
The Mobilome-Enriched Genome of the Competence-Deficient Streptococcus pneumoniae BM6001, the Original Host of Integrative Conjugative Element Tn5253, Is Phylogenetically Distinct from Historical Pneumococcal Genomes
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Growth Conditions
2.2. Genomic DNA Purification
2.3. Illumina Sequencing
2.4. Nanopore Sequencing
2.5. Genome Assembly and Annotation
2.6. Mobilome Analysis
2.7. Bacterial Lysis for qPCR Analysis
2.8. PCR, qPCR and Direct PCR Sequencing
2.9. Competent Cells Preparation and Transformation
2.10. Phylogenetic Analysis and Nucleotide Homology Searching
2.11. Nucleotide Sequences Accession Numbers
3. Results
3.1. The Genome of S. pneumoniae BM6001
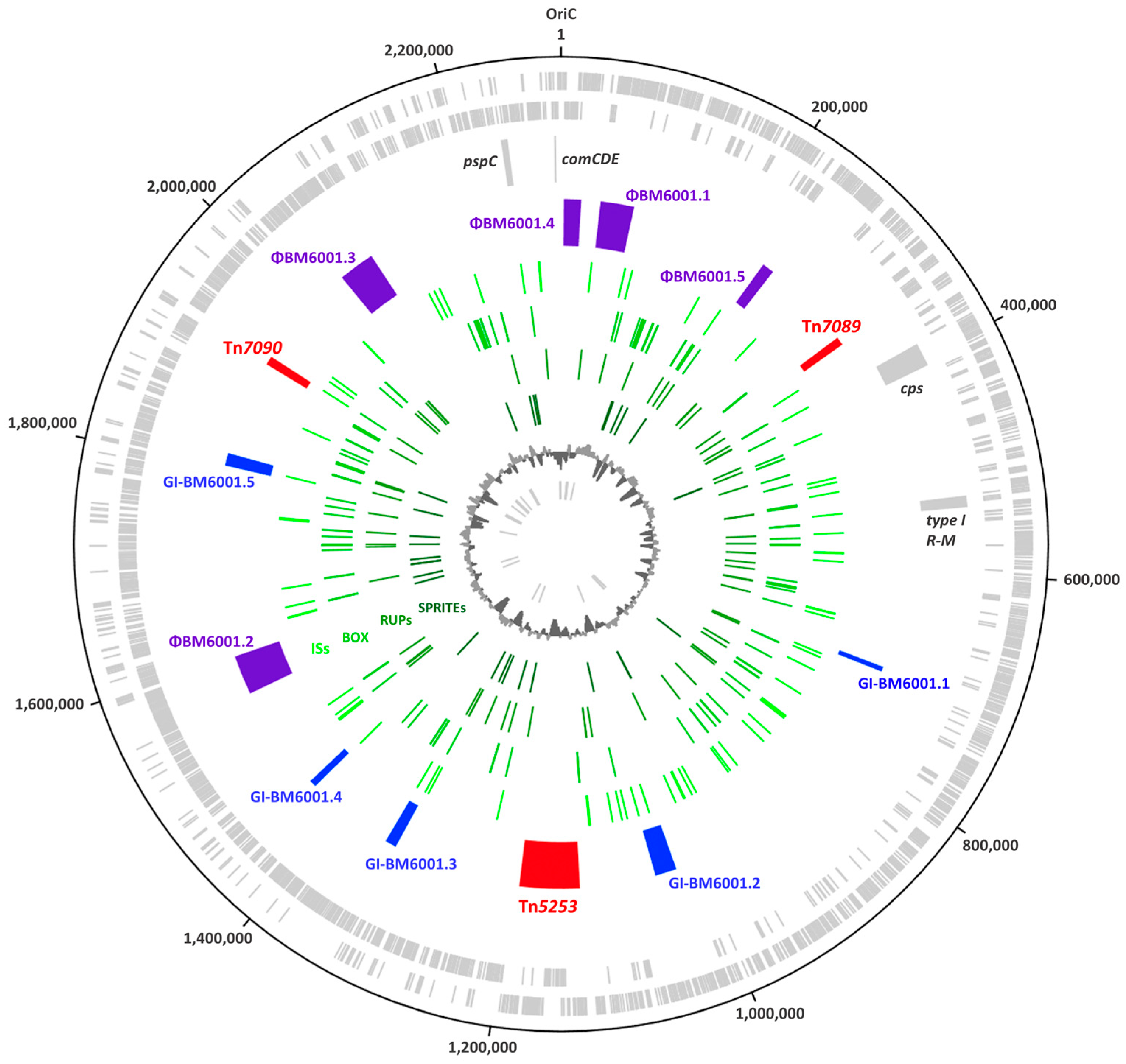
3.2. The Mobilome of BM6001
3.3. IME Tn7089
3.4. Transposon Tn7090
3.5. Prophages
3.6. Genomic Islands
3.7. Excision and Attachment Sites’ Reconstitution of BM6001 MGEs
3.8. Impact of ΦBM6001.3 on Genetic Transformation
3.9. Phylogenetic Analysis of BM6001
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Tonder, A.J.; Bray, J.E.; Jolley, K.A.; Van Rensburg, M.J.; Quirk, S.J.; Haraldsson, G.; Maiden, M.C.J.; Bentley, S.D.; Haraldsson, Á.; Erlendsdóttir, H.; et al. Genomic analyses of >3100 nasopharyngeal pneumococci revealed significant differences between pneumococci recovered in four different geographical regions. Front. Microbiol. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Bentley, S.D.; Aanensen, D.M.; Mavroidi, A.; Saunders, D.; Rabbinowitsch, E.; Collins, M.; Donohoe, K.; Harris, D.; Murphy, L.; Quail, M.A.; et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006, 2, e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannelli, F.; Oggioni, M.R.; Pozzi, G. Allelic variation in the highly polymorphic locus pspC of Streptococcus pneumoniae. Gene 2002, 284, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Chiavolini, D.; Ricci, S.; Oggioni, M.R.; Pozzi, G. Pneumococcal surface protein C contributes to sepsis caused by Streptococcus pneumoniae in mice. Infect. Immun. 2004, 72, 3077–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingshead, S.K.; Becker, R.; Briles, D.E. Diversity of PspA: Mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 2000, 68, 5889–5900. [Google Scholar] [CrossRef] [Green Version]
- Santoro, F.; Iannelli, F.; Pozzi, G. Genomics and genetics of Streptococcus pneumoniae. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Carr, V.R.; Shkoporov, A.; Hill, C.; Mullany, P.; Moyes, D.L. Probing the mobilome: Discoveries in the dynamic microbiome. Trends Microbiol. 2021, 29, 158–170. [Google Scholar] [CrossRef]
- Blomberg, C.; Dagerhamn, J.; Dahlberg, S.; Browall, S.; Fernebro, J.; Albiger, B.; Morfeldt, E.; Normark, S.; Henriques-Normark, B. Pattern of accessory regions and invasive disease potential in Streptococcus pneumoniae. J. Infect. Dis. 2009, 199, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.; Croucher, N.J.; Hiller, N.L.; Hu, F.Z.; Ehrlich, G.D.; Bentley, S.D.; García, E.; Mitchell, T.J. Comparative genomic analysis of ten Streptococcus pneumoniae temperate bacteriophages. J. Bacteriol. 2009, 191, 4854–4862. [Google Scholar] [CrossRef] [Green Version]
