

Design of a Multi-Epitope Vaccine against Tuberculosis from Mycobacterium tuberculosis PE_PGRS49 and PE_PGRS56 Proteins by Reverse Vaccinology
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Amino Acid Sequences of PE_PGRS49 and PE_PGRS56
2.2. Prediction of B-Cell Epitopes
2.3. Selection of Frequent Alleles in the World’s Population
2.4. Prediction of Epitopes of T Cells (HLA Classes I and II)
2.5. Prediction of Antigenicity
2.6. Characterization of Selected B- and T-Cell Epitopes
2.7. Prediction of Docked Epitopes to MHC Protein
2.8. Conservation of Selected Epitopes
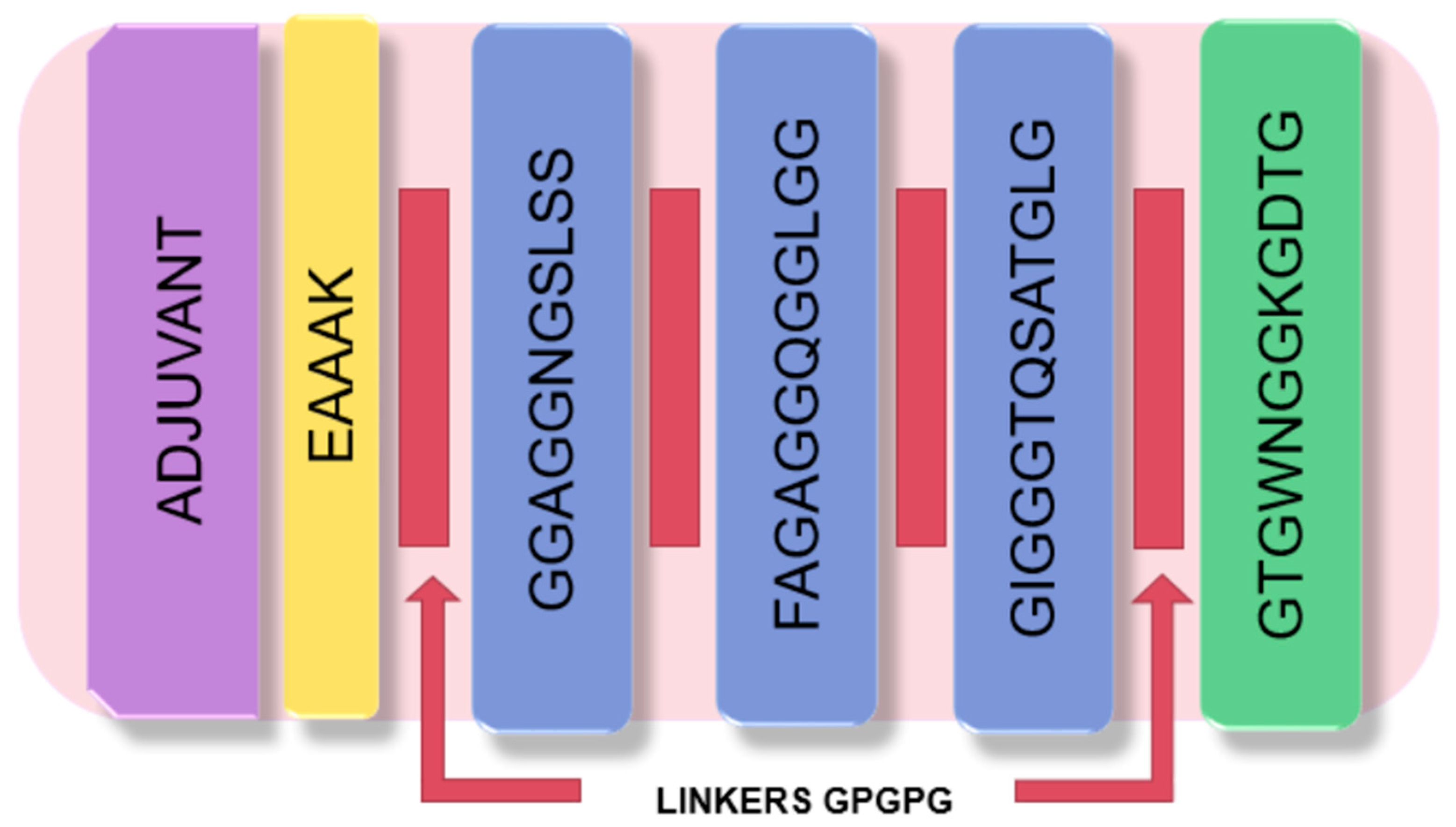
2.9. Construction of Vaccine Candidate
2.10. Vaccine Candidate Molecular Docking with an Immune Receptor (TLR-4)
2.11. Molecular Dynamics Simulation
3. Results
3.1. Prediction of B-Cell Epitopes
3.2. Prediction of T-Cell Epitopes
3.3. Selection of Epitopes
3.4. Prediction of Epitope Antigenicity
3.5. Characterization of Selected B- and T-Cell Epitopes
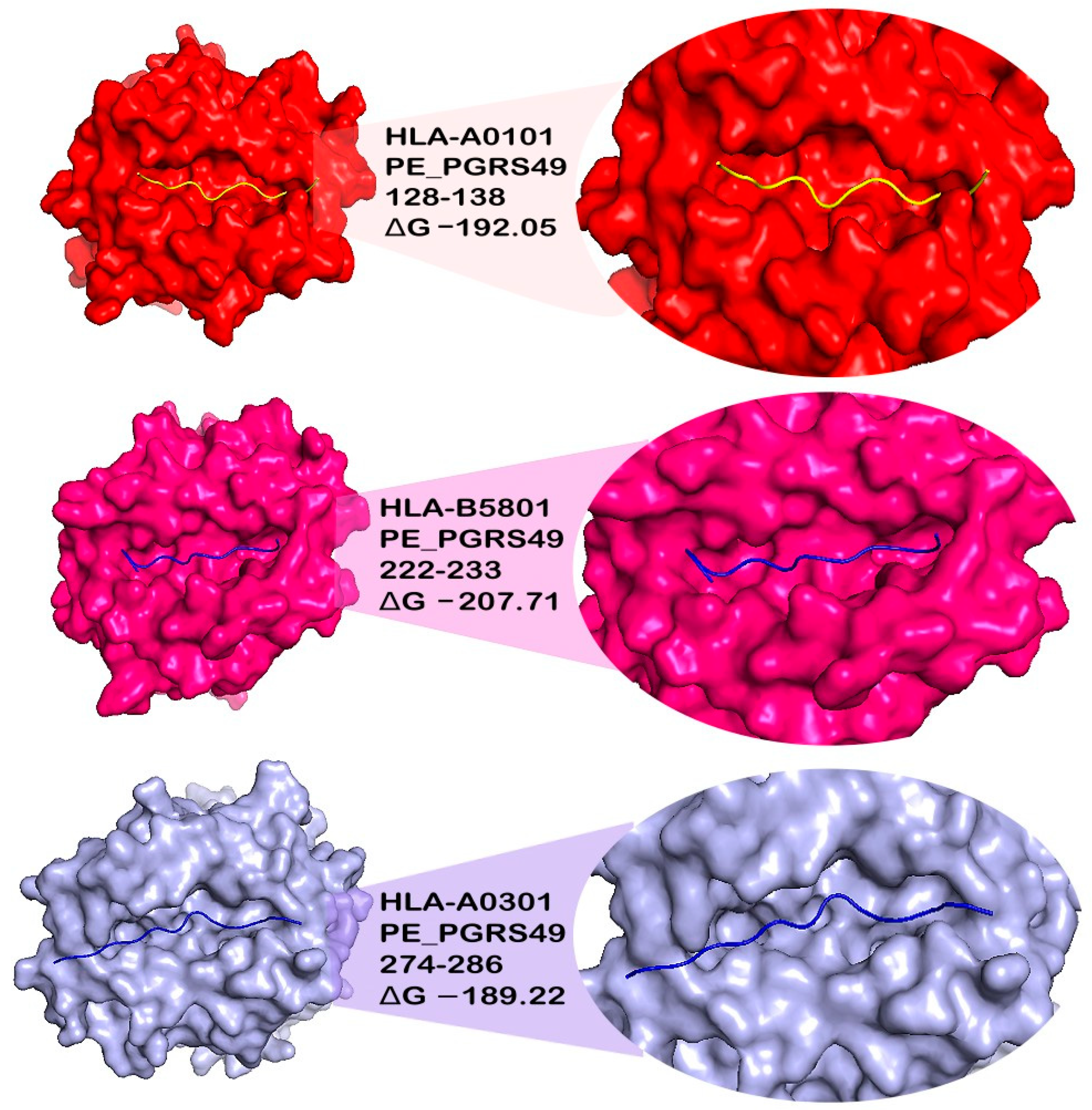
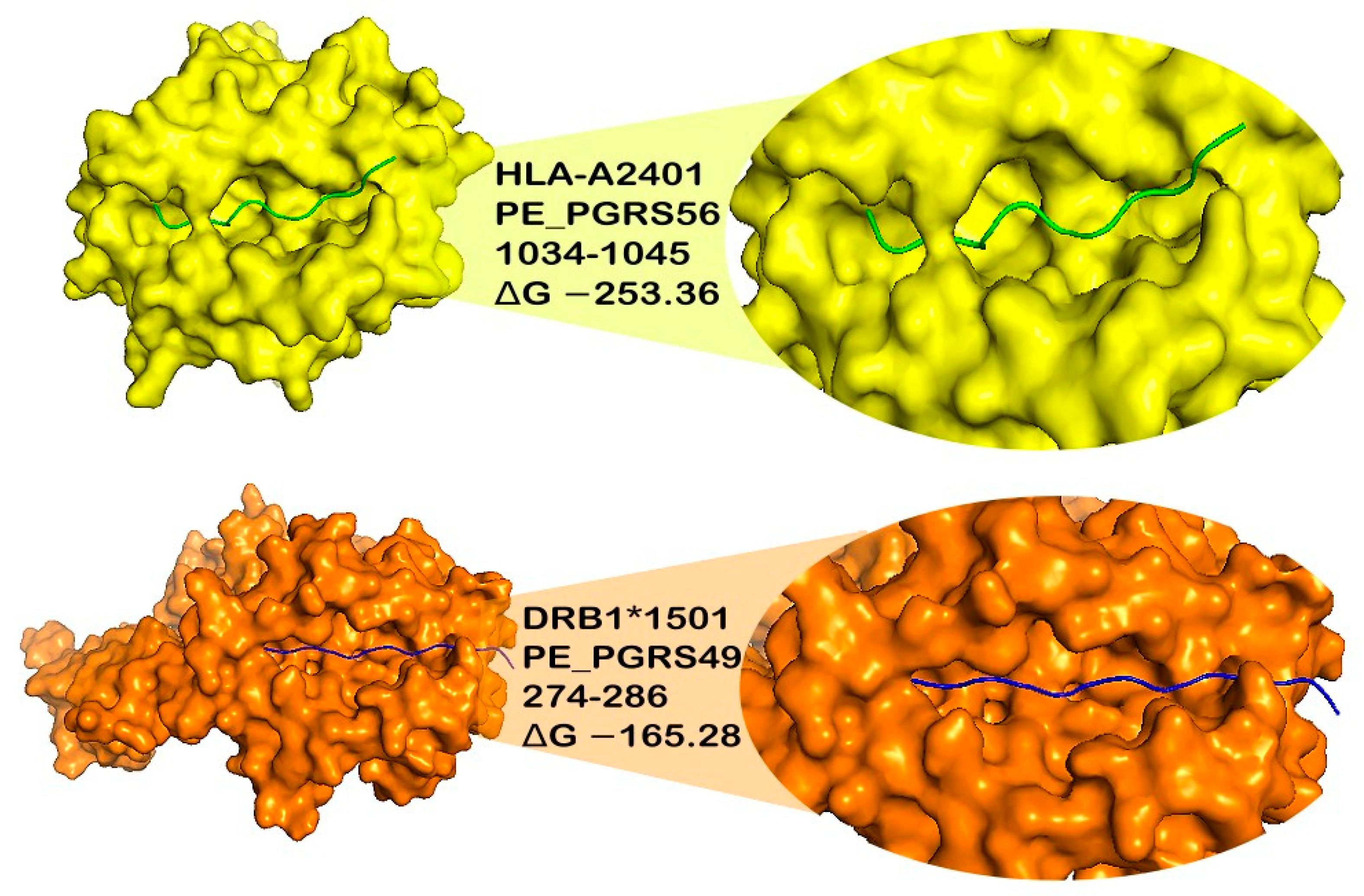
3.6. Docking of Epitopes to MHC Class I and II Proteins
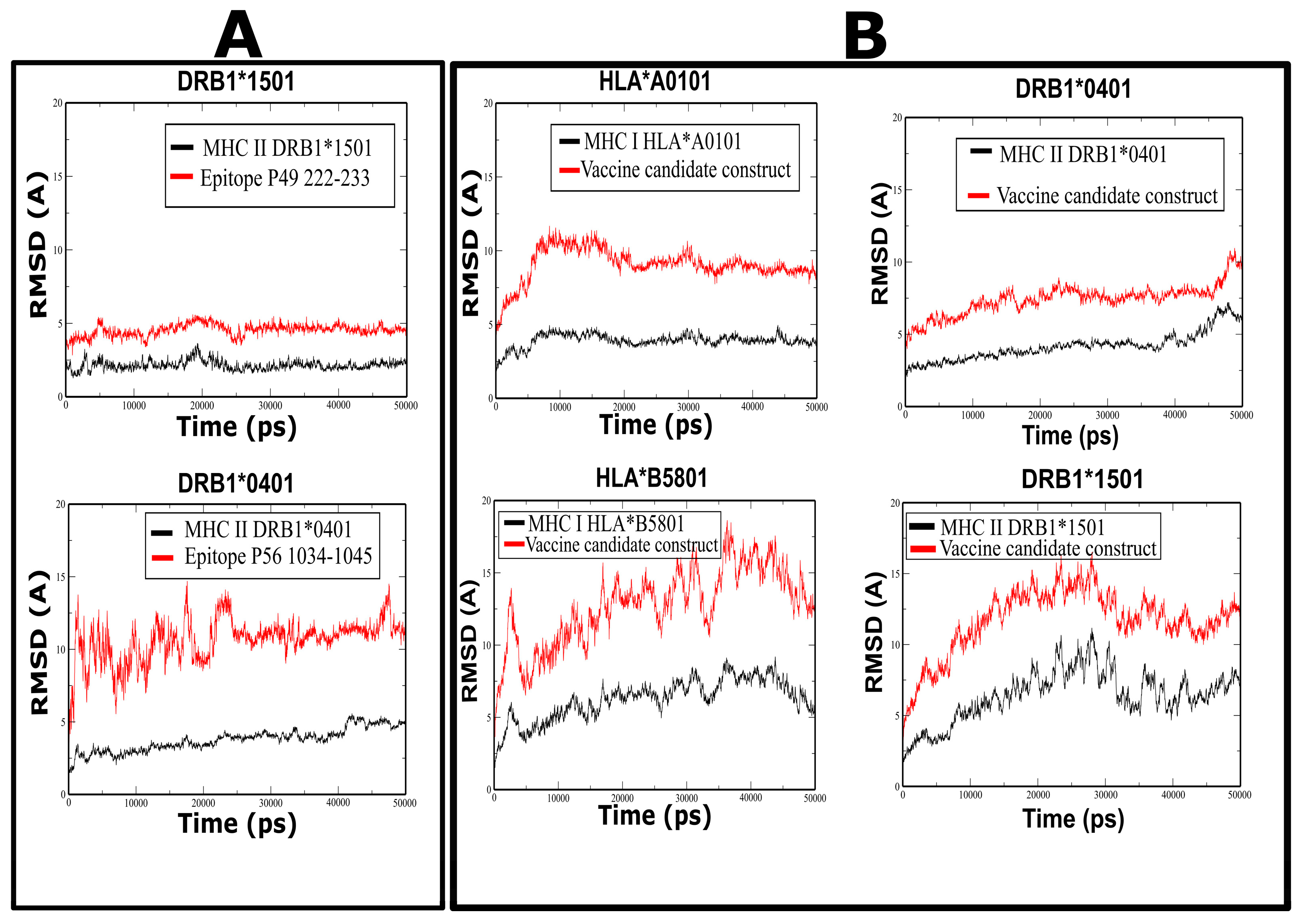
3.7. Molecular Dynamics Simulations of Peptide Complexes
3.8. Vaccine Candidate
3.9. Vaccine Candidate Molecular Docking with an Immune Receptor (TLR-4)
3.10. Molecular Dynamic Prediction for the Vaccine Candidate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2020. Available online: https://www.who.int/publications/i/item/9789240013131 (accessed on 23 December 2022).
- Ahmed, F.; Andrews, K.; Arshad, D.; Nekuri, P. A New FDA-approved Antibiotic for Drug-resistant Tuberculosis Treatment. J. Coll. Physicians Surg. Pak. 2020, 30, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug resistant Tuberculosis: A review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Lo, B.; Son, J. Phylogenomics and Comparative Genomic Studies Robustly Support Division of the Genus Mycobacterium into an Emended Genus Mycobacterium and Four Novel Genera. Front. Microbiol. 2018, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabin, S.; Herbig, A.; Vågene, Å.J.; Ahlström, T.; Bozovic, G.; Arcini, C.; Kühnert, D.; Bos, K.I. A seventeenth-century Mycobacterium tuberculosis genome supports a Neolithic emergence of the Mycobacterium tuberculosis complex. Genome Biol. 2020, 21, 201. [Google Scholar] [CrossRef]
