

Endogenous Retroviruses as Modulators of Innate Immunity
Abstract
:1. Introduction
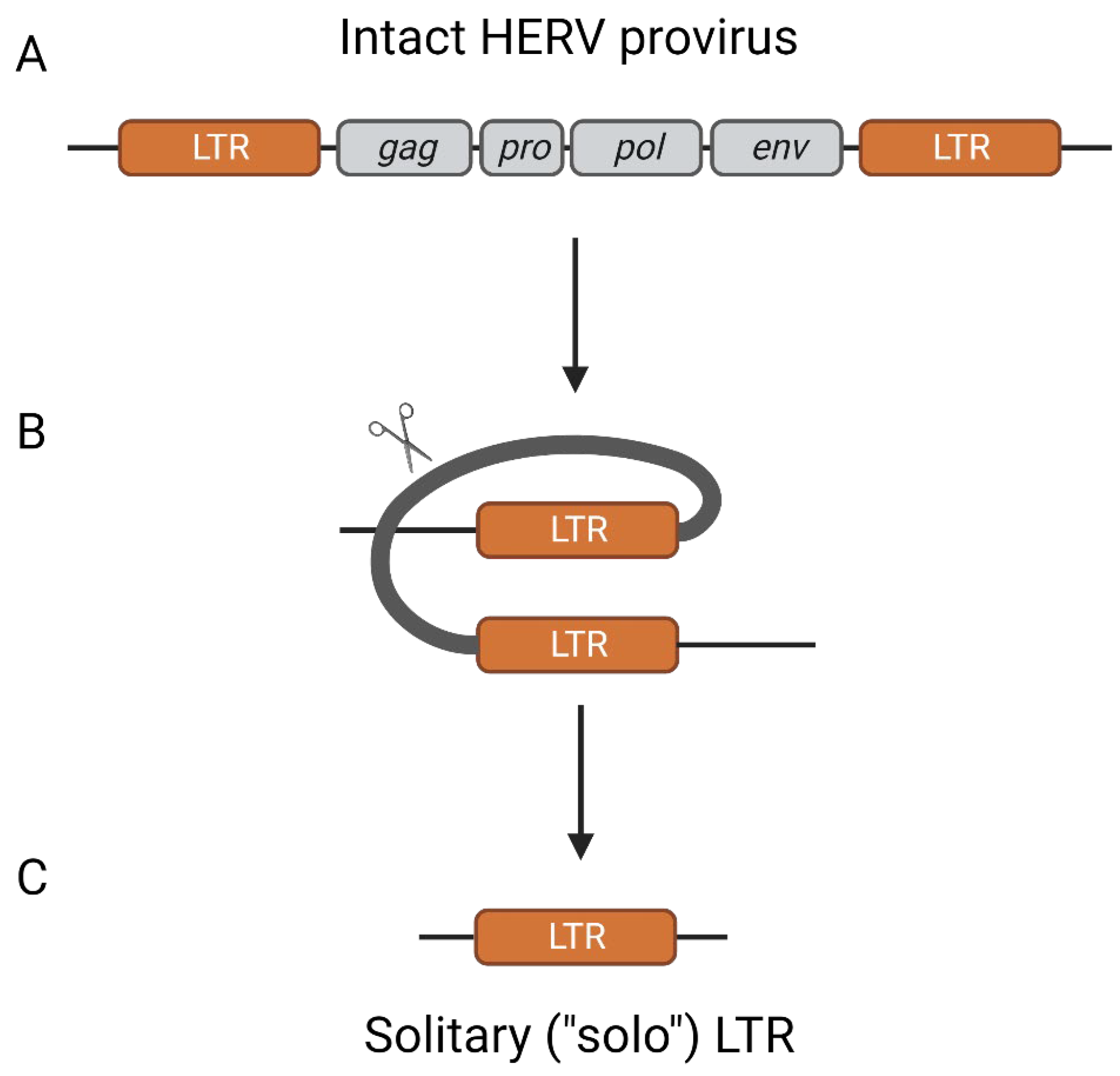
2. Origins of Endogenous Retroviruses
