
Sequential Accumulation of ‘Driver’ Pathway Mutations Induces the Upregulation of Hydrogen-Sulfide-Producing Enzymes in Human Colonic Epithelial Cell Organoids
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents and Antibodies
2.3. Western Blotting
2.4. Quantification of H2S Production
2.5. Proliferation Assays
2.6. Cellular Bioenergetic Measurements
2.7. Statistical Analysis
3. Results and Discussion
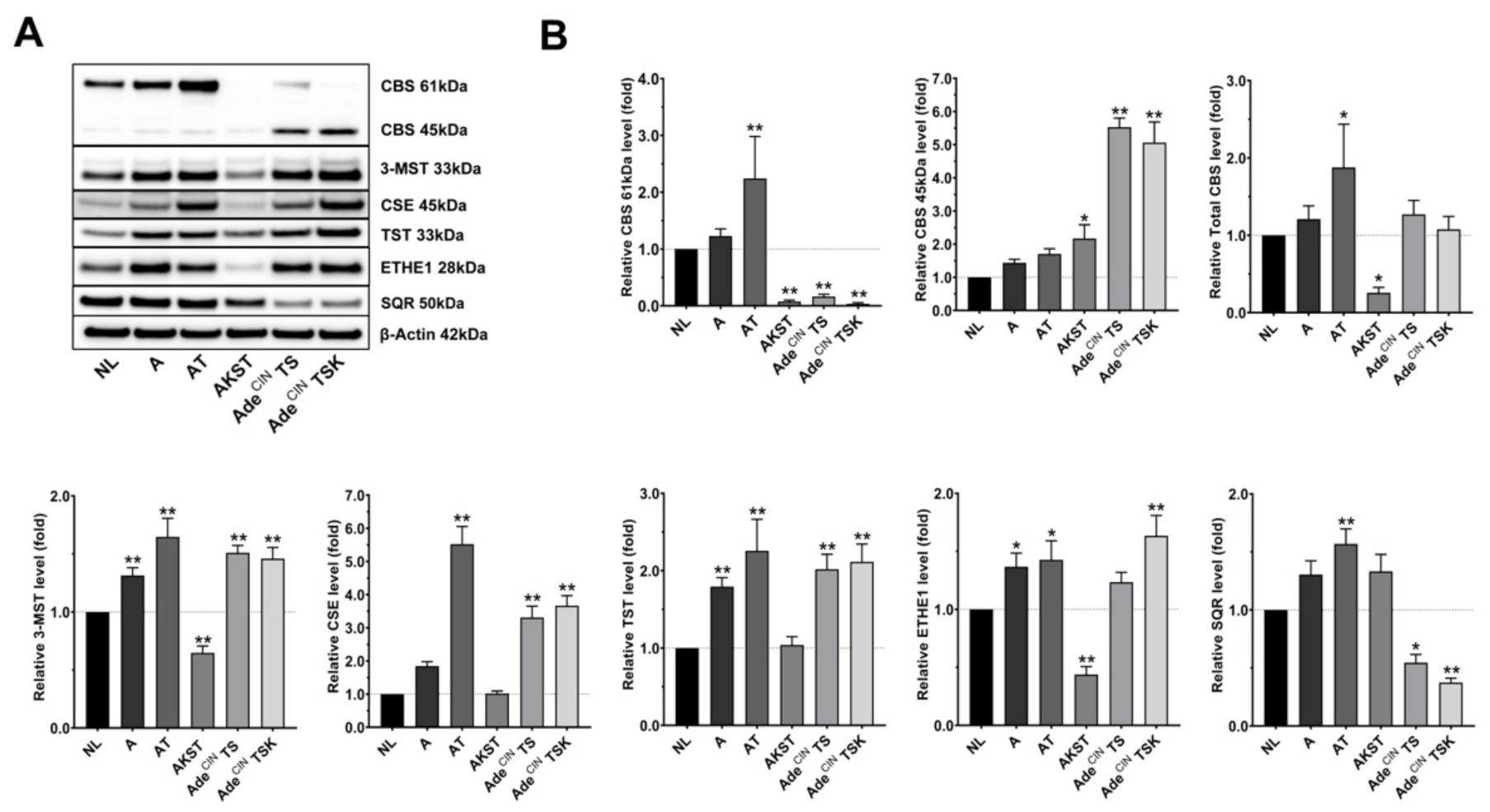
3.1. Upregulation of Hydrogen-Sulfide-Producing Enzymes Occurs during the Accumulation of ‘Driver’ Pathway Mutations
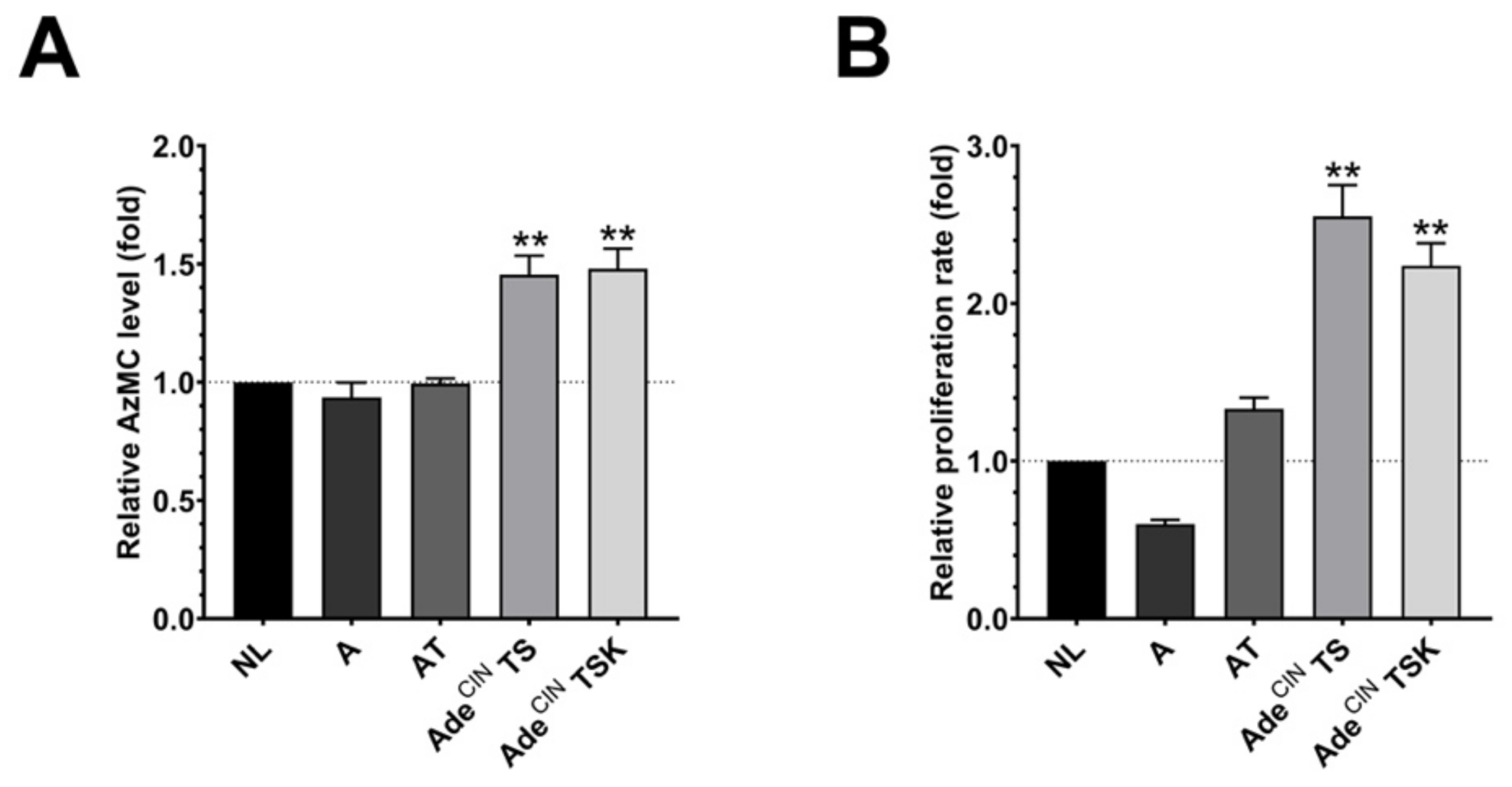
3.2. H2S Production Is Increased in AdeCIN TS and AdeCIN TSK Organoids and Correlates with Increased Proliferation
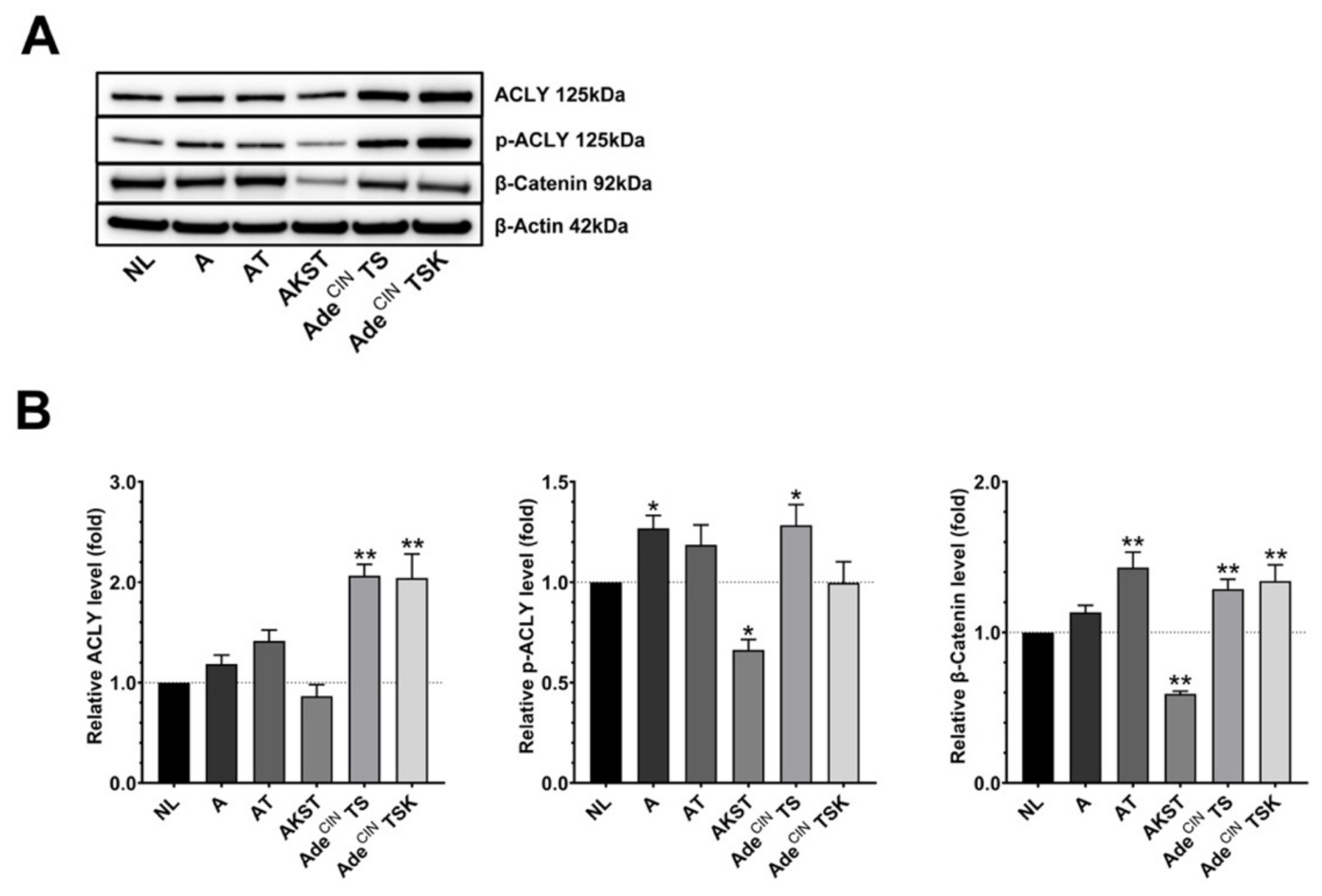
3.3. Upregulation of the Wnt/β-Catenin Pathway Occurs during the Accumulation of ‘Driver’ Pathway Mutations
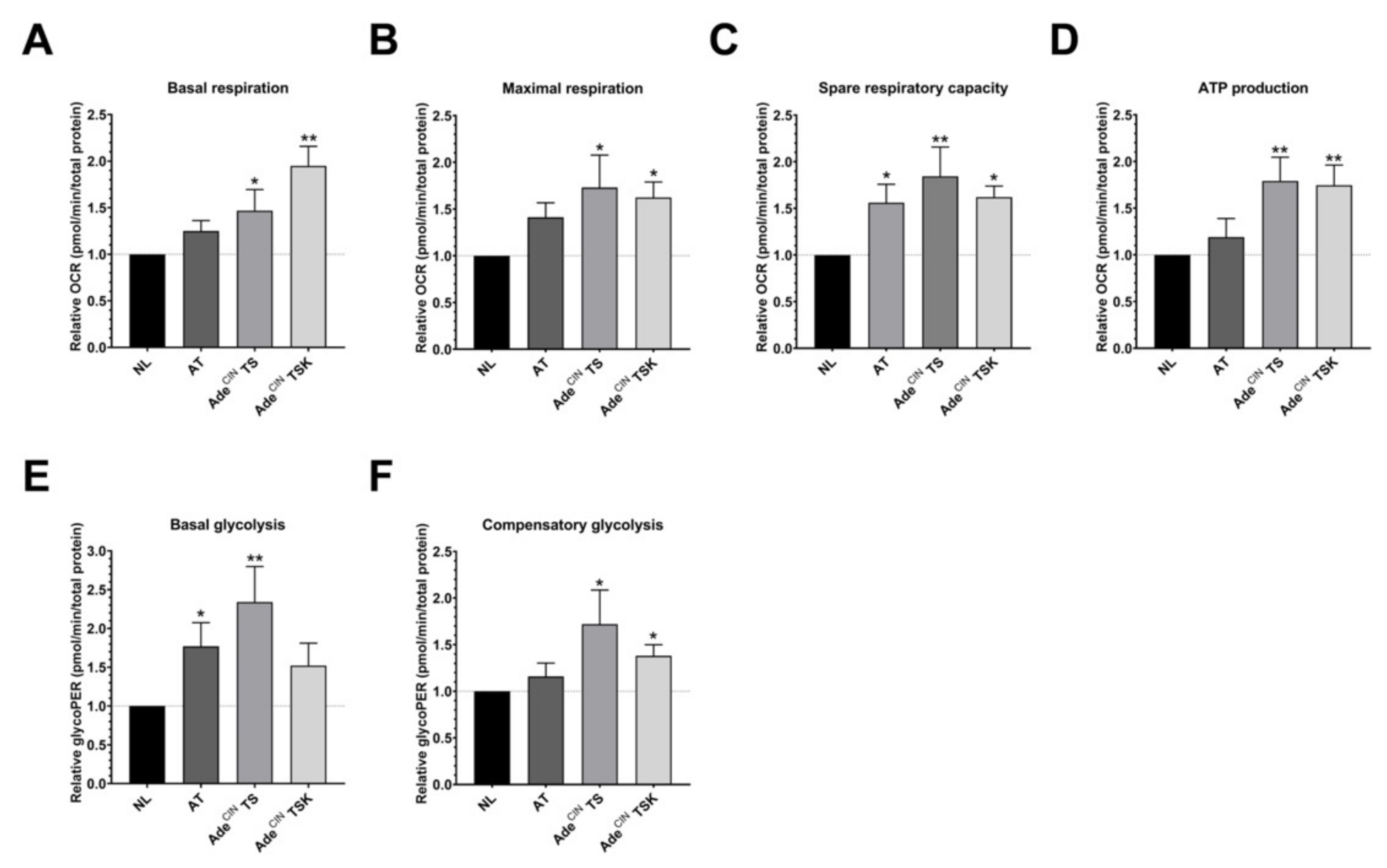
3.4. Upregulation of Cellular Bioenergetics and Glycolysis in AdeCIN TS and AdeCIN TSK Organoids
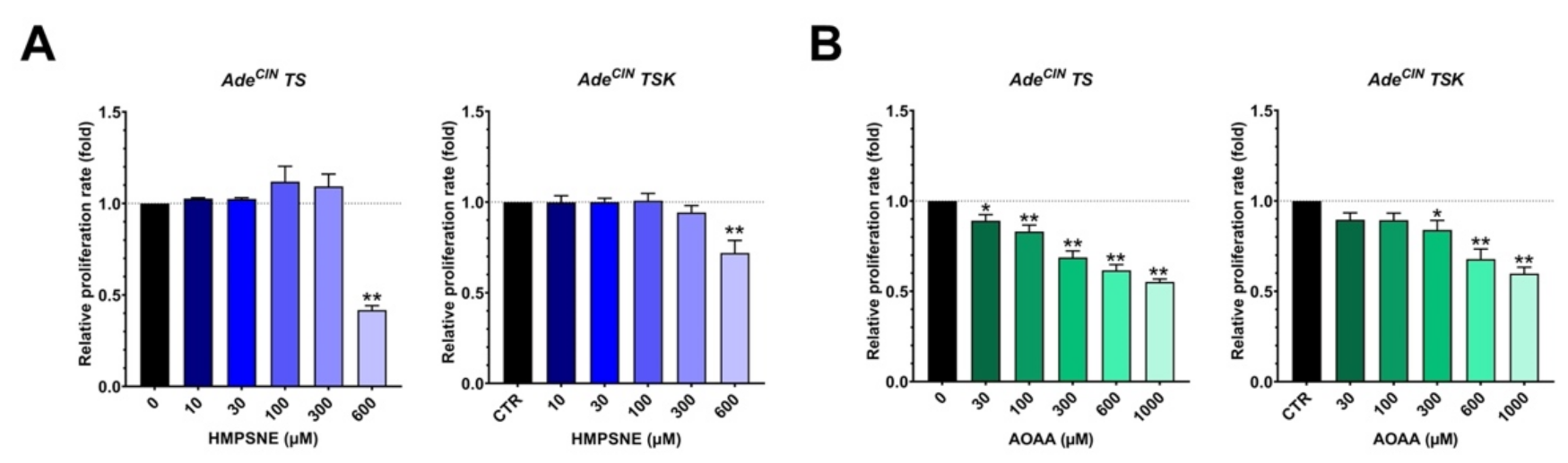
3.5. Pharmacological Inhibition of H2S Biosynthesis in AdeCIN TS and AdeCIN TSK Suppresses Cell Proliferation
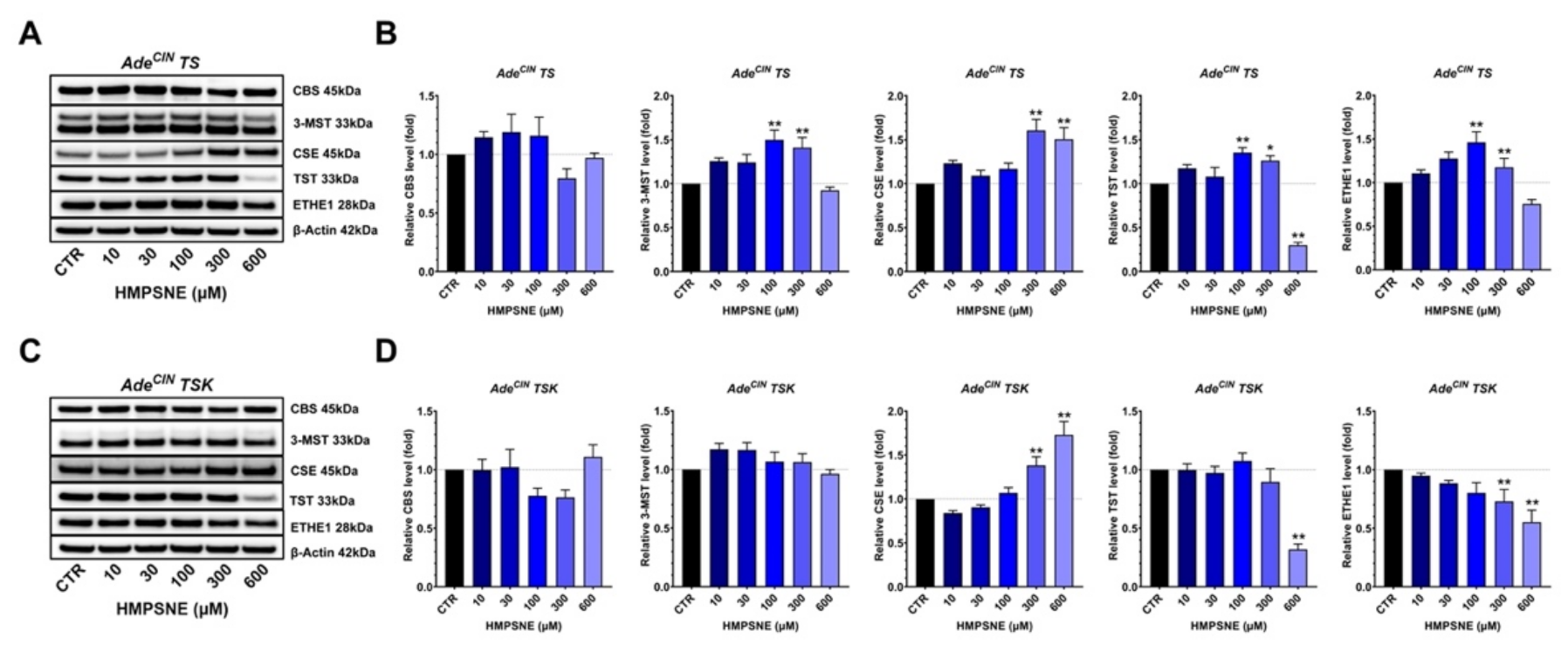
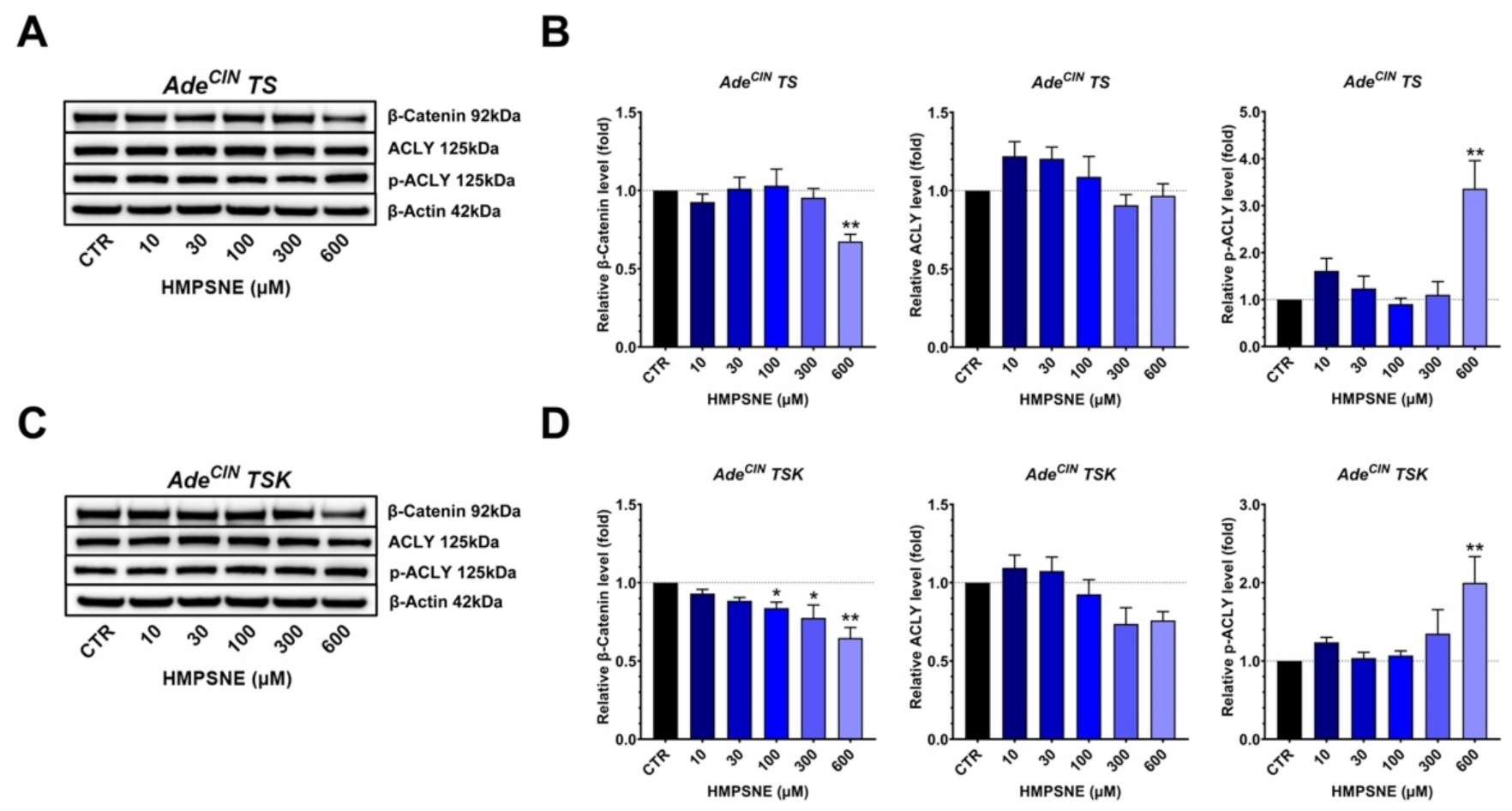
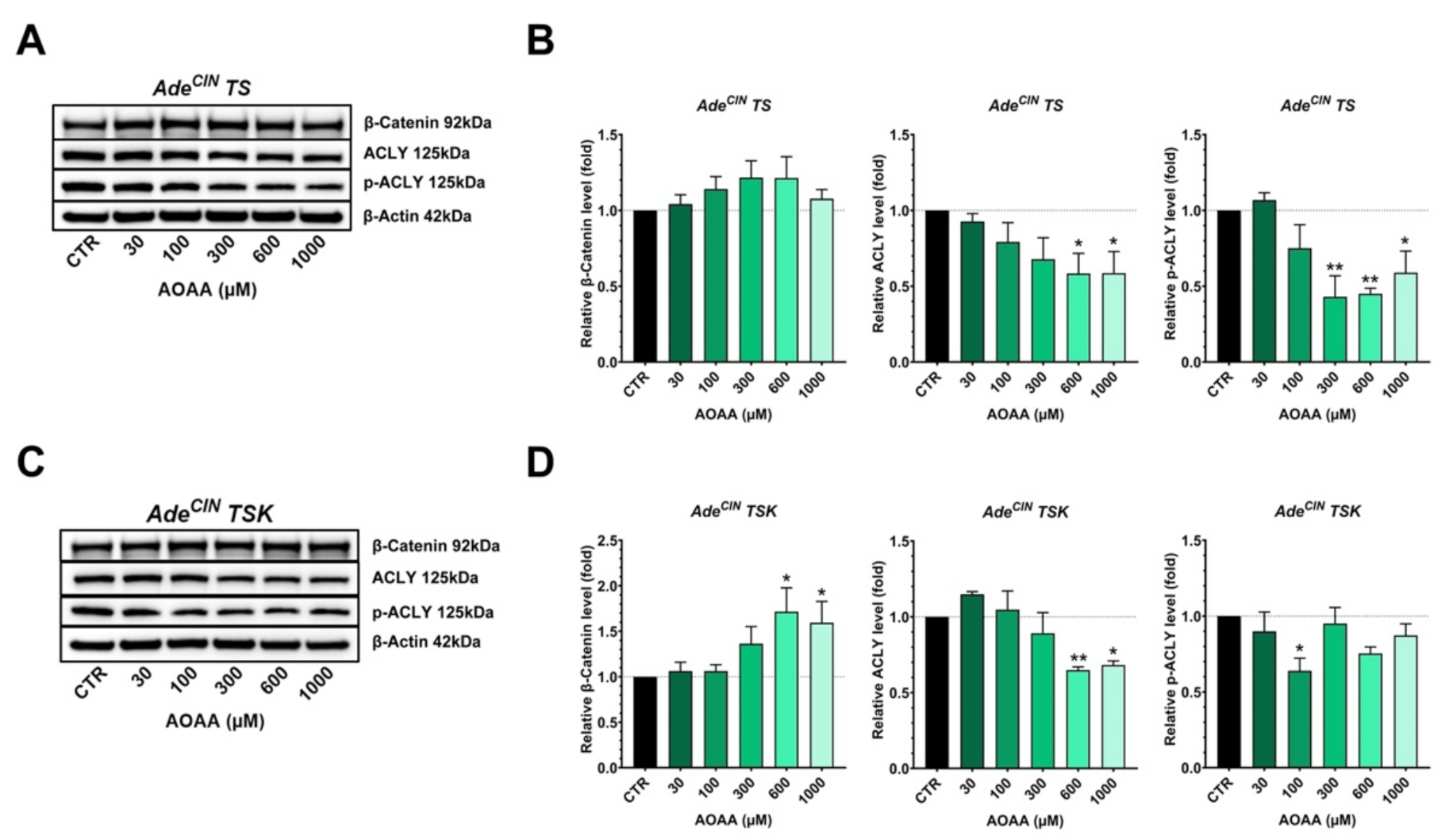
3.6. Effect of 3-MST Inhibition on the Expression of Various H2S-Producing and H2S-Metabolizing Enzymes and on the Wnt/β-Catenin Pathway in AdeCIN TS and AdeCIN TSK Organoids
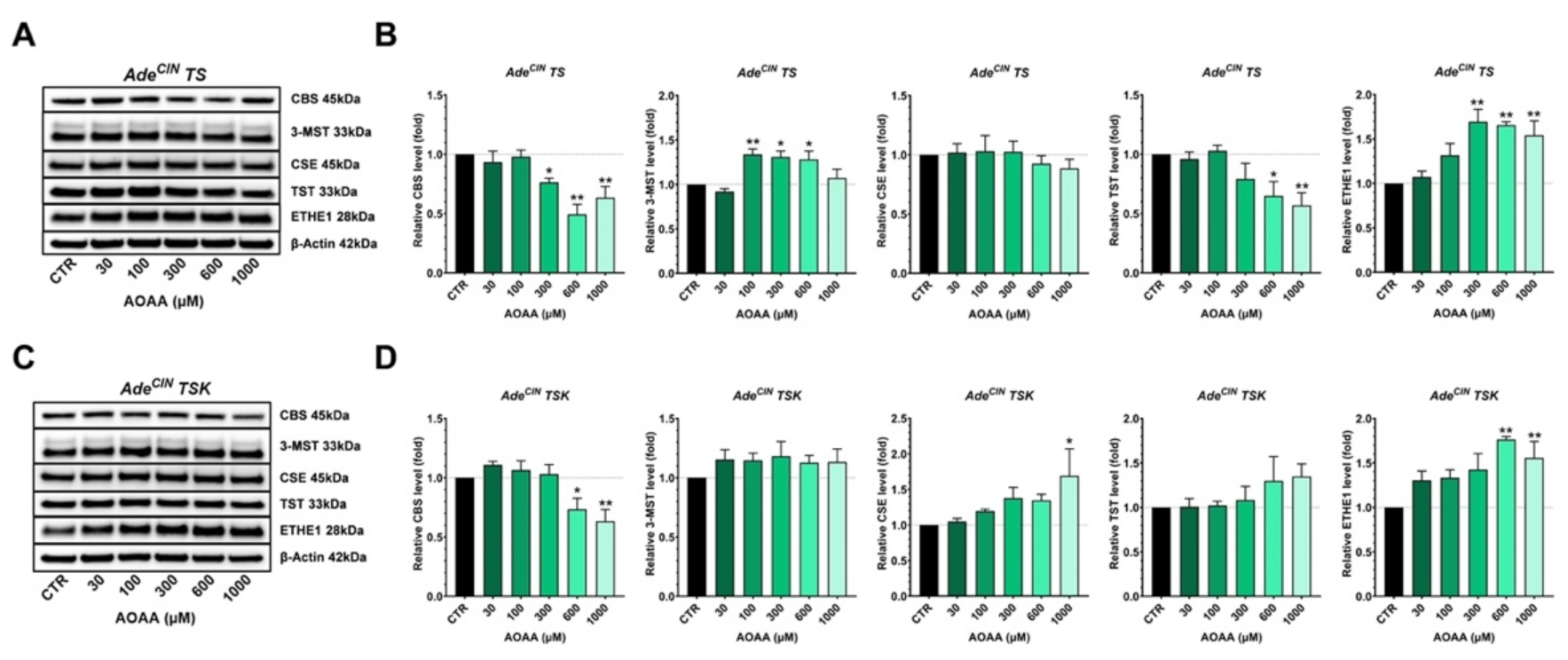
