

Knockout and Inhibition of Ape1: Roles of Ape1 in Base Excision DNA Repair and Modulation of Gene Expression
Abstract
:1. Introduction
2. Ape1 Overview
2.1. Base Excision Repair and DNA End Processing
2.2. Redox Signaling and Oxidative G-Quadruplex Formation in Gene Expression
2.3. RNA Processing
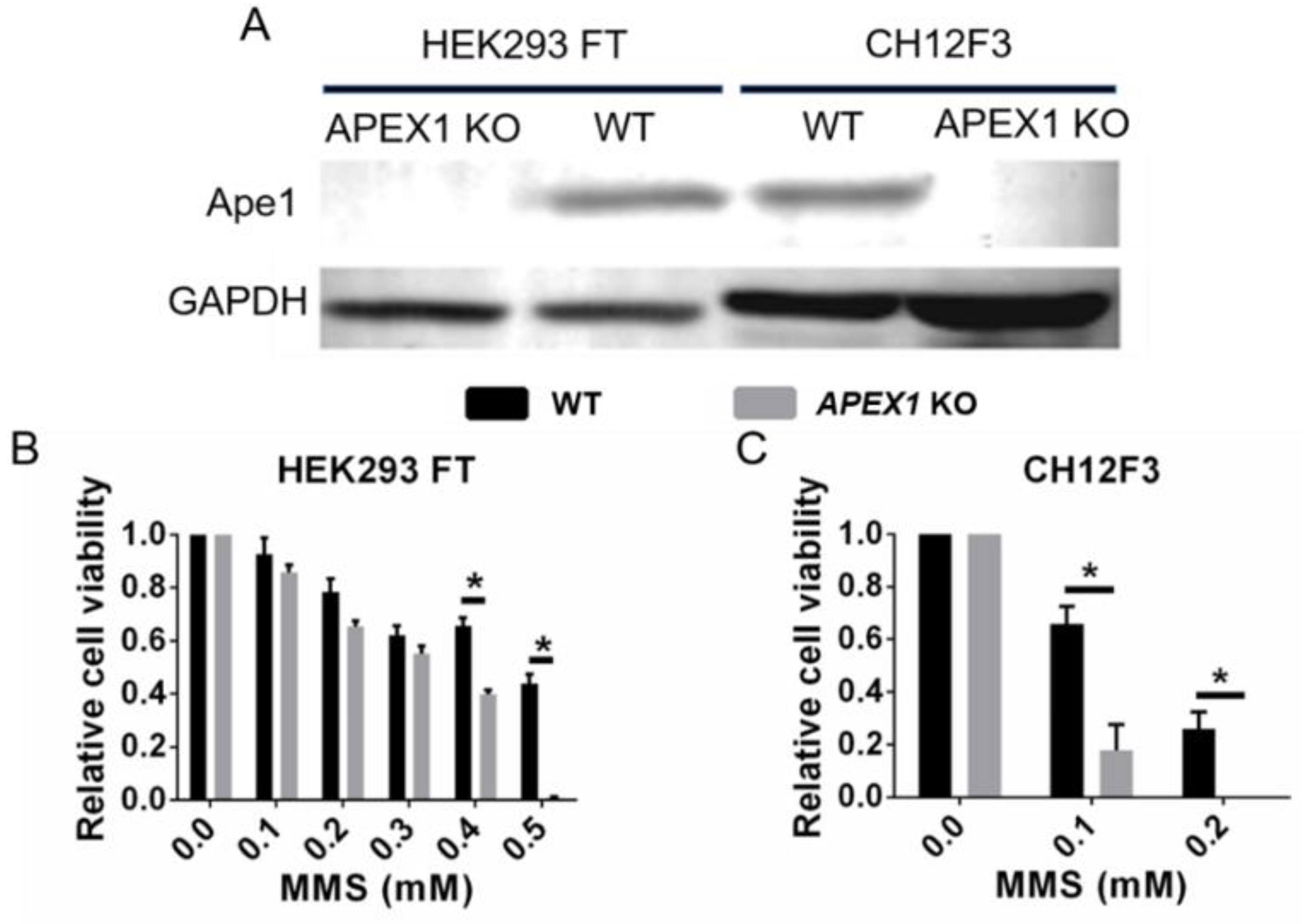
2.4. Ape1 Knockout Cell Lines: How Do They Survive?
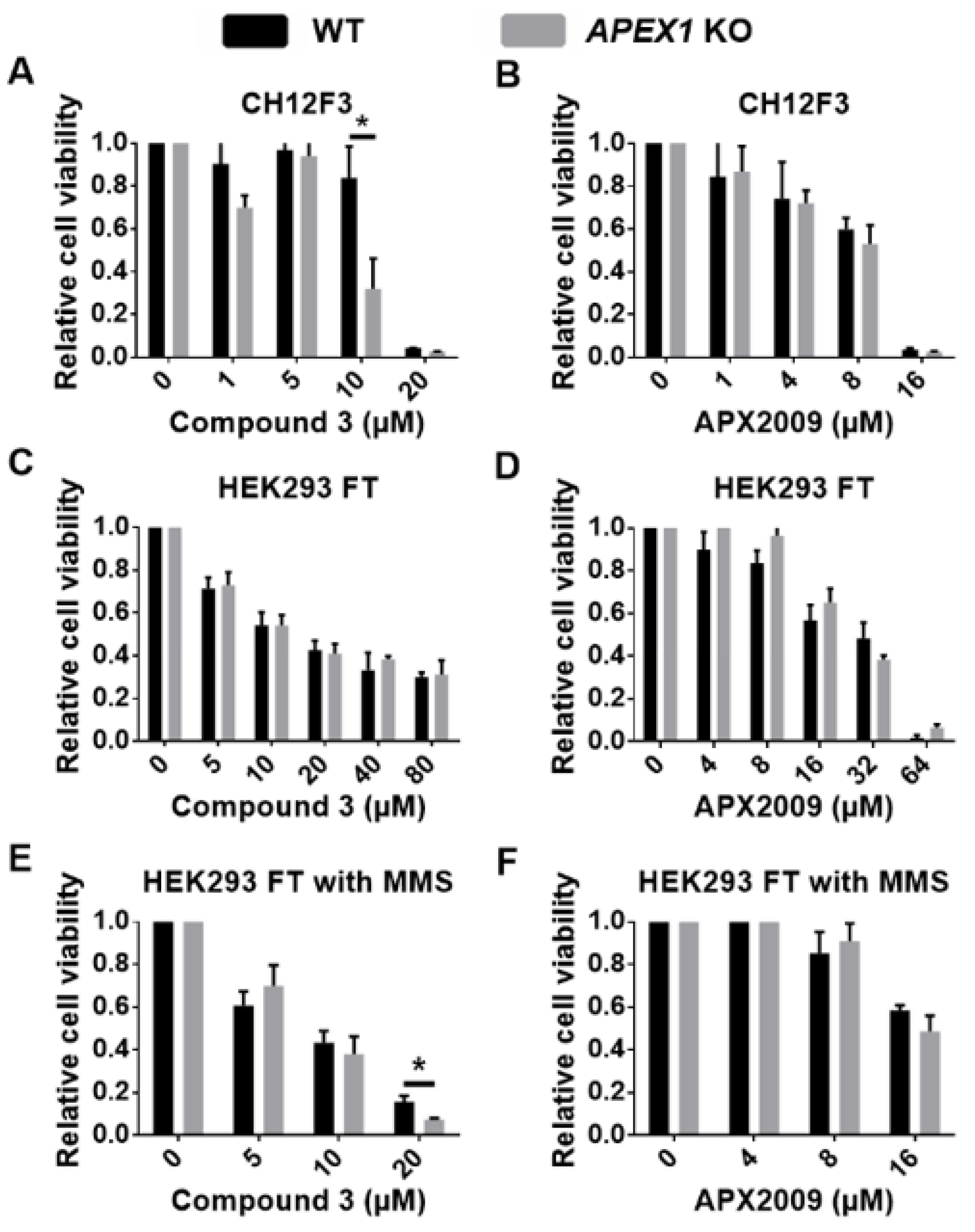
2.5. Off-Target Effects in Cell Killing by Ape1 Inhibitors
3. Materials and Methods
3.1. MTT Cell Viability Assay
3.2. Western Blotting
4. Evaluation of Compound 3 and APX2009 for Possible Off-Target Effects
5. Discussion: Mechanism of Off-Target Effects of Ape1 Inhibitors
6. Concluding Points
- Discovered as a DNA repair enzyme, Ape1 has been associated with multiple other roles, including both redox and non-redox activation of transcription factors;
- Ape1 can stabilize G-quadruplexes by binding but not cleaving AP sites in certain positions, which can mediate some transcriptional effects;
- Ape1 is essential for embryonic development in mice and probably for mammals in general;
- Genetic knockdown and knockout experiments indicate that the DNA repair function is essential in most cell types in culture;
- Inhibitors have been developed to target either the nuclease activity of Ape1 or its redox activity;
- Two viable cell lines have been developed with the Ape1-coding gene APEX1 deleted; these lines have mild phenotypes, the basis of which is unknown;
- The Ape1 inhibitors show similar toxic effects in APEX1-knockout cells and their APEX1+ counterparts, indicating that the compounds have significant off-target effects.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demple, B. Special problems for base excision repair in coping with oxidatively-induced DNA damage. In DNA Damage, DNA Repair, and Disease; Dizdaroglu, M., Lloyd, R.S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2021; Volume 1, pp. 204–219. [Google Scholar]
- Dutta, S.; Chowdhury, G.; Gates, K.S. Interstrand cross-links generated by abasic sites in duplex DNA. J. Am. Chem. Soc. 2007, 129, 1852–1853. [Google Scholar] [CrossRef]
- Pachva, M.C.; Kisselev, A.F.; Matkarimov, B.T.; Saparbaev, M.; Groisman, R. DNA-histone cross-links: Formation and repair. Front. Cell Dev. Biol. 2020, 8, 607045. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Du, H.; Liu, F.; Lu, J.C.; Yang, X.C.; Cui, W. Apurinic/apyrimidinic endonuclease/redox factor 1 (Ape1) alleviates myocardial hypoxia-reoxygenation injury by inhibiting oxidative stress and ameliorating mitochondrial dysfunction. Exp. Ther. Med. 2019, 17, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Kim, C.S.; Joo, H.K.; Lee, Y.R.; Kang, G.; Kim, S.J.; Choi, S.; Lee, S.D.; Park, J.B.; Jeon, B.H. Cytoplasmic localization and redox cysteine residue of Ape1/Ref-1 are associated with its anti-inflammatory activity in cultured endothelial cells. Mol. Cells 2013, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Meira, L.B.; Devaraj, S.; Kisby, G.E.; Burns, D.K.; Daniel, R.L.; Hammer, R.E.; Grundy, S.; Jialal, I.; Friedberg, E.C. Heterozygosity for the mouse APEX gene results in phenotypes associated with oxidative stress. Cancer Res. 2001, 61, 5552–5557. [Google Scholar]
- Izumi, T.; Brown, D.B.; Naidu, C.V.; Bhakat, K.K.; Macinnes, M.A.; Saito, H.; Chen, D.J.; Mitra, S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 5739–5743. [Google Scholar] [CrossRef]
- Osheroff, W.P.; Jung, H.K.; Beard, W.A.; Wilson, S.H.; Kunkel, T.A. The fidelity of DNA polymerase beta during distributive and processive DNA synthesis. J. Biol. Chem. 1999, 274, 3642–3650. [Google Scholar] [CrossRef]
- Chou, K.M.; Cheng, Y.C. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature 2002, 415, 655–659. [Google Scholar] [CrossRef]
- Liu, T.C.; Lin, C.T.; Chang, K.C.; Guo, K.W.; Wang, S.; Chu, J.W.; Hsiao, Y.Y. Ape1 distinguishes DNA substrates in exonucleolytic cleavage by induced space-filling. Nat. Commun. 2021, 12, 601. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Liu, T.C.; Guo, K.W.; Chu, J.W.; Hsiao, Y.Y. Understanding Ape1 cellular functions by the structural preference of exonuclease activities. Comput. Struct. Biotechnol. J. 2021, 19, 3682–3691. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Raj, J.; Li, J.; Ha, A.; Hossain, M.A.; Richardson, C.; Mukherjee, P.; Yan, S. Ape1 senses DNA single-strand breaks for repair and signaling. Nucleic Acids Res. 2020, 48, 1925–1940. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Quilon, A.; Wojtaszek, J.L.; Mathieu, M.C.; Patel, T.; Appel, C.D.; Hustedt, N.; Rossi, S.E.; Wallace, B.D.; Setiaputra, D.; Adam, S.; et al. Endogenous DNA 3′ blocks are vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the Ape2 nuclease. Mol. Cell 2020, 78, 1152–1165.e8. [Google Scholar] [CrossRef]
