
Chelerythrine-Induced Apoptotic Cell Death in HepG2 Cells Involves the Inhibition of Akt Pathway and the Activation of Oxidative Stress and Mitochondrial Apoptotic Pathway
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Regents
2.2. Cell Cultures
2.3. Measurement of Cell Viability
2.4. Measurement of Cell Apoptosis
2.5. Measurements of ATP Levels
2.6. Determination of Intracellular ROS Generation, Malondialdehyde (MDA), Catalase (CAT) and Superoxide Dismutase (SOD)
2.7. Determination of Mitochondrial Membrane Potential (MMP)
2.8. Determination of Caspase-9 and Caspase-3 Activities
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
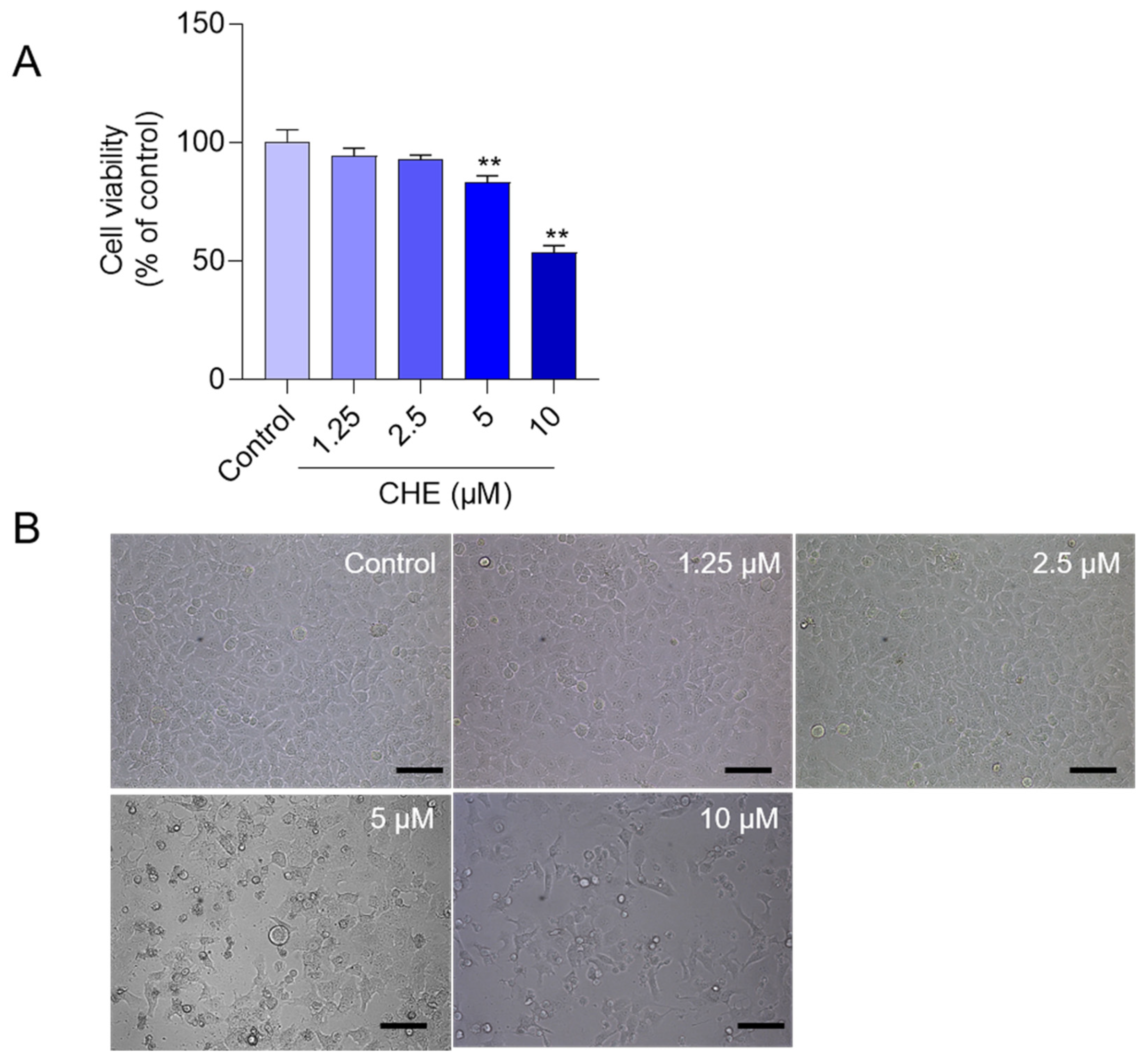
3.1. CHE Treatment Induces the Loss of Cell Viabilities in HepG2 Cells
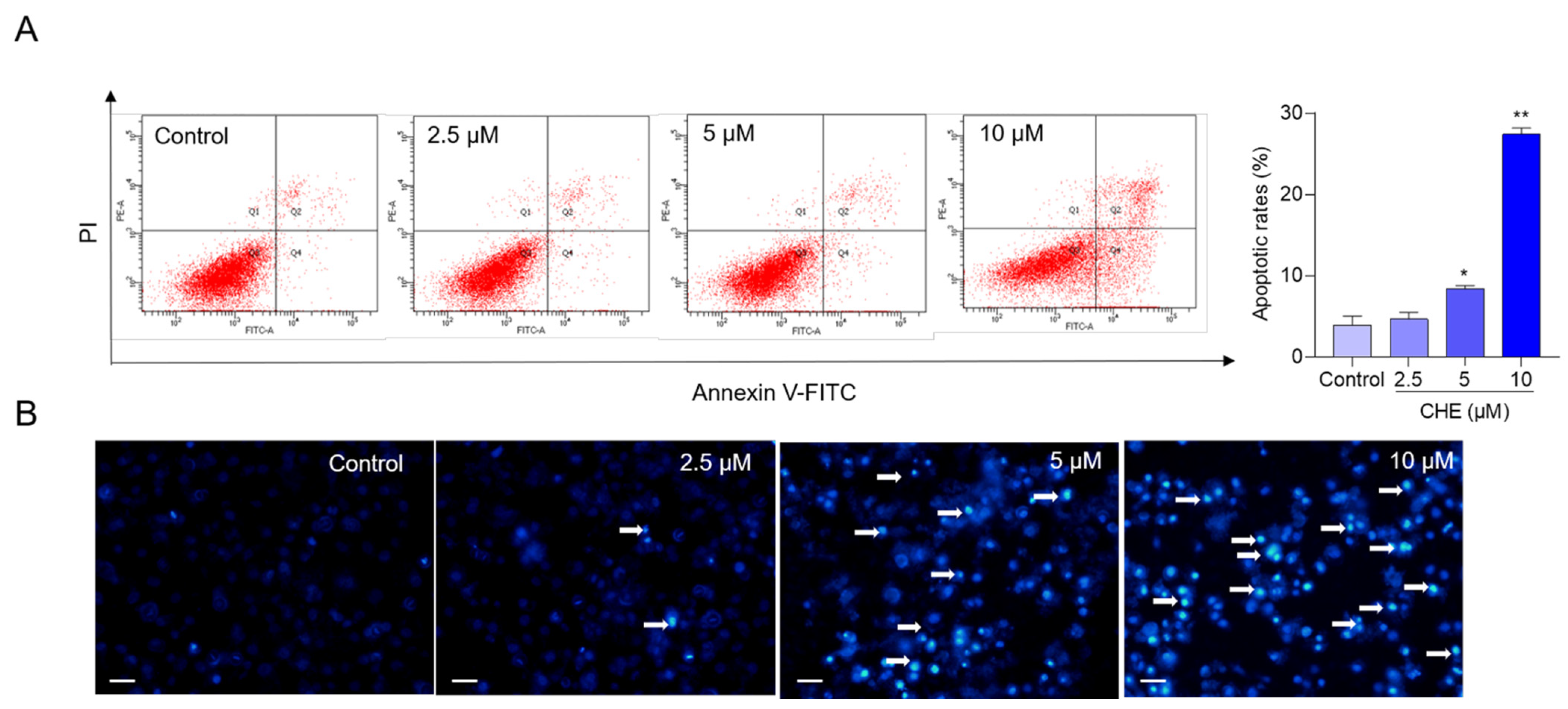
3.2. CHE Treatment Induces Cell Apoptosis
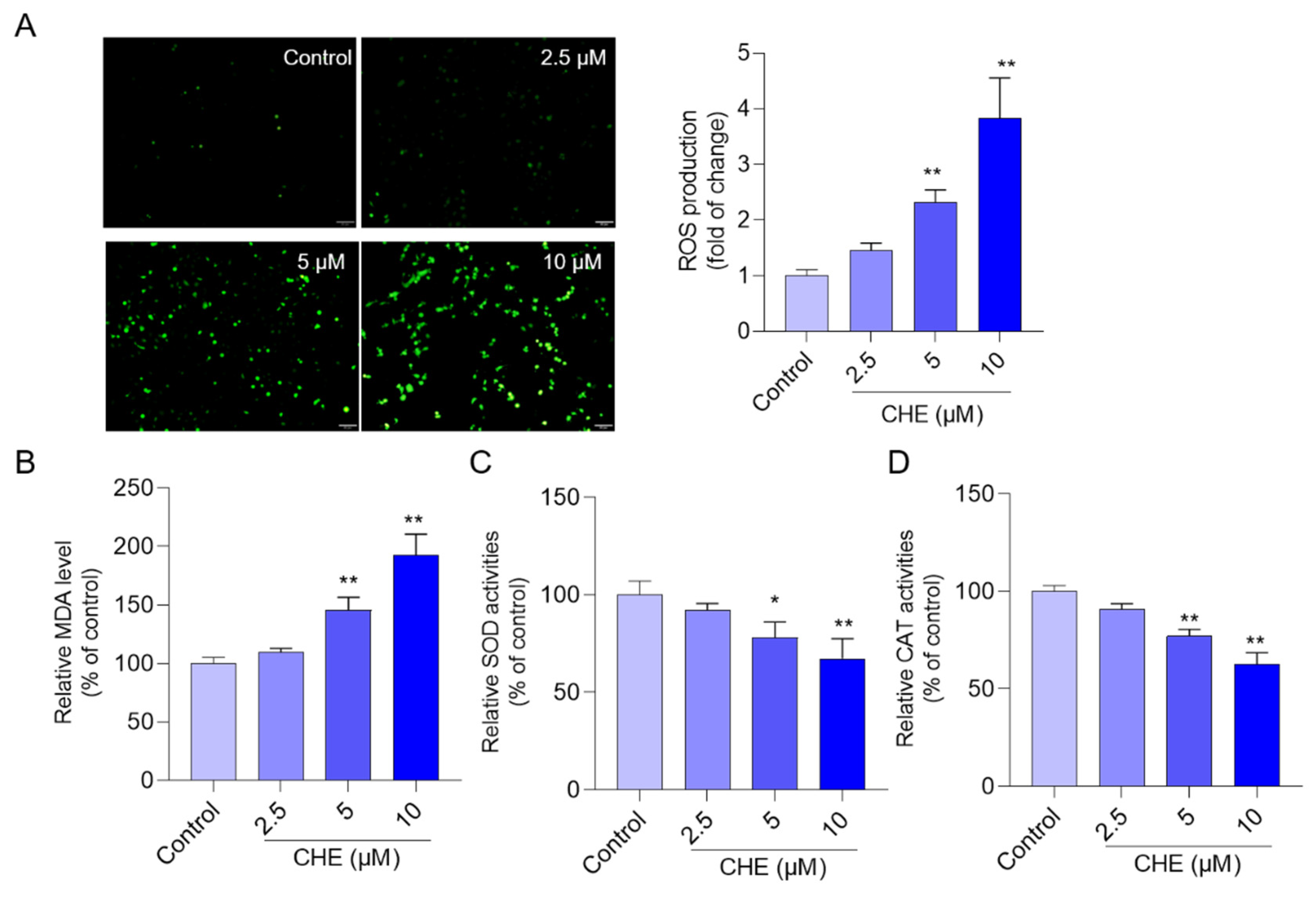
3.3. CHE Treatment Induces Oxidative Stress
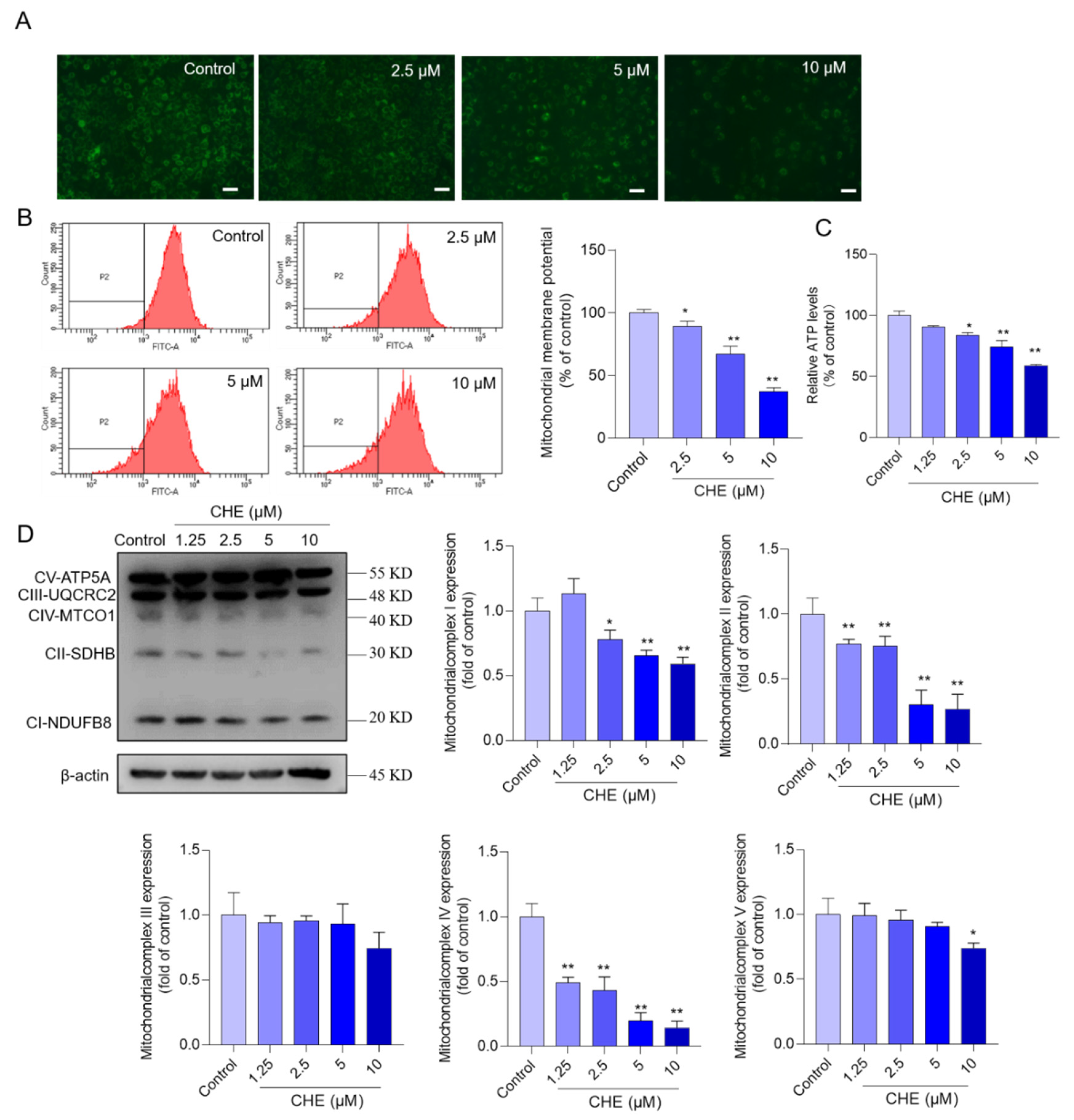
3.4. CHE Treatment Induces Mitochondrial Dysfunction
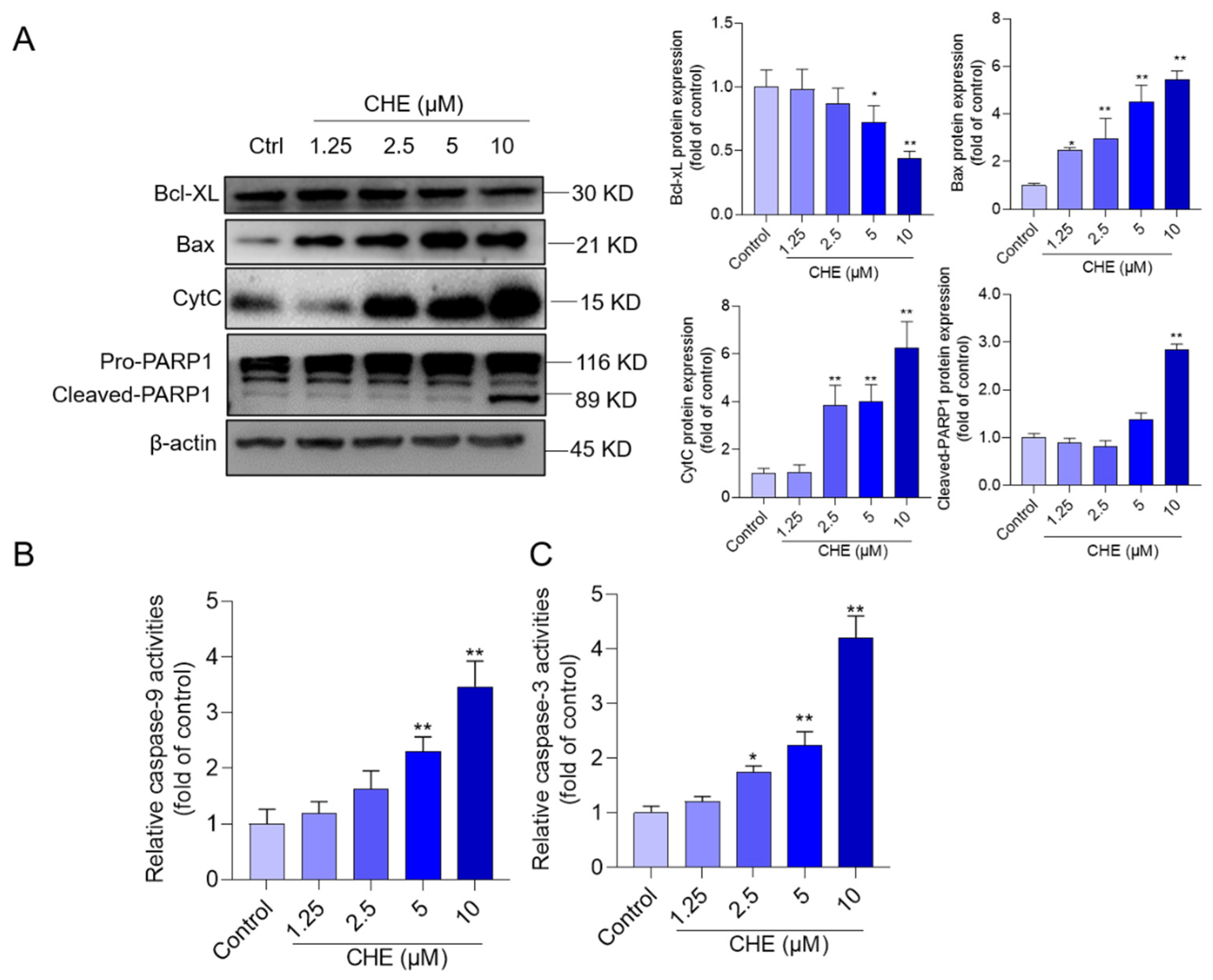
3.5. CHE Treatment Activates Mitochondrial Apoptotic Pathway
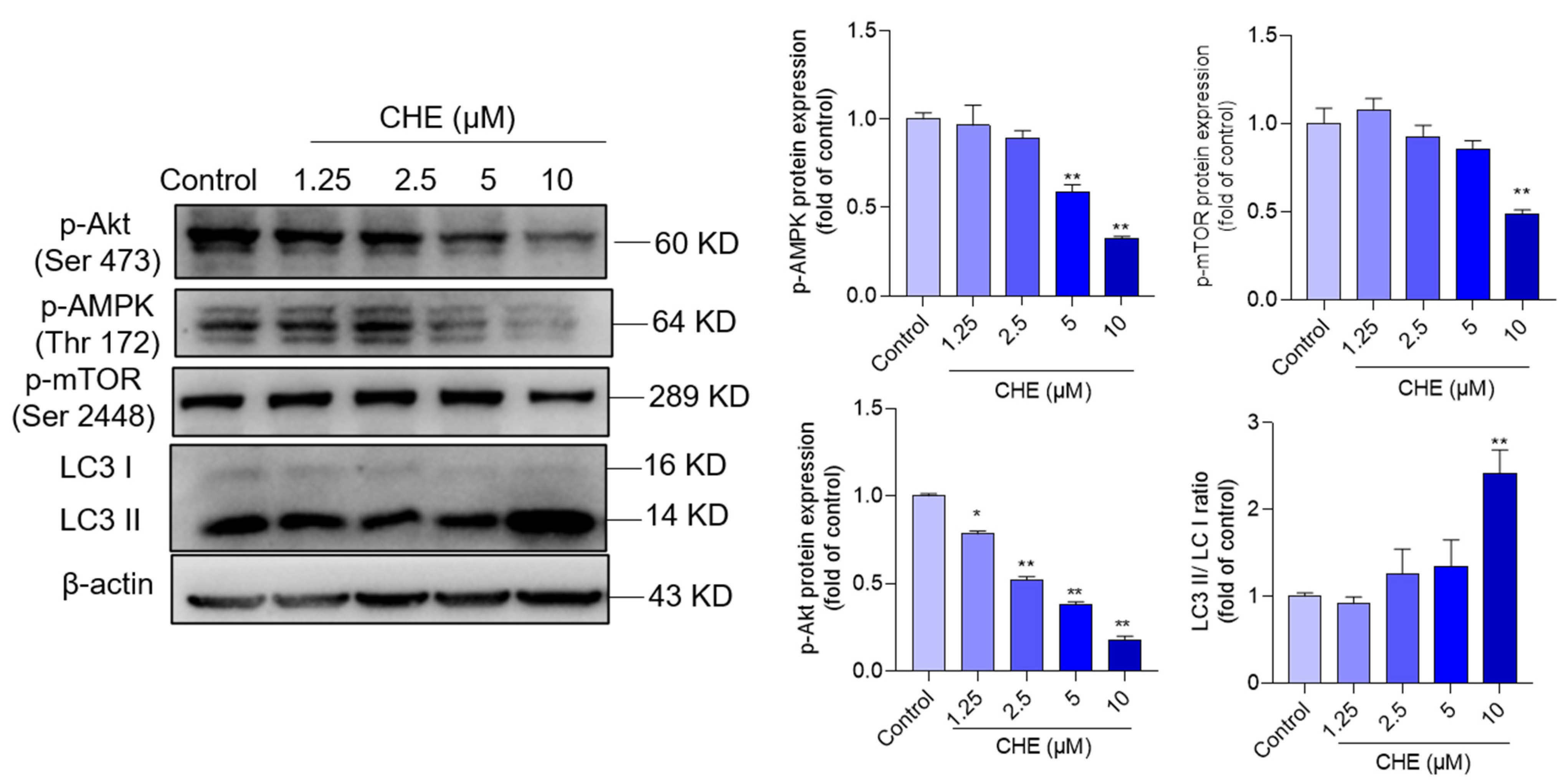
3.6. CHE Treatment Inhibits AMPK/Akt Pathway
3.7. NAC Supplementation Improved CHE-Induced the Production of ROS and Apoptosis
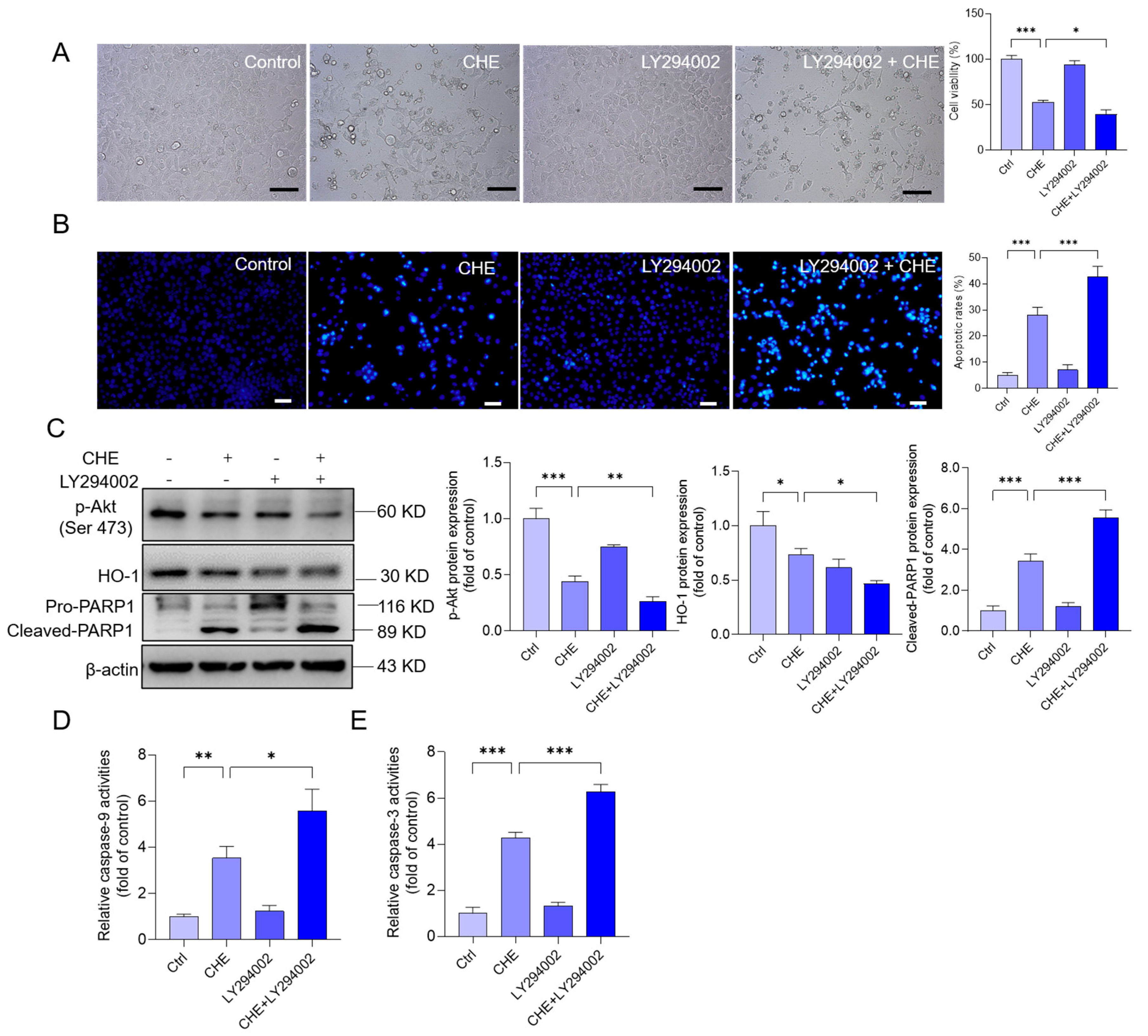
3.8. Inhibition of Akt Pathway Promotes CHE-Induced Cytotoxicity and Apoptosis
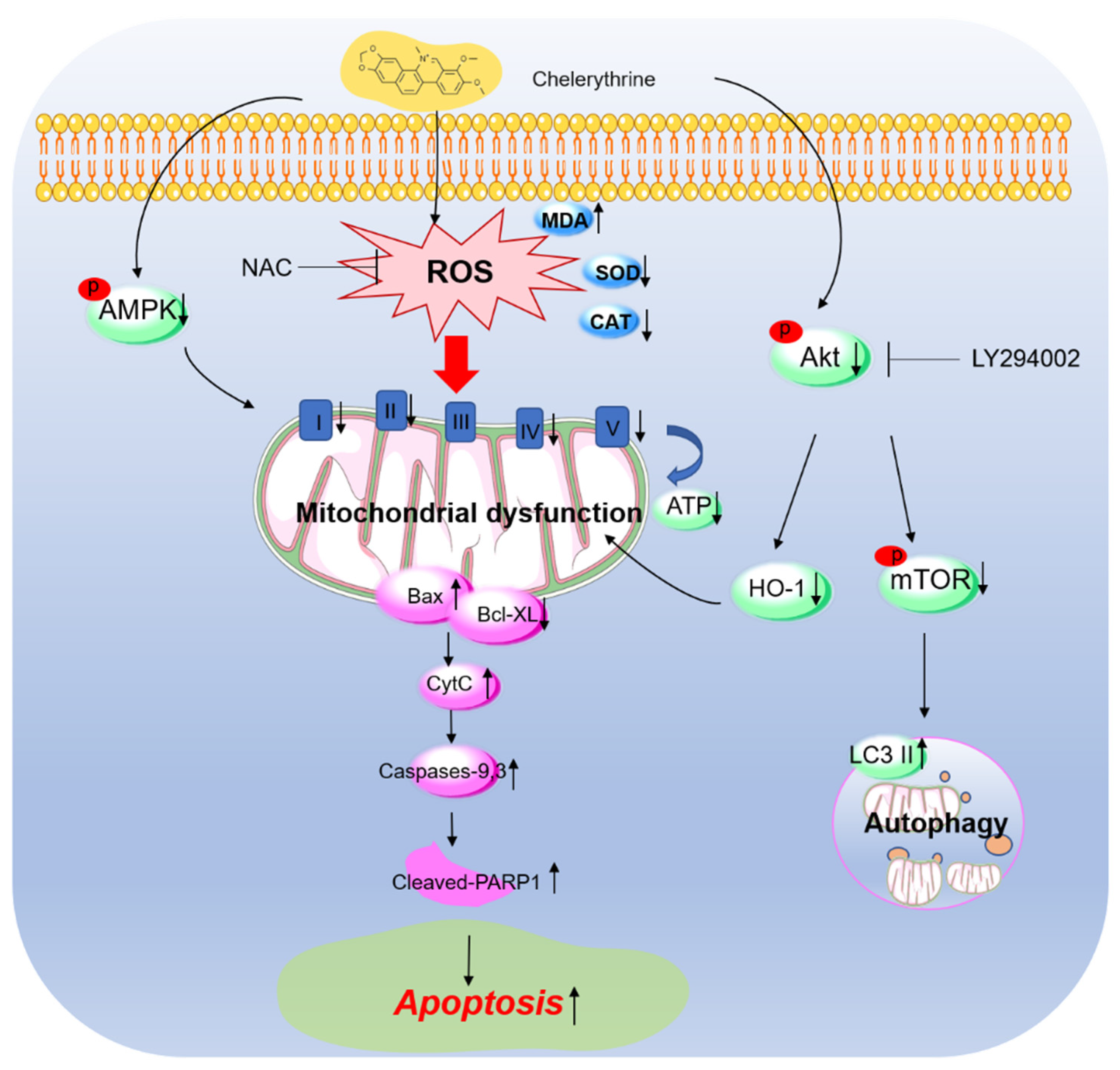
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cai, P.; Qiu, H.; Qi, F.; Zhang, X. The toxicity and safety of traditional Chinese medicines: Please treat with rationality. Biosci. Trends 2019, 13, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Yip, T.C.; Lee, H.W.; Chan, W.K.; Wong, G.L.; Wong, V.W. Asian perspective on NAFLD-associated HCC. J. Hepatol. 2022, 76, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Xie, Y.; Guo, M.; Rosner, M.H.; Yang, H.; Ronco, C. Nephrotoxicity and Chinese Herbal Medicine. Clin. J. Am. Soc. Nephrol. 2018, 13, 1605–1611. [Google Scholar] [CrossRef]