- Ehtram, A.; Shariq, M.; Ali, S.; Quadir, N.; Sheikh, J.A.; Ahmad, F.; Sharma, T.; Ehtesham, N.Z.; Hasnain, S.E. Teleological cooption of Mycobacterium tuberculosis PE/PPE proteins as porins: Role in molecular immigration and emigration. Int. J. Med. Microbiol. 2021, 311, 151495. [Google Scholar] [CrossRef]
- Yang, Z.; Zeng, X.; Tsui, S.K. Investigating function roles of hypothetical proteins encoded by the Mycobacterium tuberculosis H37Rv genome. BMC Genom. 2019, 20, 394. [Google Scholar] [CrossRef] [Green Version]
- de Maio, F.; Berisio, R.; Manganelli, R.; Delogu, G. PE_PGRS proteins of Mycobacterium tuberculosis: A specialized molecular task force at the forefront of host-pathogen interaction. Virulence 2020, 11, 898–915. [Google Scholar] [CrossRef]
- Ates, L.S. New insights into the mycobacterial PE and PPE proteins provide a framework for future research. Mol. Microbiol. 2020, 113, 4–21. [Google Scholar] [CrossRef] [Green Version]
- Minerva, M.; de Maio, F.; Camassa, S.; Battah, B.; Ivana, P.; Manganelli, R.; Sanguinetti, M.; Sali, M.; Delogu, G. Evaluation of PE_PGRS33 as a potential surface target for humoral responses against Mycobacterium tuberculosis. Pathog. Dis. 2017, 75, ftx100. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.K.; Zhen, G.; Nzungize, L.; Stojkoska, A.; Duan, X.; Li, C.; Duan, W.; Xu, J.; Xie, J. Mycobacterium tuberculosis PE31 (Rv3477) Attenuates Host Cell Apoptosis and Promotes Recombinant M. smegmatis Intracellular Survival via Up-regulating GTPase Guanylate Binding Protein-1. Front. Cell. Infect. Microbiol. 2020, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xie, Y.; Luo, W.; Dou, Y.; Xiong, H.; Xiao, Z.; Zhang, X.L. PE_PGRS31-S100A9 Interaction Promotes Mycobacterial Survival in Macrophages through the Regulation of NF-κB-TNF-α Signaling and Arachidonic Acid Metabolism. Front. Microbiol. 