- Yuan, C.L.; He, F.; Ye, J.Z.; Wu, H.N.; Zhang, J.Y.; Liu, Z.H.; Li, Y.Q.; Luo, X.L.; Lin, Y.; Liang, R. Ape1 overexpression is associated with poor survival in patients with solid tumors: A meta-analysis. Oncotarget 2017, 8, 59720–59728. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.; Demple, B. Distinct roles of Ape1 protein in the repair of DNA damage induced by ionizing radiation or bleomycin. J. Biol. Chem. 2011, 286, 4968–4977. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional regulatory functions of mammalian AP-endonuclease (Ape1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 2009, 11, 621–638. [Google Scholar] [CrossRef]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y.; Yu, Z.; Zyromski, N.; Schipani, E.; Carta, F.; et al. Regulation of HIF1alpha under hypoxia by Ape1/Ref-1 impacts Ca9 expression: Dual targeting in patient-derived 3d pancreatic cancer models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef]
- Ando, K.; Hirao, S.; Kabe, Y.; Ogura, Y.; Sato, I.; Yamaguchi, Y.; Wada, T.; Handa, H. A new Ape1/Ref-1-dependent pathway leading to reduction of Nf-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008, 36, 4327–4336. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Flynn, T.S.; Freudenthal, B.D. Molecular snapshots of Ape1 proofreading mismatches and removing DNA damage. Nat. Commun. 2018, 9, 399. [Google Scholar] [CrossRef]
- Walker, L.J.; Robson, C.N.; Black, E.; Gillespie, D.; Hickson, I.D. Identification of residues in the human DNA repair enzyme Hap1 (Ref-1) that are essential for redox regulation of jun DNA binding. Mol. Cell. Biol. 1993, 13, 5370–5376. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L., II; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, M.M.; Luo, M.; Gaur, R.K.; Delaplane, S.; Li, X.; Kelley, M.R. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 2008, 643, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, J.; He, H.; Su, D.; Chen, Q.; Gross, M.L.; Kelley, M.R.; Georgiadis, M.M. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry 2012, 51, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Ordway, J.M.; Eberhart, D.; Curran, T. Cysteine 64 of Ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol. Cell Biol. 2003, 23, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernandez, C.E.; Close, D.M.; Gorb, L.; Leszczynski, J. Determination of redox potentials for the watson-crick base pairs, DNA nucleosides, and relevant nucleoside analogues. J. Phys. Chem. B 2007, 111, 5386–5395. [Google Scholar] [CrossRef]
- Kuraoka, I.; Endou, M.; Yamaguchi, Y.; Wada, T.; Handa, H.; Tanaka, K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J. Biol. Chem. 2003, 278, 7294–7299. [Google Scholar] [CrossRef]
- Tolentino, J.H.; Burke, T.J.; Mukhopadhyay, S.; McGregor, W.G.; Basu, A.K. Inhibition of DNA replication fork progression and mutagenic potential of 1, n6-ethenoadenine and 8-oxoguanine in human cell extracts. Nucleic Acids Res. 2008, 36, 1300–1308. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Oxidative stress-mediated epigenetic regulation by G-quadruplexes. NAR Cancer 2021, 3, zcab038. [Google Scholar] [CrossRef]