- Brueggemann, A.B.; Harrold, C.L.; Rezaei Javan, R.; van Tonder, A.J.; McDonnell, A.J.; Edwards, B.A. Pneumococcal prophages are diverse, but not without structure or history. Sci. Rep. 2017, 7, 42976. [Google Scholar] [CrossRef] [Green Version]
- Rezaei Javan, R.; Ramos-Sevillano, E.; Akter, A.; Brown, J.; Brueggemann, A.B. Prophages and satellite prophages are widespread in Streptococcus and may play a role in pneumococcal pathogenesis. Nat. Commun. 2019, 10, 4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.D.; Guild, W.R. A plasmid in Streptococcus pneumoniae. J. Bacteriol. 1979, 137, 735–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oggioni, M.R.; Iannelli, F.; Pozzi, G. Characterization of cryptic plasmids pDP1 and pSMB1 of Streptococcus pneumoniae. Plasmid 1999, 41, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, N.B.; Smith, M.D.; Guild, W.R. Organization and transfer of heterologous chloramphenicol and tetracycline resistance genes in pneumococcus. J. Bacteriol. 1979, 139, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumar, M.N.; Priebe, S.D.; Pozzi, G.; Hageman, J.M.; Guild, W.R. Cloning and physical characterization of chromosomal conjugative elements in streptococci. J. Bacteriol. 1986, 166, 972–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannelli, F.; Santoro, F.; Oggioni, M.R.; Pozzi, G. Nucleotide sequence analysis of integrative conjugative element Tn5253 of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2014, 58, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Santoro, F.; Oggioni, M.R.; Pozzi, G.; Iannelli, F. Nucleotide sequence and functional analysis of the tet(M)-carrying conjugative transposon Tn5251 of Streptococcus pneumoniae: Tn5251 of Streptococcus pneumoniae. FEMS Microbiol. Lett. 2010, 308, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Santoro, F.; Romeo, A.; Pozzi, G.; Iannelli, F. Excision and circularization of integrative conjugative element Tn5253 of Streptococcus pneumoniae. Front. Microbiol. 2018, 9, 1779. [Google Scholar] [CrossRef]
- Santoro, F.; Fox, V.; Romeo, A.; Lazzeri, E.; Pozzi, G.; Iannelli, F. Chromosomal integration of Tn5253 occurs downstream of a conserved 11-bp sequence of the rbgA gene in Streptococcus pneumoniae and in all the other known hosts of this integrative conjugative element (ICE). Mob. DNA 2021, 12, 25. [Google Scholar] [CrossRef]
- Brown, J.S.; Gilliland, S.M.; Spratt, B.G.; Holden, D.W. A locus contained within a variable region of pneumococcal pathogenicity island 1 contributes to virulence in mice. Infect. Immun. 2004, 72, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Gilliland, S.M.; Holden, D.W. A Streptococcus pneumoniae pathogenicity island encoding an abc transporter involved in iron uptake and virulence: Iron uptake by Streptococcus pneumoniae. Mol. Microbiol. 2001, 40, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.M.; Stroeher, U.H.; Ogunniyi, A.D.; Smith-Vaughan, H.C.; Leach, A.J.; Paton, J.C. A variable region within the genome of Streptococcus pneumoniae contributes to strain-strain variation in virulence. PLoS ONE 2011, 6, e19650. [Google Scholar] [CrossRef] [Green Version]