3.7. Effect of CBS Inhibition on the Expression of Various H2S-Producing and H2S-Metabolizing Enzymes and on the Wnt/β-catenin Pathway in AdeCIN TS and AdeCIN TSK Organoids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Ascenção, K.; Szabo, C. Emerging roles of cystathionine β-synthase in various forms of cancer. Redox. Biol. 2022, 53, 102331. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, A.; Zulfiqar, M.; Weindel, M.; Shidham, V.B. Molecular diagnostics in colorectal carcinoma. Clin. Lab. Med. 2013, 33, 835–859. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus molecular subtypes of colorectal cancer and their clinical implications. Int. Biol. Biomed. J. 2017, 3, 105–111. [Google Scholar] [PubMed]
- Smit, W.L.; Spaan, C.N.; Johannes de Boer, R.; Ramesh, P.; Martins Garcia, T.; Meijer, B.J.; Vermeulen, J.L.M.; Lezzerini, M.; MacInnes, A.W.; Koster, J.; et al. Driver mutations of the adenoma-carcinoma sequence govern the intestinal epithelial global translational capacity. Proc. Natl. Acad. Sci. USA 2020, 117, 25560–25570. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef]
- Hanaoka, K.; Sasakura, K.; Suwanai, Y.; Toma-Fukai, S.; Shimamoto, K.; Takano, Y.; Shibuya, N.; Terai, T.; Komatsu, T.; Ueno, T.; et al. Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide. Sci. Rep. 2017, 7, 40227. [Google Scholar] [CrossRef] [Green Version]
- Ascencao, K.; Dilek, N.; Augsburger, F.; Panagaki, T.; Zuhra, K.; Szabo, C. Pharmacological induction of mesenchymal-epithelial transition via inhibition of H2S biosynthesis and consequent suppression of ACLY activity in colon cancer cells. Pharmacol. Res. 2021, 165, 105393. [Google Scholar] [CrossRef]
- Augsburger, F.; Randi, E.B.; Jendly, M.; Ascencao, K.; Dilek, N.; Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation, migration, and bioenergetics in murine colon cancer cells. Biomolecules 2020, 10, 447. [Google Scholar] [CrossRef]