- Fleming, A.M.; Manage, S.A.H.; Burrows, J.C. Binding of AP endonuclease-1 to G-quadruplex DNA depends on the N-terminal domain, Mg(2+) and ionic strength. ACS Bio Med Chem Au 2021, 1, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, S.; Pramanik, S.; Harris, H.L.; Tarpley, M.; Sarkar, A.; Spagnol, G.; Sorgen, P.L.; Chowdhury, D.; Band, V.; Klinkebiel, D.; et al. Endogenous oxidized DNA bases and Ape1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl. Acad. Sci. USA 2020, 117, 11409–11420. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Dale, A.G.; Lombardo, C.M.; Valentine, H.; de la Fuente, M.; Schatzlein, A.; Neidle, S. Inhibition of the hypoxia-inducible factor pathway by a G-quadruplex binding small molecule. Sci. Rep. 2013, 3, 2799. [Google Scholar] [CrossRef]
- Agrawal, P.; Hatzakis, E.; Guo, K.; Carver, M.; Yang, D. Solution structure of the major G-quadruplex formed in the human VEGF promoter in K+: Insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 2013, 41, 10584–10592. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Bhattacharya, S.; Dash, J.; Bhattacharya, S. Recent update on targeting c-MYC G-quadruplexes by small molecules for anticancer therapeutics. J. Med. Chem. 2021, 64, 42–70. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Liu, Z.; Ye, W.; Li, L.; Qian, L.; Ding, H.; Li, P.; Aung, L.H.H. Recent advances: Molecular mechanism of RNA oxidation and its role in various diseases. Front. Mol. Biosci. 2020, 7, 184. [Google Scholar] [CrossRef]
- Shan, X.; Chang, Y.; Lin, C.L. Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J. 2007, 21, 2753–2764. [Google Scholar] [CrossRef]
- Tanaka, M.; Chock, P.B.; Stadtman, E.R. Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. USA 2007, 104, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Barzilay, G.; Walker, L.J.; Robson, C.N.; Hickson, I.D. Site-directed mutagenesis of the human DNA repair enzyme Hap1: Identification of residues important for ap endonuclease and RNase H activity. Nucleic Acids Res. 1995, 23, 1544–1550. [Google Scholar] [CrossRef]
- Chohan, M.; Mackedenski, S.; Li, W.M.; Lee, C.H. Human apurinic/apyrimidinic endonuclease 1 (Ape1) has 3′ RNA phosphatase and 3′ exoribonuclease activities. J. Mol. Biol. 2015, 427, 298–311. [Google Scholar] [CrossRef]