- Dang-Van, A.; Tiraby, G.; Acar, J.F.; Shaw, W.V.; Bouanchaud, D.H. Chloramphenicol resistance in Streptococcus pneumoniae: Enzymatic acetylation and possible plasmid linkage. Antimicrob. Agents Chemother. 1978, 13, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzauti, D.; Iannelli, F.; Pozzi, G.; Santoro, F. DNA isolation methods for nanopore sequencing of the Streptococcus mitis genome. Microb. Genom. 2022, 8, 000764. [Google Scholar] [CrossRef]
- Cuppone, A.M.; Colombini, L.; Fox, V.; Pinzauti, D.; Santoro, F.; Pozzi, G.; Iannelli, F. Complete genome sequence of Streptococcus pneumoniae strain Rx1, a Hex mismatch repair-deficient standard transformation recipient. Microbiol. Resour. Announc. 2021, 10, e00799-21. [Google Scholar] [CrossRef] [PubMed]
- Teodori, L.; Colombini, L.; Cuppone, A.M.; Lazzeri, E.; Pinzauti, D.; Santoro, F.; Iannelli, F.; Pozzi, G. Complete genome sequence of Lactobacillus crispatus type strain ATCC 33820. Microbiol. Resour. Announc. 2021, 10, e0063421. [Google Scholar] [CrossRef]
- Iannelli, F.; Giunti, L.; Pozzi, G. Direct sequencing of long polymerase chain reaction fragments. Mol. Biotechnol. 1998, 10, 183–185. [Google Scholar] [CrossRef]
- Iannelli, F.; Pozzi, G. Method for introducing specific and unmarked mutations into the chromosome of Streptococcus pneumoniae. Mol. Biotechnol. 2004, 26, 81–86. [Google Scholar] [CrossRef]
- Iannelli, F.; Santoro, F.; Fox, V.; Pozzi, G. A mating procedure for genetic transfer of integrative and conjugative elements (ICEs) of streptococci and enterococci. Methods Protoc. 2021, 4, 59. [Google Scholar] [CrossRef]
- Bogaardt, C.; Van Tonder, A.J.; Brueggemann, A.B. Genomic analyses of pneumococci reveal a wide diversity of bacteriocins—Including pneumocyclicin, a novel circular bacteriocin. BMC Genom. 2015, 16, 554. [Google Scholar] [CrossRef] [Green Version]
- Wyres, K.L.; Van Tonder, A.; Lambertsen, L.M.; Hakenbeck, R.; Parkhill, J.; Bentley, S.D.; Brueggemann, A.B. Evidence of antimicrobial resistance-conferring genetic elements among pneumococci isolated prior to 1974. BMC Genom. 2013, 14, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morona, J.K.; Morona, R.; Paton, J.C. Characterization of the locus encoding the Streptococcus pneumoniae type 19F capsular polysaccharide biosynthetic pathway. Mol. Microbiol. 1997, 23, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Pearce, B.J.; Pozzi, G. The type 2 capsule locus of Streptococcus pneumoniae. J. Bacteriol. 1999, 181, 2652–2654. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, G.; Masala, L.; Iannelli, F.; Manganelli, R.; Havarstein, L.S.; Piccoli, L.; Simon, D.; Morrison, D.A. Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: Two allelic variants of the peptide pheromone. J. Bacteriol. 1996, 178, 6087–6090. [Google Scholar] [CrossRef] [Green Version]