- Panagaki, T.; Randi, E.B.; Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation and cellular bioenergetics in human Down syndrome fibroblasts. Biomolecules 2020, 10, 653. [Google Scholar] [CrossRef]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef]
- Kery, V.; Poneleit, L.; Kraus, J.P. Trypsin cleavage of human cystathionine beta-synthase into an evolutionarily conserved active core: Structural and functional consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martínez-Cruza, L.A. Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration. Proc. Natl. Acad. Sci. USA 2013, 110, E3790–E3799. [Google Scholar] [CrossRef]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular regulation and pharmacological inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide 2014, 41, 146–156. [Google Scholar] [CrossRef]
- Panagaki, T.; Lozano-Montes, L.; Janickova, L.; Zuhra, K.; Szabo, M.P.; Majtan, T.; Rainer, G.; Maréchal, D.; Herault, Y.; Szabo, C. Overproduction of hydrogen sulfide, generated by cystathionine β-synthase, disrupts brain wave patterns and contributes to neurobehavioral dysfunction in a rat model of Down syndrome. Redox. Biol. 2022, 51, 102233. [Google Scholar] [CrossRef]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of cystathionine-β-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular portrait of metastasis-competent circulating tumor cells in colon cancer reveals the crucial role of genes regulating energy metabolism and DNA repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Oláh, G.; Módis, K.; Törö, G.; Hellmich, M.R.; Szczesny, B.; Szabo, C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem. Pharmacol. 2018, 149, 186–204. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Chao, C.; Módis, K.; Ding, Y.; Zatarain, J.R.; Thanki, K.; Maskey, M.; Druzhyna, N.; Untereiner, A.A.; Ahmad, A.; et al. Efficacy of novel aminooxyacetic acid prodrugs in colon cancer models: Towards clinical translation of the cystathionine β-synthase inhibition concept. Biomolecules 2021, 11, 1073. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Li, J.; Huang, Z.; Yue, T.; Zhu, J.; Wang, X.; Liu, Y.; Wang, P.; Chen, S. The CBS-H2S axis promotes liver metastasis of colon cancer by upregulating VEGF through AP-1 activation. Br. J. Cancer 2022, 126, 1055–1066. [Google Scholar] [CrossRef]
- Zuhra, K.; Tomé, C.S.; Masi, L.; Giardina, G.; Paulini, G.; Malagrinò, F.; Forte, E.; Vicente, J.B.; Giuffrè, A. N-acetylcysteine serves as substrate of 3-mercaptopyruvate sulfurtransferase and stimulates sulfide metabolism in colon cancer cells. Cells 2019, 8, 828. [Google Scholar] [CrossRef]
- Bantzi, M.; Augsburger, F.; Loup, J.; Berset, Y.; Vasilakaki, S.; Myrianthopoulos, V.; Mikros, E.; Szabo, C.; Bochet, C.G. Novel aryl-substituted pyrimidones as inhibitors of 3-mercaptopyruvate sulfurtransferase with antiproliferative efficacy in colon cancer. J. Med. Chem. 2021, 64, 6221–6240. [Google Scholar] [CrossRef]
- Ramasamy, S.; Singh, S.; Taniere, P.; Langman, M.J.; Eggo, M.C. Sulfide-detoxifying enzymes in the human colon are decreased in cancer and upregulated in differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G288–G296. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef] [Green Version]
- Malagrinò, F.; Zuhra, K.; Mascolo, L.; Mastronicola, D.; Vicente, J.B.; Forte, E.; Giuffrè, A. Hydrogen sulfide oxidation: Adaptive changes in mitochondria of SW480 colorectal cancer cells upon exposure to hypoxia. Oxid. Med. Cell. Longev. 2019, 2019, 8102936. [Google Scholar] [CrossRef]