- Barnes, T.; Kim, W.C.; Mantha, A.K.; Kim, S.E.; Izumi, T.; Mitra, S.; Lee, C.H. Identification of apurinic/apyrimidinic endonuclease 1 (Ape1) as the endoribonuclease that cleaves c-MYC mRNA. Nucleic Acids Res. 2009, 37, 3946–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.C.; King, D.; Lee, C.H. RNA-cleaving properties of human apurinic/apyrimidinic endonuclease 1 (Ape1). Int. J. Biochem. Mol. Biol. 2010, 1, 12–25. [Google Scholar]
- Fantini, D.; Vascotto, C.; Marasco, D.; D′Ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F.; et al. Critical lysine residues within the overlooked N-terminal domain of human Ape1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D′Ambrosio, C.; Scaloni, A.; et al. Ape1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed]
- Barchiesi, A.; Bazzani, V.; Jabczynska, A.; Borowski, L.S.; Oeljeklaus, S.; Warscheid, B.; Chacinska, A.; Szczesny, R.J.; Vascotto, C. DNA repair protein Ape1 degrades dysfunctional abasic mRNA in mitochondria affecting oxidative phosphorylation. J. Mol. Biol. 2021, 433, 167125. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, R.; Wiederhold, L.; Szczesny, B.; Boldogh, I.; Hazra, T.K.; Izumi, T.; Mitra, S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006, 34, 2067–2076. [Google Scholar] [CrossRef]
- Tomkinson, A.E.; Bonk, R.T.; Linn, S. Mitochondrial endonuclease activities specific for apurinic/apyrimidinic sites in DNA from mouse cells. J. Biol. Chem. 1988, 263, 12532–12537. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.L.; MacInnes, M.A.; Takiguchi, Y.; Purtymun, P.E.; Henrie, M.; Flannery, M.; Meneses, J.; Pedersen, R.A.; Chen, D.J. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat. Res./DNA Repair 1998, 409, 17–29. [Google Scholar] [CrossRef]
- Dumitrache, L.C.; Shimada, M.; Downing, S.M.; Kwak, Y.D.; Li, Y.; Illuzzi, J.L.; Russell, H.R.; Wilson, D.M., III; McKinnon, P.J. Apurinic endonuclease-1 preserves neural genome integrity to maintain homeostasis and thermoregulation and prevent brain tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E12285–E12294. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, X.; Lu, X.; Dai, N.; Zhang, S.; Cheng, Y.; Zhang, L.; Yang, Y.; Liu, Y.; Yang, Z.; et al. Ape1 deficiency promotes cellular senescence and premature aging features. Nucleic Acids Res. 2018, 46, 5664–5677. [Google Scholar] [CrossRef] [PubMed]
- Cheo, D.L.; Meira, L.B.; Burns, D.K.; Reis, A.M.; Issac, T.; Friedberg, E.C. Ultraviolet B radiation-induced skin cancer in mice defective in the xpc, trp53, and APEX (HAP1) genes: Genotype-specific effects on cancer predisposition and pathology of tumors. Cancer Res. 2000, 60, 1580–1584. [Google Scholar] [PubMed]