2020, 11, 845. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Balaji, K.N. The PE and PPE proteins of Mycobacterium tuberculosis. Tuberculosis 2011, 91, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Meena, L.S. An overview to understand the role of PE_PGRS family proteins in Mycobacterium tuberculosis H37Rv and their potential as new drug targets. Biotechnol. Appl. Biochem. 2015, 62, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhou, Y.; Liu, S.; Zhang, X.L. PE_PGRS: Vital proteins in promoting mycobacterial survival and modulating host immunity and metabolism. Cell. Microbiol. 2021, 23, e13290. [Google Scholar] [CrossRef]
- Moxon, R.; Reche, P.A.; Rappuoli, R. Editorial: Reverse Vaccinology. Front. Immunol. 2019, 10, 2776. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Sharma, G.; Lee, S.S. Immunoinformatics Approach for the Identification and Characterization of T Cell and B Cell Epitopes towards the Peptide-Based Vaccine against SARS-CoV-2. Arch. Med. Res. 2021, 52, 362–370. [Google Scholar] [CrossRef]
- Monterrubio-López, G.P.; González-Y-Merchand, J.A.; Ribas-Aparicio, R.M. Identification of Novel Potential Vaccine Candidates against Tuberculosis Based on Reverse Vaccinology. Biomed Res. Int. 2015, 2015, 483150. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.J.; Delogu, G. The PE multigene family: A ‘molecular mantra’ for mycobacteria. Trends Microbiol. 2002, 10, 246–249. [Google Scholar] [CrossRef]
- Banu, S.; Honoré, N.; Saint-Joanis, B.; Philpott, D.; Prévost, M.C.; Cole, S.T. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol. Microbiol. 2002, 44, 9–19. [Google Scholar] [CrossRef]
- Espitia, C.; Laclette, J.P.; Mondragón-Palomino, M.; Amador, A.; Campuzano, J.; Martens, A.; Singh, M.; Cicero, R.; Zhang, Y.; Moreno, C. The PE-PGRS glycine-rich proteins of Mycobacterium tuberculosis: A new family of fibronectin-binding proteins? Microbiology 1999, 145 Pt 12, 3487–3495. [Google Scholar] [CrossRef] [Green Version]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Galarza, F.F.; McCabe, A.; Melo