- Koj, A.; Frendo, J.; Janik, Z. [35S]thiosulphate oxidation by rat liver mitochondria in the presence of glutathione. Biochem. J. 1967, 103, 791–795. [Google Scholar] [CrossRef]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Ogasawara, Y.; Kimura, H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 2011, 439, 479–485. [Google Scholar] [CrossRef]
- Seiler, N.; Schneider, Y.; Gossé, F.; Schleiffer, R.; Raul, F. Polyploidisation of metastatic colon carcinoma cells by microtubule and tubulin interacting drugs: Effect on proteolytic activity and invasiveness. Int. J. Oncol. 2004, 25, 1039–1048. [Google Scholar]
- Solakidi, S.; Tiniakos, D.G.; Petraki, K.; Stathopoulos, G.P.; Markaki, I.; Androulakis, G.; Sekeris, C.E. Co-expression of trypsin and tumour-associated trypsin inhibitor (TATI) in colorectal adenocarcinomas. Histol. Histopathol. 2003, 18, 1181–1188. [Google Scholar]
- Ji, K.; Mayernik, L.; Moin, K.; Sloane, B.F. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 103–112. [Google Scholar] [CrossRef]
- Buttacavoli, M.; Di Cara, G.; Roz, E.; Pucci-Minafra, I.; Feo, S.; Cancemi, P. Integrated multi-omics investigations of metalloproteinases in colon cancer: Focus on MMP2 and MMP9. Int. J. Mol. Sci. 2021, 22, 12389. [Google Scholar] [CrossRef]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular mechanisms of colon cancer progression and metastasis: Recent insights and advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef]
- Fan, K.; Li, N.; Qi, J.; Yin, P.; Zhao, C.; Wang, L.; Li, Z.; Zha, X. Wnt/β-catenin signaling induces the transcription of cystathionine-γ-lyase, a stimulator of tumor in colon cancer. Cell Signal. 2014, 26, 2801–2808. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen sulfide, an endogenous stimulator of mitochondrial function in cancer cells. Cells 2021, 10, 220. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Mukai, K.; Hishiki, T.; Kubo, A.; Ohmura, M.; Sugiura, Y.; Matsuura, T.; Nagahata, Y.; Hayakawa, N.; Yamamoto, T.; et al. Energy management by enhanced glycolysis in G1-phase in human colon cancer cells in vitro and in vivo. Mol. Cancer Res. 2013, 11, 973–985. [Google Scholar] [CrossRef]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol. Cell Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Ding, L.; Xie, Z.Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A review of hydrogen sulfide synthesis, metabolism, and measurement: Is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef]
- Shackelford, R.E.; Mohammad, I.Z.; Meram, A.T.; Kim, D.; Alotaibi, F.; Patel, S.; Ghali, G.E.; Kevil, C.G. Molecular functions of hydrogen sulfide in cancer. Pathophysiology 2021, 28, 437–456. [Google Scholar] [CrossRef]
- Khattak, S.; Rauf, M.A.; Khan, N.H.; Zhang, Q.Q.; Chen, H.J.; Muhammad, P.; Ansari, M.A.; Alomary, M.N.; Jahangir, M.; Zhang, C.Y.; et al. Hydrogen sulfide biology and its role in cancer. Molecules 2022, 27, 3389. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues and organs. Physiol. Rev. 2022, in press. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. International union of basic and clinical pharmacology. CII: Pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [Green Version]
- Maclean, K.N.; Janosík, M.; Kraus, E.; Kozich, V.; Allen, R.H.; Raab, B.K.; Kraus, J.P. Cystathionine beta-synthase is coordinately regulated with proliferation through a redox-sensitive mechanism in cultured human cells and Saccharomyces cerevisiae. J. Cell Physiol. 2002, 192, 81–92. [Google Scholar] [CrossRef]