- Brown, A.C. Kidney toxicity related to herbs and dietary supplements: Online table of case reports. Part 3 of 5 series. Food Chem. Toxicol. 2017, 107, 502–519. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.L.; Bosisio, E. Pharmacological activities of Chelidonium majus L. (Papaveraceae). Pharmacol. Res. 1996, 33, 127–134. [Google Scholar] [CrossRef]
- Zielińska, S.; Wójciak-Kosior, M.; Dziągwa-Becker, M.; Gleńsk, M.; Sowa, I.; Fijałkowski, K.; Rurańska-Smutnicka, D.; Matkowski, A.; Junka, A. The Activity of Isoquinoline Alkaloids and Extracts from Chelidonium majus against Pathogenic Bacteria and Candida sp. Toxins 2019, 11, 406. [Google Scholar] [CrossRef] [PubMed]
- Nile, S.H.; Wang, H.; Nile, A.; Lin, X.; Dong, H.; Venkidasamy, B.; Sieniawska, E.; Enkhtaivan, G.; Kai, G. Comparative analysis of metabolic variations, antioxidant potential and cytotoxic effects in different parts of Chelidonium majus L. Food Chem. Toxicol. 2021, 156, 112483. [Google Scholar] [CrossRef] [PubMed]
- Gardin, N.E.; Braga, A.J. Greater celandine (Chelidonium majus L.) for COVID-19: A twenty-case series. Phytother. Res. 2021, 35, 3792–3798. [Google Scholar] [CrossRef] [PubMed]
- Capistrano, I.R.; Wouters, A.; Lardon, F.; Gravekamp, C.; Apers, S.; Pieters, L. In vitro and in vivo investigations on the antitumour activity of Chelidonium majus. Phytomedicine Int. J. Phytother. Phytopharm. 2015, 22, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, S.; Czerwińska, M.E.; Dziągwa-Becker, M.; Dryś, A.; Kucharski, M.; Jezierska-Domaradzka, A.; Płachno, B.J.; Matkowski, A. Modulatory Effect of Chelidonium majus Extract and Its Alkaloids on LPS-Stimulated Cytokine Secretion in Human Neutrophils. Molecules 2020, 25, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warowicka, A.; Popenda, Ł.; Bartkowiak, G.; Musidlak, O.; Litowczenko-Cybulska, J.; Kuźma, D.; Nawrot, R.; Jurga, S.; Goździcka-Józefiak, A. Protoberberine compounds extracted from Chelidonium majus L. as novel natural photosensitizers for cancer therapy. Phytomedicine Int. J. Phytother. Phytopharm. 2019, 64, 152919. [Google Scholar] [CrossRef] [PubMed]