- Havarstein, L.S.; Coomaraswamy, G.; Morrison, D.A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 1995, 92, 11140–11144. [Google Scholar] [CrossRef] [Green Version]
- Iannelli, F.; Oggioni, M.R.; Pozzi, G. Sensor domain of histidine kinase ComD confers competence pherotype specificity in Streptoccoccus pneumoniae. FEMS Microbiol. Lett. 2005, 252, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Manso, A.S.; Chai, M.H.; Atack, J.M.; Furi, L.; De Ste Croix, M.; Haigh, R.; Trappetti, C.; Ogunniyi, A.D.; Shewell, L.K.; Boitano, M.; et al. A random six-phase switch regulates pneumococcal virulence via global epigenetic changes. Nat. Commun. 2014, 5, 5055. [Google Scholar] [CrossRef] [Green Version]
- Oggioni, M.R.; Claverys, J.P. Repeated extragenic sequences in prokaryotic genomes: A proposal for the origin and dynamics of the RUP element in Streptococcus pneumoniae. Microbiology 1999, 145, 2647–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, B.; Humbert, O.; Camara, M.; Guenzi, E.; Walker, J.; Mitchell, T.; Andrew, P.; Prudhomme, M.; Alloing, G.; Hakenbeck, R.; et al. A highly conserved repeated DNA element located in the chromosome of Streptococcus pneumoniae. Nucleic Acids Res. 1992, 20, 3479–3483. [Google Scholar] [CrossRef] [Green Version]
- Croucher, N.J.; Vernikos, G.S.; Parkhill, J.; Bentley, S.D. Identification, variation and transcription of pneumococcal repeat sequences. BMC Genom. 2011, 12, 120. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Koonin, E.V. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998, 26, 3746–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, T.J.; Ehrmann, M.; Boos, W. Periplasmic glycerophosphodiester phosphodiesterase of Escherichia coli, a new enzyme of the glp Regulon. J. Biol. Chem. 1983, 258, 5428–5432. [Google Scholar] [CrossRef]
- Corda, D.; Mosca, M.G.; Ohshima, N.; Grauso, L.; Yanaka, N.; Mariggiò, S. The emerging physiological roles of the glycerophosphodiesterase family. FEBS J. 2014, 281, 998–1016. [Google Scholar] [CrossRef] [PubMed]
- Rang, C.; Alix, E.; Felix, C.; Heitz, A.; Tasse, L.; Blanc-Potard, A.-B. Dual role of the MgtC virulence factor in host and non-host environments: Dual role of the MgtC virulence factor. Mol. Microbiol. 2007, 63, 605–622. [Google Scholar] [CrossRef]
- Koonin, E.V.; Tatusov, R.L. Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity. Application of an iterative approach to database search. J. Mol. Biol. 1994, 244, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viboud, G.I.; Bliska, J.B. Yersinia outer proteins: Role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 2005, 59, 69–89. [Google Scholar] [CrossRef]