Dos Santos, E.J.; Takeshita, L.; Ghattaoraya, G.; Jones, A.R.; Middleton, D. Allele Frequency Net Database. Methods Mol. Biol. 2018, 1802, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Can, H.; Erkunt Alak, S.; Köseoğlu, A.E.; Döşkaya, M.; Ün, C. Do Toxoplasma gondii apicoplast proteins have antigenic potential? An in silico study. Comput. Biol. Chem. 2020, 84, 107158. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, C.; Richard, G.; Salverda, M.L.M.; Hindocha, P.; Martin, W.D.; de Groot, A.S.; Van Riet, E. In silico identification and modification of T cell epitopes in pertussis antigens associated with tolerance. Hum. Vaccines Immunother. 2020, 16, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.; Raghava, G.P. ProPred1: Prediction of promiscuous MHC Class-I binding sites. Bioinformatics 2003, 19, 1009–1014. [Google Scholar] [CrossRef] [Green Version]
- Reche, P.A.; Glutting, J.P.; Zhang, H.; Reinherz, E.L. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics 2004, 56, 405–419. [Google Scholar] [CrossRef] [Green Version]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P. Open Source Drug Discovery Consortium. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Jiang, Y.; Gong, T.; Zhang, Z.; Sun, X. Intranasal Vaccination against HIV-1 with Adenoviral Vector-Based Nanocomplex Using Synthetic TLR-4 Agonist Peptide as Adjuvant. Mol. Pharm. 2016, 13, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.O.; Al-Khafaji, K.; Sarker, M.T.; Taskin-Tok, T.; Rana, A.S.; Rahman, M.S. Design of a multi-epitope vaccine against SARS-CoV-2: Immunoinformatic and computational methods. RSC Adv. 2022, 12, 4288–4310. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Whitlow, E.; Mustafa, A.S.; Hanif, S.N.M. An Overview of the Development of New Vaccines for Tuberculosis. Vaccines 2020, 8, 586. [Google Scholar] [CrossRef]
- Meza, B.; Ascencio, F.; Sierra-Beltrán, A.P.; Torres, J.; Angulo, C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: An in silico approach. Infect. Genet. Evol. 2017, 49, 309–317. [Google Scholar] [CrossRef]