- Pantano, F.; Mannocchi, G.; Marinelli, E.; Gentili, S.; Graziano, S.; Busardò, F.P.; di Luca, N.M. Hepatotoxicity induced by greater celandine (Chelidonium majus L.): A review of the literature. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 46–52. [Google Scholar] [PubMed]
- Teschke, R.; Glass, X.; Schulze, J. Herbal hepatotoxicity by Greater Celandine (Chelidonium majus): Causality assessment of 22 spontaneous reports. Regul. Toxicol. Pharmacol. 2011, 61, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, X.; Xu, M.; Liu, Y.; Di, X. Intracellular Accumulation as an Indicator of Cytotoxicity to Screen Hepatotoxic Components of Chelidonium majus L. by LC-MS/MS. Molecules 2019, 24, 2410. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhu, C.; Wang, Y.; Xu, J.; Xue, R.; Ji, F.; Wu, Y.; Wu, Z.; Zhang, W.; Zheng, Z.; et al. Evaluation the binding of chelerythrine, a potentially harmful toxin, with bovine serum albumin. Food Chem. Toxicol. 2020, 135, 110933. [Google Scholar] [CrossRef] [PubMed]
- Chmura, S.J.; Dolan, M.E.; Cha, A.; Mauceri, H.J.; Kufe, D.W.; Weichselbaum, R.R. In vitro and in vivo activity of protein kinase C inhibitor chelerythrine chloride induces tumor cell toxicity and growth delay in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 737–742. [Google Scholar]
- Wan, K.F.; Chan, S.L.; Sukumaran, S.K.; Lee, M.C.; Yu, V.C. Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. J. Biol. Chem. 2008, 283, 8423–8433. [Google Scholar] [CrossRef]
- Yamamoto, S.; Seta, K.; Morisco, C.; Vatner, S.F.; Sadoshima, J. Chelerythrine rapidly induces apoptosis through generation of reactive oxygen species in cardiac myocytes. J. Mol. Cell. Cardiol. 2001, 33, 1829–1848. [Google Scholar] [CrossRef] [PubMed]
- Medvetz, D.; Sun, Y.; Li, C.; Khabibullin, D.; Balan, M.; Parkhitko, A.; Priolo, C.; Asara, J.M.; Pal, S.; Yu, J.; et al. High-throughput drug screen identifies chelerythrine as a selective inducer of death in a TSC2-null setting. Mol. Cancer Res. 2015, 13, 50–62. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zhuo, R.; Dai, J.; Wang, X.; Huang, X.; Wang, H.; Xu, D. Chelerythrine induces apoptosis via ROS-mediated endoplasmic reticulum stress and STAT3 pathways in human renal cell carcinoma. J. Cell. Mol. Med. 2020, 24, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Vrba, J.; Dolezel, P.; Vicar, J.; Modrianský, M.; Ulrichová, J. Chelerythrine and dihydrochelerythrine induce G1 phase arrest and bimodal cell death in human leukemia HL-60 cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2008, 22, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.P.; Hagihara, M.; Nakatani, K.; Jiang, Z.H. Recognition of chelerythrine to human telomeric DNA and RNA G-quadruplexes. Sci. Rep. 2014, 4, 6767. [Google Scholar] [CrossRef]
- Tang, Z.H.; Cao, W.X.; Wang, Z.Y.; Lu, J.H.; Liu, B.; Chen, X.; Lu, J.J. Induction of reactive oxygen species-stimulated distinctive autophagy by chelerythrine in non-small cell lung cancer cells. Redox Biol. 2017, 12, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Li, H.; Wang, Y.; Tang, S.; Velkov, T.; Shen, J. Inhibition of Oxidative Stress and ALOX12 and NF-κB Pathways Contribute to the Protective Effect of Baicalein on Carbon Tetrachloride-Induced Acute Liver Injury. Antioxidants 2021, 10, 976. [Google Scholar] [CrossRef]
- Dai, C.; Li, M.; Sun, T.; Zhang, Y.; Wang, Y.; Shen, Z.; Velkov, T.; Tang, S.; Shen, J. Colistin-induced pulmonary toxicity involves the activation of NOX4/TGF-β/mtROS pathway and the inhibition of Akt/mTOR pathway. Food Chem. Toxicol. 2022, 163, 112966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, T.; Li, M.; Lin, Y.; Liu, Y.; Tang, S.; Dai, C. Ivermectin-Induced Apoptotic Cell Death in Human SH-SY5Y Cells Involves the Activation of Oxidative Stress and Mitochondrial Pathway and Akt/mTOR-Pathway-Mediated Autophagy. Antioxidants 2022, 11, 908. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Ciccotosto, G.D.; Cappai, R.; Tang, S.; Li, D.; Xie, S.; Xiao, X.; Velkov, T. Curcumin Attenuates Colistin-Induced Neurotoxicity in N2a Cells via Anti-inflammatory Activity, Suppression of Oxidative Stress, and Apoptosis. Mol. Neurobiol. 2018, 55, 421–434. [Google Scholar] [CrossRef]