- Unnikrishnan, A.; Raffoul, J.J.; Patel, H.V.; Prychitko, T.M.; Anyangwe, N.; Meira, L.B.; Friedberg, E.C.; Cabelof, D.C.; Heydari, A.R. Oxidative stress alters base excision repair pathway and increases apoptotic response in apurinic/apyrimidinic endonuclease 1/redox factor-1 haploinsufficient mice. Free Radic. Biol. Med. 2009, 46, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Ballista-Hernandez, J.; Martinez-Ferrer, M.; Velez, R.; Climent, C.; Sanchez-Vazquez, M.M.; Torres, C.; Rodriguez-Munoz, A.; Ayala-Pena, S.; Torres-Ramos, C.A. Mitochondrial DNA integrity is maintained by Ape1 in carcinogen-induced colorectal cancer. Mol. Cancer Res. 2017, 15, 831–841. [Google Scholar] [CrossRef]
- Fung, H.; Demple, B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 2005, 17, 463–470. [Google Scholar] [CrossRef]
- Chen, T.; Liu, C.; Lu, H.; Yin, M.; Shao, C.; Hu, X.; Wu, J.; Wang, Y. The expression of Ape1 in triple-negative breast cancer and its effect on drug sensitivity of olaparib. Tumour Biol. 2017, 39, 1010428317713390. [Google Scholar] [CrossRef] [PubMed]
- Illuzzi, J.L.; McNeill, D.R.; Bastian, P.; Brenerman, B.; Wersto, R.; Russell, H.R.; Bunz, F.; McKinnon, P.J.; Becker, K.G.; Wilson, D.M., III. Tumor-associated Ape1 variant exhibits reduced complementation efficiency but does not promote cancer cell phenotypes. Environ. Mol. Mutagen. 2017, 58, 84–98. [Google Scholar] [CrossRef]
- Kim, D.V.; Kulishova, L.M.; Torgasheva, N.A.; Melentyev, V.S.; Dianov, G.L.; Medvedev, S.P.; Zakian, S.M.; Zharkov, D.O. Mild phenotype of knockouts of the major apurinic/apyrimidinic endonuclease Apex1 in a non-cancer human cell line. PLoS ONE 2021, 16, e0257473. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.J.; Craig, R.B.; Harris, A.L.; Hickson, I.D. A role for the human DNA repair enzyme Hap1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994, 22, 4884–4889. [Google Scholar] [CrossRef]
- Masani, S.; Han, L.; Yu, K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol. Cell. Biol. 2013, 33, 1468–1473. [Google Scholar] [CrossRef]
- Raffoul, J.J.; Cabelof, D.C.; Nakamura, J.; Meira, L.B.; Friedberg, E.C.; Heydari, A.R. Apurinic/apyrimidinic endonuclease (Ape/Ref-1) haploinsufficient mice display tissue-specific differences in DNA polymerase beta-dependent base excision repair. J. Biol. Chem. 2004, 279, 18425–18433. [Google Scholar] [CrossRef]
- Rai, G.; Vyjayanti, V.N.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., III; Maloney, D.J. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J. Med. Chem. 2012, 55, 3101–3112. [Google Scholar] [CrossRef] [Green Version]