- Ahmad, I.; Ali, S.S.; Zafar, B.; Hashmi, H.F.; Shah, I.; Khan, S.; Suleman, M.; Khan, M.; Ullah, S.; Ali, S.; et al. Development of multi-epitope subunit vaccine for protection against the norovirus’ infections based on computational vaccinology. J. Biomol. Struct. Dyn. 2020, 7, 3098–3109. [Google Scholar] [CrossRef]
- Jagadeb, M.; Pattanaik, K.P.; Rath, S.N.; Sonawane, A. Identification and evaluation of immunogenic MHC-I and MHC-II binding peptides from Mycobacterium tuberculosis. Comput. Biol. Med. 2021, 130, 104203. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.B.; Niu, S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. 2021, 11, 1249. [Google Scholar] [CrossRef] [PubMed]
- Albutti, A. An integrated computational framework to design a multi-epitopes vaccine against Mycobacterium tuberculosis. Sci. Rep. 2021, 11, 21929. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Alam, A.; Ehtram, A.; Rani, A.; Grover, S.; Ehtesham, N.Z.; Hasnain, S.E. The Mycobacterium tuberculosis PE_PGRS Protein Family Acts as an Immunological Decoy to Subvert Host Immune Response. Int. J. Mol. Sci. 2022, 23, 525. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Tirado, D.; Arvizu-Flores, A.A.; Velazquez, C.; Garibay-Escobar, A. The role of immunoinformatics in the development of T-cell peptide-based vaccines against Mycobacterium tuberculosis. Expert Rev. Vaccines 2020, 19, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Vakili, B.; Nezafat, N.; Zare, B.; Erfani, N.; Akbari, M.; Ghasemi, Y.; Rahbar, M.R.; Hatam, G.R. A new multi-epitope peptide vaccine induces immune responses and protection against Leishmania infantum in BALB/c mice. Med. Microbiol. Immunol. 2020, 209, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Oladipo, E.K.; Ajayi, A.F.; Onile, O.S.; Ariyo, O.E.; Jimah, E.M.; Ezediuno, L.O.; Adebayo, O.I.; Adebayo, E.T.; Odeyemi, A.N.; Oyeleke, M.O.; et al. Designing a conserved peptide-based subunit vaccine against SARS-CoV-2 using immunoinformatics approach. Silico Pharmacol. 