- Dai, C.; Li, B.; Zhou, Y.; Li, D.; Zhang, S.; Li, H.; Xiao, X.; Tang, S. Curcumin attenuates quinocetone induced apoptosis and inflammation via the opposite modulation of Nrf2/HO-1 and NF-kB pathway in human hepatocyte L02 cells. Food Chem. Toxicol. 2016, 95, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; He, X.; Yi, Z.; Xiang, W.; Ding, Y. Chelidonine suppresses LPS-Induced production of inflammatory mediators through the inhibitory of the TLR4/NF-κB signaling pathway in RAW264.7 macrophages. Biomed. Pharmacother. 2018, 107, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Moro, P.A.; Cassetti, F.; Giugliano, G.; Falce, M.T.; Mazzanti, G.; Menniti-Ippolito, F.; Raschetti, R.; Santuccio, C. Hepatitis from Greater celandine (Chelidonium majus L.): Review of literature and report of a new case. J. Ethnopharmacol. 2009, 124, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Frenzel, C.; Glass, X.; Schulze, J.; Eickhoff, A. Greater Celandine hepatotoxicity: A clinical review. Ann. Hepatol. 2012, 11, 838–848. [Google Scholar] [CrossRef]
- Ulrichová, J.; Walterová, D.; Vavrecková, C.; Kamarád, V.; Símánek, V.m. Cytotoxicity of Benzo[c]phenanthridinium Alkaloids in Isolated Rat Hepatocytes. Phytother. Res. 1996, 10, 220–223. [Google Scholar] [CrossRef]
- Yang, T.; Xu, R.; Su, Q.; Wang, H.; Liu, F.; Dai, B.; Wang, B.; Zhang, Y. Chelerythrine hydrochloride inhibits proliferation and induces mitochondrial apoptosis in cervical cancer cells via PI3K/BAD signaling pathway. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2020, 68, 104965. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Q.; Li, M.; Tang, S.; Dai, C. T-2 Toxin Induces Apoptotic Cell Death and Protective Autophagy in Mouse Microglia BV2 Cells. J. Fungi 2022, 8, 761. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Tang, S.; Dai, C.; Zhou, Y.; Yang, X.; Li, D.; Xiao, X. P21(Waf1/Cip1) plays a critical role in furazolidone-induced apoptosis in HepG2 cells through influencing the caspase-3 activation and ROS generation. Food Chem. Toxicol. 2016, 88, 1–12. [Google Scholar] [CrossRef]