- Naidu, M.D.; Agarwal, R.; Pena, L.A.; Cunha, L.; Mezei, M.; Shen, M.; Wilson, D.M., III; Liu, Y.; Sanchez, Z.; Chaudhary, P.; et al. Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (Ape1) by direct protein binding. PLoS ONE 2011, 6, e23679. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Kelley, M.R. Inhibition of the human apurinic/apyrimidinic endonuclease (Ape1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 2004, 24, 2127–2134. [Google Scholar] [PubMed]
- Madhusudan, S.; Smart, F.; Shrimpton, P.; Parsons, J.L.; Gardiner, L.; Houlbrook, S.; Talbot, D.C.; Hammonds, T.; Freemont, P.A.; Sternberg, M.J.; et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005, 33, 4711–4724. [Google Scholar] [CrossRef]
- Bapat, A.; Glass, L.S.; Luo, M.; Fishel, M.L.; Long, E.C.; Georgiadis, M.M.; Kelley, M.R. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J. Pharmacol. Exp. Ther. 2010, 334, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Wikel, J.H.; Guo, C.; Pollok, K.E.; Bailey, B.J.; Wireman, R.; Fishel, M.L.; Vasko, M.R. Identification and characterization of new chemical entities targeting apurinic/apyrimidinic endonuclease 1 for the prevention of chemotherapy-induced peripheral neuropathy. J. Pharmacol. Exp. Ther. 2016, 359, 300–309. [Google Scholar] [CrossRef]
- Sardar Pasha, S.P.B.; Sishtla, K.; Sulaiman, R.S.; Park, B.; Shetty, T.; Shah, F.; Fishel, M.L.; Wikel, J.H.; Kelley, M.R.; Corson, T.W. Ref-1/Ape1 inhibition with novel small molecules blocks ocular neovascularization. J. Pharmacol. Exp. Ther. 2018, 367, 108–118. [Google Scholar] [CrossRef]
- Shimizu, N.; Sugimoto, K.; Tang, J.; Nishi, T.; Sato, I.; Hiramoto, M.; Aizawa, S.; Hatakeyama, M.; Ohba, R.; Hatori, H.; et al. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000, 18, 877–881. [Google Scholar] [CrossRef]
- Zhong, C.; Xu, M.; Wang, Y.; Xu, J.; Yuan, Y. An Ape1 inhibitor reveals critical roles of the redox function of Ape1 in KSHV replication and pathogenic phenotypes. PLoS Pathog. 2017, 13, e1006289. [Google Scholar] [CrossRef]
- Xu, J.; Husain, A.; Hu, W.; Honjo, T.; Kobayashi, M. Ape1 is dispensable for S-region cleavage but required for its repair in class switch recombination. Proc. Natl. Acad. Sci. USA 2014, 111, 17242–17247. [Google Scholar] [CrossRef] [PubMed]
- Kharat, S.S.; Ding, X.; Swaminathan, D.; Suresh, A.; Singh, M.; Sengodan, S.K.; Burkett, S.; Marks, H.; Pamala, C.; He, Y.; et al. Degradation of 5hmC-marked stalled replication forks by Ape1 causes genomic instability. Sci. Signal. 2020, 13, eaba8091. [Google Scholar] [CrossRef]