- Maclean, K.N.; Kraus, E.; Kraus, J.P. The dominant role of Sp1 in regulating the cystathionine beta-synthase -1a and -1b promoters facilitates potential tissue-specific regulation by Kruppel-like factors. J. Biol. Chem. 2004, 279, 8558–8566. [Google Scholar] [CrossRef]
- Gupta, S.; Wang, L.; Anderl, J.; Slifker, M.J.; Kirk, C.; Kruger, W.D. Correction of cystathionine β-synthase deficiency in mice by treatment with proteasome inhibitors. Hum. Mutat. 2013, 34, 1085–1093. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, L.; Ren, Z.; Zhu, G.; Wang, W.; Jia, Y.; Peng, A.; Wang, X. Glucose-induced decrease of cystathionine β-synthase mediates renal injuries. FASEB J. 2021, 35, e21576. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xia, M.; Lin, M.; Yang, H.; Kuhlenkamp, J.; Li, T.; Sodir, N.M.; Chen, Y.H.; Josef-Lenz, H.; Laird, P.W.; et al. Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells. Gastroenterology 2007, 133, 207–218. [Google Scholar] [CrossRef]
- Zuhra, K.; Tomé, C.S.; Forte, E.; Vicente, J.B.; Giuffrè, A. The multifaceted roles of sulfane sulfur species in cancer-associated processes. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148338. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Hipólito, A.; Nunes, S.C.; Vicente, J.B.; Serpa, J. Cysteine aminotransferase (CAT): A pivotal sponsor in metabolic remodeling and an ally of 3-mercaptopyruvate sulfurtransferase (MST) in cancer. Molecules 2020, 25, 3984. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clones | Mutation(s) & Chromosomal Instability | Cancer Stage | CBS 45 kDa 1 | CBS 61 kDa 1 | 3-MST 1 | CSE 1 |
|---|---|---|---|---|---|---|
| NL | Normal control | Healthy | 100 | 100 | 100 | 100 |
| A | Loss of APC gene | Early adenoma | 143 ± 11 | 123 ± 12 | 131 ± 7 | 185 ± 13 |