- Alamos, P.; Tello, M.; Bustamante, P.; Gutiérrez, F.; Shmaryahu, A.; Maldonado, J.; Levicán, G.; Orellana, O. Functionality of tRNAs encoded in a mobile genetic element from an acidophilic bacterium. RNA Biol. 2018, 15, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Croucher, N.J.; Mostowy, R.; Wymant, C.; Turner, P.; Bentley, S.D.; Fraser, C. Horizontal DNA transfer mechanisms of bacteria as weapons of intragenomic conflict. PLoS Biol. 2016, 14, e1002394. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Nelson, K.E.; Paulsen, I.T.; Eisen, J.A.; Read, T.D.; Peterson, S.; Heidelberg, J.; DeBoy, R.T.; Haft, D.H.; Dodson, R.J.; et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 2001, 293, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Brückner, R.; Nuhn, M.; Reichmann, P.; Weber, B.; Hakenbeck, R. Mosaic genes and mosaic chromosomes–genomic variation in Streptococcus pneumoniae. Int. J. Med. Microbiol. 2004, 294, 157–168. [Google Scholar] [CrossRef]
- Obert, C.; Sublett, J.; Kaushal, D.; Hinojosa, E.; Barton, T.; Tuomanen, E.I.; Orihuela, C.J. Identification of a candidate Streptococcus pneumoniae core genome and regions of diversity correlated with invasive pneumococcal disease. Infect. Immun. 2006, 74, 4766–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, N.A.; McCluskey, J.; Jefferies, J.M.C.; Hinds, J.; Smith, A.; Clarke, S.C.; Mitchell, T.J.; Paterson, G.K. Genomic diversity between strains of the same serotype and multilocus sequence type among pneumococcal clinical isolates. Infect. Immun. 2006, 74, 3513–3518. [Google Scholar] [CrossRef] [Green Version]
- McAllister, L.J.; Ogunniyi, A.D.; Stroeher, U.H.; Paton, J.C. Contribution of a genomic accessory region encoding a putative cellobiose phosphotransferase system to virulence of Streptococcus pneumoniae. PLoS ONE 2012, 7, e32385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.V.; Kelleher, N.L.; Kinsland, C.; Chiu, H.J.; Costello, C.A.; Backstrom, A.D.; McLafferty, F.W.; Begley, T.P. Thiamin biosynthesis in Escherichia coli. J. Biol. Chem. 1998, 273, 16555–16560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, F.; Vianna, M.E.; Roberts, A.P. Variation on a theme; an overview of the Tn916/Tn1545 family of mobile genetic elements in the oral and nasopharyngeal streptococci. Front. Microbiol. 2014, 5, 535. [Google Scholar] [CrossRef]
- Croucher, N.J.; Walker, D.; Romero, P.; Lennard, N.; Paterson, G.K.; Bason, N.C.; Mitchell, A.M.; Quail, M.A.; Andrew, P.W.; Parkhill, J.; et al. Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcus pneumoniaeSpain23F ST81. J. Bacteriol. 2009, 191, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Severi, E.; Hood, D.W.; Thomas, G.H. Sialic acid utilization by bacterial pathogens. Microbiology 2007, 153, 2817–2822. [Google Scholar] [CrossRef] [Green Version]
- Garriss, G.; Henriques-Normark, B. Lysogeny in Streptococcus pneumoniae. Microorganisms 2020, 8, 1546. [Google Scholar] [CrossRef]
- Albano, M.; Dubnau, D.A. Cloning and characterization of a cluster of linked Bacillus subtilis late competence mutations. J. Bacteriol. 1989, 171, 5376–5385. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.; Dubnau, D. DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2004, 2, 241–249. [Google Scholar] [CrossRef]