- Cunniffe, S.M.; Lomax, M.E.; O′Neill, P. An ap site can protect against the mutagenic potential of 8-oxoG when present within a tandem clustered site in E. coli. DNA Repair 2007, 6, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair 2004, 3, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Bennett, P.V.; Wilson, D.M., III; Sutherland, B.M. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res. 2004, 32, 5609–5620. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Inhibitor Name | Ape1 AP Endonuclease Assay | AP Site Reactivity of Compound | Ape1 Redox Activity | Other Ape1 Activity | Other DNA Repair Pathways |
|---|---|---|---|---|---|
| Compound 3 [63] | HeLa WCE * incision assay | NA * | NA | Ape1 AP site binding not affected | NA |
| Lucanthone [64,65] | U251-MG glioblastoma multiforme cell WCE incision assay | Enzyme digestion assay; no binding | No effect | Did not affect exonuclease activity | NA |
| CRT0044876 [66] | Recombinant Ape1 incision assay | Enzyme digestion assay; no binding | NA | 3′-phosphatase and 3′-phosphoglycolate diesterase activities not affected | Did not potentiate the cytotoxicity of ionizing radiation or UV light |
| AR03 (Synonym: BMH-23) [67] | SF767 cell WCE incision assay | Fluorescence intercalation displacement assay; no binding | Did not affect AP-1 DNA binding in vitro | NA | NA |
| Inhibitor Name | Transcription Factor Target | Ape1 Endo Activity | |||
|---|---|---|---|---|---|
| NF-kB | AP-1 | HIF-1a | |||
| APX2009 [68,69] | Transactivation in a cell-based reporter assay system | Electrophoretic mobility shift assay (EMSA *) | NA * | In vitro AP site cleavage increased | |
| E3330 (APX3330) [23,70] | Transactivation in a cell-based reporter assay system and EMSA | Transactivation in a cell-based reporter assay system and EMSA | EMSA | In vitro AP site digestion; no effect | |
| C10 [71] | NA | EMSA; Inhibited | NA | In vitro AP site digestion; no effect | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Z.; Demple, B. Knockout and Inhibition of Ape1: Roles of Ape1 in Base Excision DNA Repair and Modulation of Gene Expression. Antioxidants 2022, 11, 1817. https://doi.org/10.3390/antiox11091817
Xue Z, Demple B. Knockout and Inhibition of Ape1: Roles of Ape1 in Base Excision DNA Repair and Modulation of Gene Expression. Antioxidants. 2022; 11(9):1817. https://doi.org/10.3390/antiox11091817
Chicago/Turabian StyleXue, Zhouyiyuan, and Bruce Demple. 2022. "Knockout and Inhibition of Ape1: Roles of Ape1 in Base Excision DNA Repair and Modulation of Gene Expression" Antioxidants 11, no. 9: 1817. https://doi.org/10.3390/antiox11091817