2021, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Kone, A.; Diarra, B.; Cohen, K.; Diabate, S.; Kone, B.; Diakite, M.T.; Diarra, H.; Sanogo, M.; Togo, A.C.G.; Sarro, Y.D.S.; et al. Differential HLA allele frequency in Mycobacterium africanum vs. Mycobacterium tuberculosis in Mali. HLA 2019, 93, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Albekairi, T.H.; Alshammari, A.; Alharbi, M.; Alshammary, A.F.; Tahir Ul Qamar, M.; Ullah, A.; Irfan, M.; Ahmad, S. Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach. Vaccines 2022, 10, 665. [Google Scholar] [CrossRef]
- Alzarea, S.I. Identification and construction of a multi-epitopes vaccine design against Klebsiella aerogenes: Molecular modeling study. Sci. Rep. 2022, 12, 14402. [Google Scholar] [CrossRef]
- Li, F.; Guo, X.; Xiang, D.; Pitt, M.E.; Bainomugisa, A.; Coin, L.J.M. Computational analysis and prediction of PE_PGRS proteins using machine learning. Comput. Struct. Biotechnol. J. 2022, 20, 662–674. [Google Scholar] [CrossRef]
- Sunita; Singhvi, N.; Singh, Y.; Shukla, P. Computational approaches in epitope design using DNA binding proteins as vaccine candidate in Mycobacterium tuberculosis. Infect. Genet. Evol. 2020, 83, 104357. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, J.; Aguilar, D.; Arriaga, K.; León, J.C.; Salas-Rangel, L.P.; González-y-Merchand, J.; Hernández-Pando, R.; Espitia, C. The PGRS domain of Mycobacterium tuberculosis PE_PGRS Rv1759c antigen is an efficient subunit vaccine to prevent reactivation in a murine model of chronic tuberculosis. Vaccine 2007, 25, 3722–3729. [Google Scholar] [CrossRef]
- Joshi, K.; Meena, S.; Meena, L.S. Analysis of predicted amino acid biosynthesis in Rv3344c in Mycobacterium tuberculosis H37 Rv using bioinformatics tools. Biotechnol. Appl. Biochem. 2020, 67, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Nebenzahl-Guimaraes, H.; van Laarhoven, A.; Farhat, M.R.; Koeken, V.A.C.M.; Mandemakers, J.J.; Zomer, A.; van Hijum, S.A.F.T.; Netea, M.G.; Murray, M.; van Crevel, R.; et al. Transmissible Mycobacterium tuberculosis strains share genetic markers and immune phenotypes. Am. J. Respir. Crit. Care Med. 2017, 195, 1519–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