- Pestova, E.V.; Morrison, D.A. Isolation and characterization of three Streptococcus pneumoniae transformation-specific loci by use of a lacZ reporter insertion vector. J. Bacteriol. 1998, 180, 2701–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claverys, J.-P.; Martin, B.; Polard, P. The genetic transformation machinery: Composition, localization, and mechanism. FEMS Microbiol. Rev. 2009, 33, 643–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, F.; Pastore, G.; Fox, V.; Petit, M.-A.; Iannelli, F.; Pozzi, G. Streptococcus pyogenes Φ1207.3 is a temperate bacteriophage carrying the macrolide resistance gene pair mef(A)-msr(D) and capable of lysogenizing different streptococci. Microbiol. Spectr. 2023, 11, e04211–e0421122. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Santagati, M.; Santoro, F.; Oggioni, M.R.; Stefani, S.; Pozzi, G. Nucleotide Sequence of Conjugative Prophage Φ1207.3 (Formerly Tn1207.3) carrying the mef(A)/msr(D) genes for efflux resistance to macrolides in Streptococcus pyogenes. Front. Microbiol. 2014, 5, 687. [Google Scholar] [CrossRef] [Green Version]
- Santagati, M.; Iannelli, F.; Cascone, C.; Campanile, F.; Oggioni, M.R.; Stefani, S.; Pozzi, G. The novel conjugative transposon Tn1207.3 carries the macrolide efflux gene mef(A) in Streptococcus pyogenes. Microb. Drug Resist. 2003, 9, 243–247. [Google Scholar] [CrossRef]
- Pozzi, G.; Iannelli, F.; Oggioni, M.; Santagati, M.; Stefani, S. Genetic elements carrying macrolide efflux genes in streptococci. Curr. Drug Targets Infect. Disord. 2004, 4, 203–206. [Google Scholar] [CrossRef]
- Chan, W.T.; Espinosa, M. The Streptococcus pneumoniae pezAT toxin–antitoxin system reduces β-Lactam resistance and genetic competence. Front. Microbiol. 2016, 7, 1322. [Google Scholar] [CrossRef] [Green Version]
- Croucher, N.J.; Harris, S.R.; Fraser, C.; Quail, M.A.; Burton, J.; van der Linden, M.; McGee, L.; von Gottberg, A.; Song, J.H.; Ko, K.S.; et al. Rapid pneumococcal evolution in response to clinical interventions. Science 2011, 331, 430–434. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF (aa) a | Predicted Protein | Homologous Protein ID/Origin Identity (%) [E Value] b | Pfam Domain c (aa) [E Value] |
|---|---|---|---|
| orf1 (406) | Tyrosine-type DNA integrase | Integrase, catalytic domain (210–387) [3.2 × 10−15] | |
| orf2 (89) | Excisionase, putative | Helix–turn–helix domain, group 17 (42–86) [7.6 × 10−11] | |
| orf3 (264) | Rep protein, putative | CBJ21467.1/S. mitis 152/261 (58%) [3.0 × 10−111] | |
| orf4 (257) | Metal-dependent phosphoesterase | CBJ21468.1/S. mitis 234/254 (92%) [1.0 × 10−179] | Calcineurin-like phosphoesterase domain, ApaH type (1–194) [3.9 × 10−9] |
| orf5 (242) | Metal-dependent phosphoesterase | Calcineurin-like phosphoesterase domain, ApaH type (4–196) [4.8 × 10−12] | |
| orf7 (439) | FtsK/SpoIIIE domain-containing protein | FtsK domain (188–371) [1.1 × 10−14] |
| ORF (aa) a | Predicted Protein | Homologous Protein ID/Origin Identity (%) [E value] b | Pfam Domain c (aa) [E value] |
|---|---|---|---|
