatin-Induced Muscle Atrophy by Regulating Inflammation and Oxidative Stress. Antioxidants 2022, 11, 1839. https://doi.org/10.3390/antiox11091839
Liu X, Xu M, Yu Y, Chen Y, Weng X, Zhu L. PD-1 Alleviates Cisplatin-Induced Muscle Atrophy by Regulating Inflammation and Oxidative Stress. Antioxidants. 2022; 11(9):1839. https://doi.org/10.3390/antiox11091839
Chicago/Turabian StyleLiu, Xiaoguang, Miaomiao Xu, Yang Yu, Yingjie Chen, Xinyu Weng, and Lin Zhu. 2022. "PD-1 Alleviates Cisplatin-Induced Muscle Atrophy by Regulating Inflammation and Oxidative Stress" Antioxidants 11, no. 9: 1839. https://doi.org/10.3390/antiox11091839