- Dai, C.; Liu, Q.; Li, D.; Sharma, G.; Xiong, J.; Xiao, X. Molecular Insights of Copper Sulfate Exposure-Induced Nephrotoxicity: Involvement of Oxidative and Endoplasmic Reticulum Stress Pathways. Biomolecules 2020, 10, 1010. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Li, D.; Gong, L.; Xiao, X.; Tang, S. Curcumin Ameliorates Furazolidone-Induced DNA Damage and Apoptosis in Human Hepatocyte L02 Cells by Inhibiting ROS Production and Mitochondrial Pathway. Molecules 2016, 21, 1061. [Google Scholar] [CrossRef]
- Xie, Y.J.; Gao, W.N.; Wu, Q.B.; Yao, X.J.; Jiang, Z.B.; Wang, Y.W.; Wang, W.J.; Li, W.; Hussain, S.; Liu, L.; et al. Chelidonine selectively inhibits the growth of gefitinib-resistant non-small cell lung cancer cells through the EGFR-AMPK pathway. Pharmacol. Res. 2020, 159, 104934. [Google Scholar] [CrossRef]
- Dai, C.; Tang, S.; Li, D.; Zhao, K.; Xiao, X. Curcumin attenuates quinocetone-induced oxidative stress and genotoxicity in human hepatocyte L02 cells. Toxicol. Mech. Methods 2015, 25, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Kemény-Beke, A.; Aradi, J.; Damjanovich, J.; Beck, Z.; Facskó, A.; Berta, A.; Bodnár, A. Apoptotic response of uveal melanoma cells upon treatment with chelidonine, sanguinarine and chelerythrine. Cancer Lett. 2006, 237, 67–75. [Google Scholar] [CrossRef]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef]
- Kumar, S.; Acharya, A. Chelerythrine induces reactive oxygen species-dependent mitochondrial apoptotic pathway in a murine T cell lymphoma. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 129–140. [Google Scholar] [CrossRef]
- Vallejos, R.H.; Rizzotto, M.G. Effect of chelerythrine on mitochondrial energy coupling. FEBS Lett. 1972, 21, 195–198. [Google Scholar] [CrossRef]
- Funakoshi, T.; Aki, T.; Nakayama, H.; Watanuki, Y.; Imori, S.; Uemura, K. Reactive oxygen species-independent rapid initiation of mitochondrial apoptotic pathway by chelerythrine. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2011, 25, 1581–1587. [Google Scholar] [CrossRef]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef]