| orf1 (286) | Glycerophosphodiester phosphodiesterase | Glycerophosphodiester phosphodiesterase domain (18–102) [4.0 × 10−7] | |
| orf2 (353) | Bacterial extracellular solute-binding protein | WP_000475761.1/Streptococcus pneumoniae 353/353 (100%) [0.0] | Bacterial extracellular solute-binding domain (88–338) [4.3 × 10−28] |
| orf3 (334) | ABC transporter ATP-binding protein | ABC transporter-like, ATP-binding domain (18–160) [2.6 × 10−36] | |
| orf4 (564) | Iron ABC transporter permease | ABC transporter type 1, transmembrane domain MetI-like (60–266) [2.9 × 10−7]; ABC transporter type 1, transmembrane domain MetI-like (375–551) [1.4 × 10−12] | |
| orf5 (233) | MgtC/SapB/SrpB/YhiD family protein | MgtC/SapB/SrpB/YhiD domain (17–140) [5.0 × 10−35] | |
| orf6 (199) | Haloacid dehalogenase-like hydrolase | Haloacid dehalogenase-like hydrolase (3–182) [1.8 × 10−13] | |
| orf7 (675) | Bacterial extracellular solute-binding protein | Bacterial extracellular solute-binding protein (334–619) [7.1 × 10−17]; LacI-type HTH domain (1–66) [1.4 × 10−11] | |
| tnp (133) | ISSpn7, family IS5, truncated | WP_001105201.1/Streptococcus pneumoniae 36/267 (29%) [5.0 × 10−23] |
| Mobile Genetic Element | Circular Forms a | Reconstituted Integration Site a |
|---|---|---|
| Tn7089 | 3.92 × 10−8 (±1.08 × 10−8) | 5.73 × 10−8 (±5.78 × 10−9) |
| Tn7090 | 4.19 × 10−4 (±1.60 × 10−4) | 3.21 × 10−4 (±3.11 × 10−5) |
| ΦBM6001.1 | 1.27 × 10−3 (±9.86 × 10−4) | 4.12 × 10−3 (±4.84 × 10−4) |
| ΦBM6001.2 | 8.34 × 10−3 (±9.78 × 10−3) | 4.74 × 10−3 (±3.5 × 10−4) |
| ΦBM6001.3 | 2.35 × 10−1 (±1.92 × 10−1) | 3.01 × 10−2 (±2.17 × 10−2) |
| ΦBM6001.4 | 2.34 × 10−2 (±2.28 × 10−3) | 1.12 × 10−2 (±1.49 × 10−2) |
| ΦBM6001.5 | 1.12 × 10−5 (±2.43 × 10−6) | 3.23 × 10−7 (±1.35 × 10−8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombini, L.; Cuppone, A.M.; Tirziu, M.; Lazzeri, E.; Pozzi, G.; Santoro, F.; Iannelli, F. The Mobilome-Enriched Genome of the Competence-Deficient Streptococcus pneumoniae BM6001, the Original Host of Integrative Conjugative Element Tn5253, Is Phylogenetically Distinct from Historical Pneumococcal Genomes. Microorganisms 2023, 11, 1646. https://doi.org/10.3390/microorganisms11071646
Colombini L, Cuppone AM, Tirziu M, Lazzeri E, Pozzi G, Santoro F, Iannelli F. The Mobilome-Enriched Genome of the Competence-Deficient Streptococcus pneumoniae BM6001, the Original Host of Integrative Conjugative Element Tn5253, Is Phylogenetically Distinct from Historical Pneumococcal Genomes. Microorganisms. 2023; 11(7):1646. https://doi.org/10.3390/microorganisms11071646
Chicago/Turabian StyleColombini, Lorenzo, Anna Maria Cuppone, Mariana Tirziu, Elisa Lazzeri, Gianni Pozzi, Francesco Santoro, and Francesco Iannelli. 2023. "The Mobilome-Enriched Genome of the Competence-Deficient Streptococcus pneumoniae BM6001, the Original Host of Integrative Conjugative Element Tn5253, Is Phylogenetically Distinct from Historical Pneumococcal Genomes" Microorganisms 11, no. 7: 1646. https://doi.org/10.3390/microorganisms11071646