| AT |
| Carcinoma | 170 ± 16 | 224 ± 74 | 165 ± 16 | 552 ± 53 |
| AKST |
| Carcinoma | 217 ± 42 | 8 ± 3 | 65 ± 6 | 102 ± 7 |
| AdeCIN TS |
| Carcinoma (non-metastatic) | 553 ± 27 | 16 ± 4 | 151 ± 6 | 331 ± 34 |
| AdeCIN TSK |
| Carcinoma (metastatic) | 506 ± 62 | 4 ± 2 | 146 ± 10 | 367 ± 30 |
| Adenoma (n = 5) | Primary Colorectal Cancer (n = 7) | Metastatic Colorectal Cancer (n = 2) | Organoid ‘A’ (n = 1) | Organoid ‘AKST’ (n = 2) | |
|---|---|---|---|---|---|
| CBS | 88 ± 24 | 118 ± 27 | 90 ± 10 | 112 | 69 ± 8 |
| 3-MST | 99 ± 7 | 95 ± 5 | 108 ± 8 | 144 | 100 ± 1 |
| CSE | 101 ± 21 | 96 ± 11 | 112 ± 25 | 95 | 80 ± 14 |
| ETHE1 | 113 ± 30 | 88 ± 18 | 59 ± 31 | 171 | 85 ± 2 |
| TST | 105 ± 7 | 81 ± 9 | 102 ± 27 | 142 | 89 ± 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ascenção, K.; Dilek, N.; Zuhra, K.; Módis, K.; Sato, T.; Szabo, C. Sequential Accumulation of ‘Driver’ Pathway Mutations Induces the Upregulation of Hydrogen-Sulfide-Producing Enzymes in Human Colonic Epithelial Cell Organoids. Antioxidants 2022, 11, 1823. https://doi.org/10.3390/antiox11091823
Ascenção K, Dilek N, Zuhra K, Módis K, Sato T, Szabo C. Sequential Accumulation of ‘Driver’ Pathway Mutations Induces the Upregulation of Hydrogen-Sulfide-Producing Enzymes in Human Colonic Epithelial Cell Organoids. Antioxidants. 2022; 11(9):1823. https://doi.org/10.3390/antiox11091823
Chicago/Turabian StyleAscenção, Kelly, Nahzli Dilek, Karim Zuhra, Katalin Módis, Toshiro Sato, and Csaba Szabo. 2022. "Sequential Accumulation of ‘Driver’ Pathway Mutations Induces the Upregulation of Hydrogen-Sulfide-Producing Enzymes in Human Colonic Epithelial Cell Organoids" Antioxidants 11, no. 9: 1823. https://doi.org/10.3390/antiox11091823