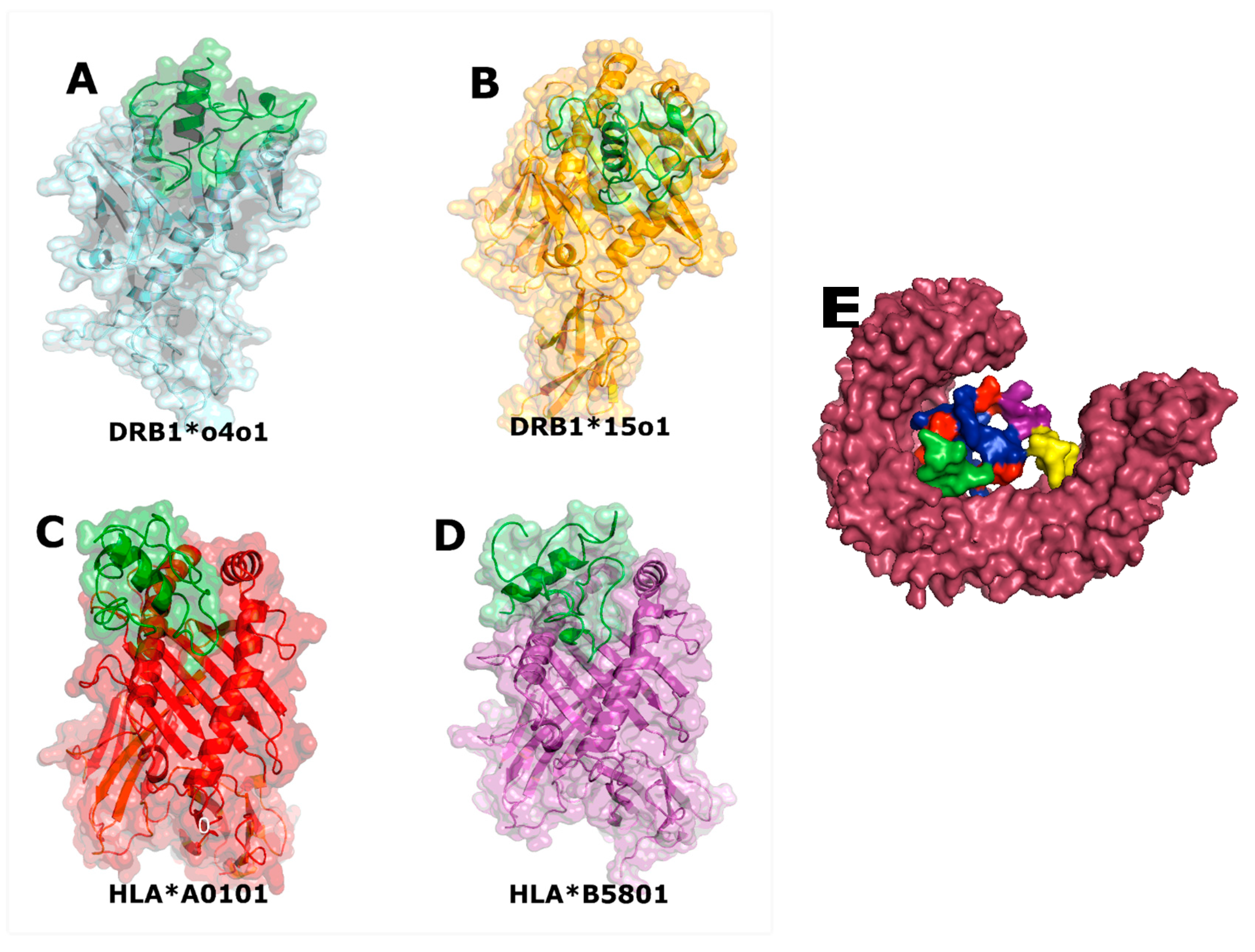{kind=link}
| PE_PGRS Protein | Epitope Sequence | Location | Size | Molecular Weight | Theoretical pI | Antigenicity | Allergenicity | Toxicity | Stability (Yes/No) | Estimated Half-Life (h) | Epitope Type |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 49 | GSFGATSGPASIGVTG | 58–73 | 16 | 1365.46 | 5.52 | 0.8794 | Probable allergen | Not toxic | Yes | 30 | B-cell |
| GSIGANSGIVGG | 109–120 | 12 | 988.07 | 5.52 | 1.0119 | Not allergen | Not toxic | Yes | 30 | B-cell, T-cell HLA I | |
| GGAGGNGSLSS | 128–138 | 12 | 919.90 | 5.52 | 1.4603 | Not allergen | Not toxic | Yes | 30 | B-cell, T-cell HLA I, II | |
| SGFFGGKGGFG | 177–187 | 11 | 1017.11 | 8.47 | 0.0638 | Probable allergen | Not toxic | Yes | 1.9 | B-cell, T-cell HLA I, II | |
| FAGAGGQGGLGG | 222–233 | 12 | 948.00 | 5.52 | 2.2969 | Not allergen | Not toxic | Yes | 1.9 | B-cell, T-cell HLA I, II | |
| GIGGGTQSATGLG | 274–286 | 13 | 1075.14 | 5.52 | 2.0843 | Not allergen | Not toxic | Yes | 30 | B-cell, T-cell HLA I, II | |
| 56 | DAGKAGTGSAPGT | 101–113 | 13 | 1089.13 | 5.84 | 1.8399 | Not allergen | Not toxic | Yes | 1.1 | B-cell, T-cell HLA II |
| GGAAGAATAG | 338–347 | 10 | 702.72 | 5.52 | 2.0568 | Probable allergen | Not toxic | Yes | 30 | B-cell, T-cell HLA II | |
| TVGGGTVPAGSGGQG | 579–593 | 15 | 1201.26 | 5.19 | 2.1006 | Not allergen | Not toxic | No | 7.2 | B-cell, T-cell HLA II | |
| NTANMTAQAG | 780–789 | 10 | 978.04 | 5.52 | 0.9916 | Probable allergen | Not toxic | Yes | 1.4 | B-cell, T-cell HLA II | |
| GTGWNGGKGDTG | 1034–1045 | 12 | 1106.12 | 5.84 | 2.2536 | Not allergen | Not toxic | Yes | 30 | T-cell HLA II |
| Protein | Epitope | HLA-A0101 | HLA-A0201 | HLA-A0301 | HLA-A2401 | HLA-B5801 | DRB1*0401 | DRB1*1501 |
|---|---|---|---|---|---|---|---|---|
| PE_PGRS49 | 58–73 | −211.89 | −226.14 | −225.61 | −219.56 | −229.65 | NB | −214.04 |
| 109–120 | −195.58 | −198.88 | −191.89 | −199.03 | −198.8 | NB | −160.73 | |