- Mbaya, E.; Oulès, B.; Caspersen, C.; Tacine, R.; Massinet, H.; Pennuto, M.; Chrétien, D.; Munnich, A.; Rötig, A.; Rizzuto, R.; et al. Calcium signalling-dependent mitochondrial dysfunction and bioenergetics regulation in respiratory chain Complex II deficiency. Cell Death Differ. 2010, 17, 1855–1866. [Google Scholar] [CrossRef]
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Dürr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, H.; Xu, A.; Ross, T.; Bowler, E.; Hu, Y.; Lesnefsky, E.J. Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria. PLoS ONE 2015, 10, e0118834. [Google Scholar] [CrossRef]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Gahl, R.F.; Dwivedi, P.; Tjandra, N. Bcl-2 proteins bid and bax form a network to permeabilize the mitochondria at the onset of apoptosis. Cell Death Dis. 2016, 7, e2424. [Google Scholar] [CrossRef]
- Chan, S.L.; Lee, M.C.; Tan, K.O.; Yang, L.K.; Lee, A.S.; Flotow, H.; Fu, N.Y.; Butler, M.S.; Soejarto, D.D.; Buss, A.D.; et al. Identification of chelerythrine as an inhibitor of BclXL function. J. Biol. Chem. 2003, 278, 20453–20456. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.F.; Guo, Y.; Zhang, J.B.; Wei, X.H. Induction of apoptosis by chelerythrine chloride through mitochondrial pathway and Bcl-2 family proteins in human hepatoma SMMC-7721 cell. Arch. Pharmacal Res. 2011, 34, 791–800. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Pi, H.; Li, M.; Zou, L.; Yang, M.; Deng, P.; Fan, T.; Liu, M.; Tian, L.; Tu, M.; Xie, J.; et al. AKT inhibition-mediated dephosphorylation of TFE3 promotes overactive autophagy independent of MTORC1 in cadmium-exposed bone mesenchymal stem cells. Autophagy 2019, 15, 565–582. [Google Scholar] [CrossRef]
- Wang, J.; Yang, C.; Yuan, Z.; Yi, J.; Wu, J. T-2 Toxin Exposure Induces Apoptosis in TM3 Cells by Inhibiting Mammalian Target of Rapamycin/Serine/Threonine Protein Kinase(mTORC2/AKT) to Promote Ca(2+)Production. Int. J. Mol. Sci. 2018, 19, 3360. [Google Scholar] [CrossRef]