| 128–138 | −192.05 | −166.43 | −181.6 | −186.36 | −182.14 | NB | −163.5 | |
| 177–187 | −220.21 | −223.45 | −240.43 | −242.92 | −262.45 | NB | −221.46 | |
| 222–233 | −191.82 | −220.33 | −207.86 | −215.65 | −207.71 | −146.49 | −183.23 | |
| 274–286 | −190.24 | −197.71 | −189.22 | −203.38 | −199.63 | NB | −165.28 | |
| PE_PGRS56 | 101–113 | −193.22 | −175.7 | −170.51 | −191.85 | −188.23 | NB | −156.42 |
| 338–347 | −175.35 | −170.81 | −162.55 | −180.24 | −176.91 | NB | −136.44 | |
| 579–593 | −203.23 | −201.54 | −182.91 | −207.04 | −197.08 | −142.88 | −179.87 | |
| 780–789 | −206.4 | −194.52 | −214.13 | −224.82 | −205.7 | NB | −167.75 | |
| 1034–1045 | −201.1 | −210.28 | −216.18 | −253.36 | −209.21 | −151.67 | −197.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruaro-Moreno, M.; Monterrubio-López, G.P.; Reyes-Gastellou, A.; Castelán-Vega, J.A.; Jiménez-Alberto, A.; Aparicio-Ozores, G.; Delgadillo-Gutiérrez, K.; González-Y-Merchand, J.A.; Ribas-Aparicio, R.M. Design of a Multi-Epitope Vaccine against Tuberculosis from Mycobacterium tuberculosis PE_PGRS49 and PE_PGRS56 Proteins by Reverse Vaccinology. Microorganisms 2023, 11, 1647. https://doi.org/10.3390/microorganisms11071647
Ruaro-Moreno M, Monterrubio-López GP, Reyes-Gastellou A, Castelán-Vega JA, Jiménez-Alberto A, Aparicio-Ozores G, Delgadillo-Gutiérrez K, González-Y-Merchand JA, Ribas-Aparicio RM. Design of a Multi-Epitope Vaccine against Tuberculosis from Mycobacterium tuberculosis PE_PGRS49 and PE_PGRS56 Proteins by Reverse Vaccinology. Microorganisms. 2023; 11(7):1647. https://doi.org/10.3390/microorganisms11071647
Chicago/Turabian StyleRuaro-Moreno, Maritriny, Gloria Paulina Monterrubio-López, Abraham Reyes-Gastellou, Juan Arturo Castelán-Vega, Alicia Jiménez-Alberto, Gerardo Aparicio-Ozores, Karen Delgadillo-Gutiérrez, Jorge Alberto González-Y-Merchand, and Rosa María Ribas-Aparicio. 2023. "Design of a Multi-Epitope Vaccine against Tuberculosis from Mycobacterium tuberculosis PE_PGRS49 and PE_PGRS56 Proteins by Reverse Vaccinology" Microorganisms 11, no. 7: 1647. https://doi.org/10.3390/microorganisms11071647