- Dai, C.; Ciccotosto, G.D.; Cappai, R.; Wang, Y.; Tang, S.; Hoyer, D.; Schneider, E.K.; Velkov, T.; Xiao, X. Rapamycin Confers Neuroprotection against Colistin-Induced Oxidative Stress, Mitochondria Dysfunction, and Apoptosis through the Activation of Autophagy and mTOR/Akt/CREB Signaling Pathways. ACS Chem. Neurosci. 2018, 9, 824–837. [Google Scholar] [CrossRef]
- Deng, S.; Tang, S.; Zhang, S.; Zhang, C.; Wang, C.; Zhou, Y.; Dai, C.; Xiao, X. Furazolidone induces apoptosis through activating reactive oxygen species-dependent mitochondrial signaling pathway and suppressing PI3K/Akt signaling pathway in HepG2 cells. Food Chem. Toxicol. 2015, 75, 173–186. [Google Scholar] [CrossRef]
- Hao, Y.; Li, Y.; Liu, J.; Wang, Z.; Gao, B.; Zhang, Y.; Wang, J. Protective Effect of Chrysanthemum morifolium cv. Fubaiju Hot-Water Extracts Against ARPE-19 Cell Oxidative Damage by Activating PI3K/Akt-Mediated Nrf2/HO-1 Signaling Pathway. Front. Nutr. 2021, 8, 648973. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L.C.; Zhao, R.J.; Pu, L.M.; Chen, K.Y.; Nasim, A.A.; Leung, E.L.; Fan, X.X. Chelerythrine ameliorates rheumatoid arthritis by modulating the AMPK/mTOR/ULK-1 signaling pathway. Phytomedicine Int. J. Phytother. Phytopharm. 2022, 104, 154140. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Zhang, Q.; Xie, B.; Jiang, H.; Shen, J.; Tang, S.; Dai, C. Chelerythrine-Induced Apoptotic Cell Death in HepG2 Cells Involves the Inhibition of Akt Pathway and the Activation of Oxidative Stress and Mitochondrial Apoptotic Pathway. Antioxidants 2022, 11, 1837. https://doi.org/10.3390/antiox11091837
Lin Y, Zhang Q, Xie B, Jiang H, Shen J, Tang S, Dai C. Chelerythrine-Induced Apoptotic Cell Death in HepG2 Cells Involves the Inhibition of Akt Pathway and the Activation of Oxidative Stress and Mitochondrial Apoptotic Pathway. Antioxidants. 2022; 11(9):1837. https://doi.org/10.3390/antiox11091837
Chicago/Turabian StyleLin, Yanling, Qinzhi Zhang, Baofu Xie, Haiyang Jiang, Jianzhong Shen, Shusheng Tang, and Chongshan Dai. 2022. "Chelerythrine-Induced Apoptotic Cell Death in HepG2 Cells Involves the Inhibition of Akt Pathway and the Activation of Oxidative Stress and Mitochondrial Apoptotic Pathway" Antioxidants 11, no. 9: 1837. https://doi.org/10.3390/antiox11091837