3. Epigenetic Regulation of HERV Activity
3.1. DNA Methylation
3.2. Histone Modifications
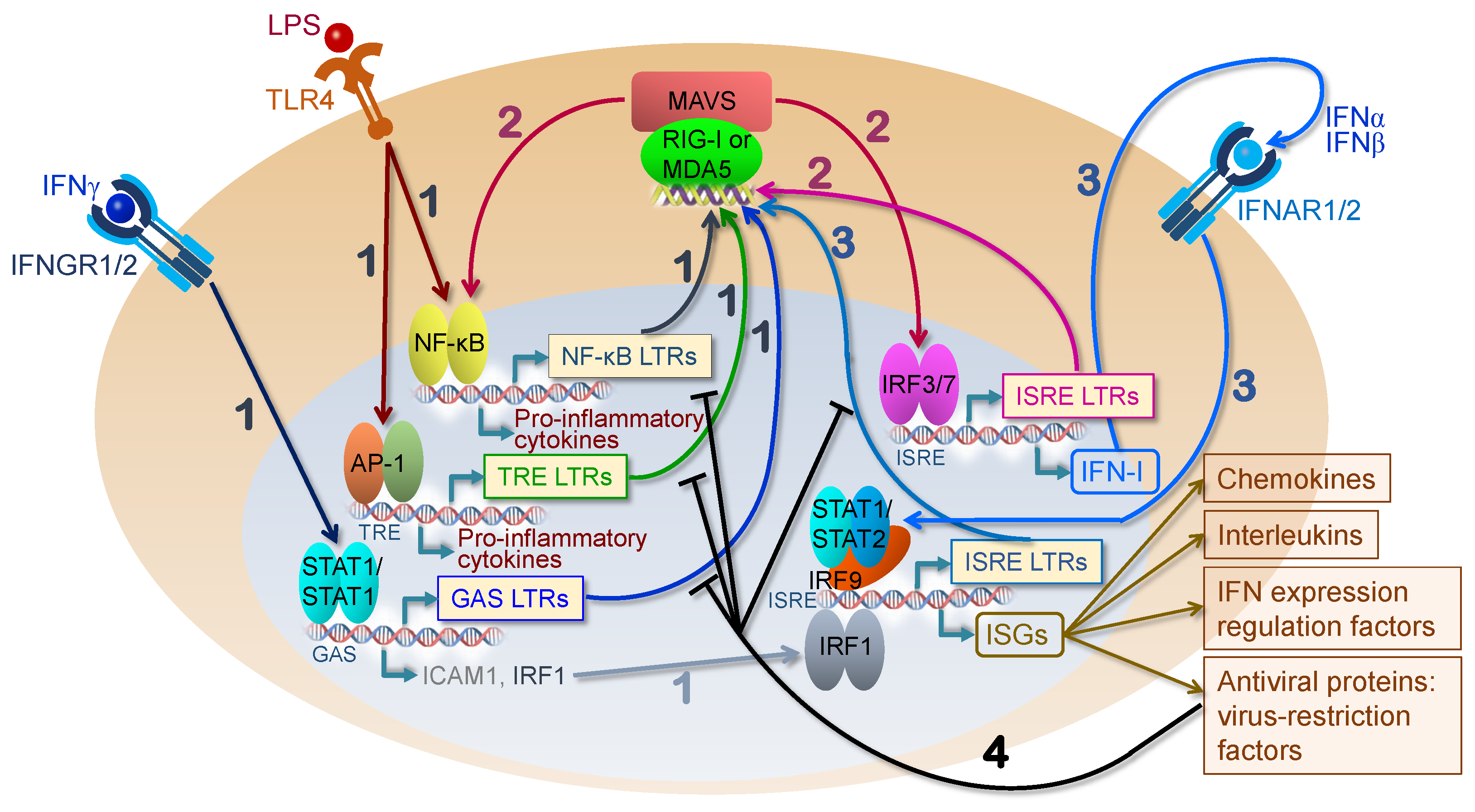
4. HERV Elements as Genomic Enhancers of Innate Immunity
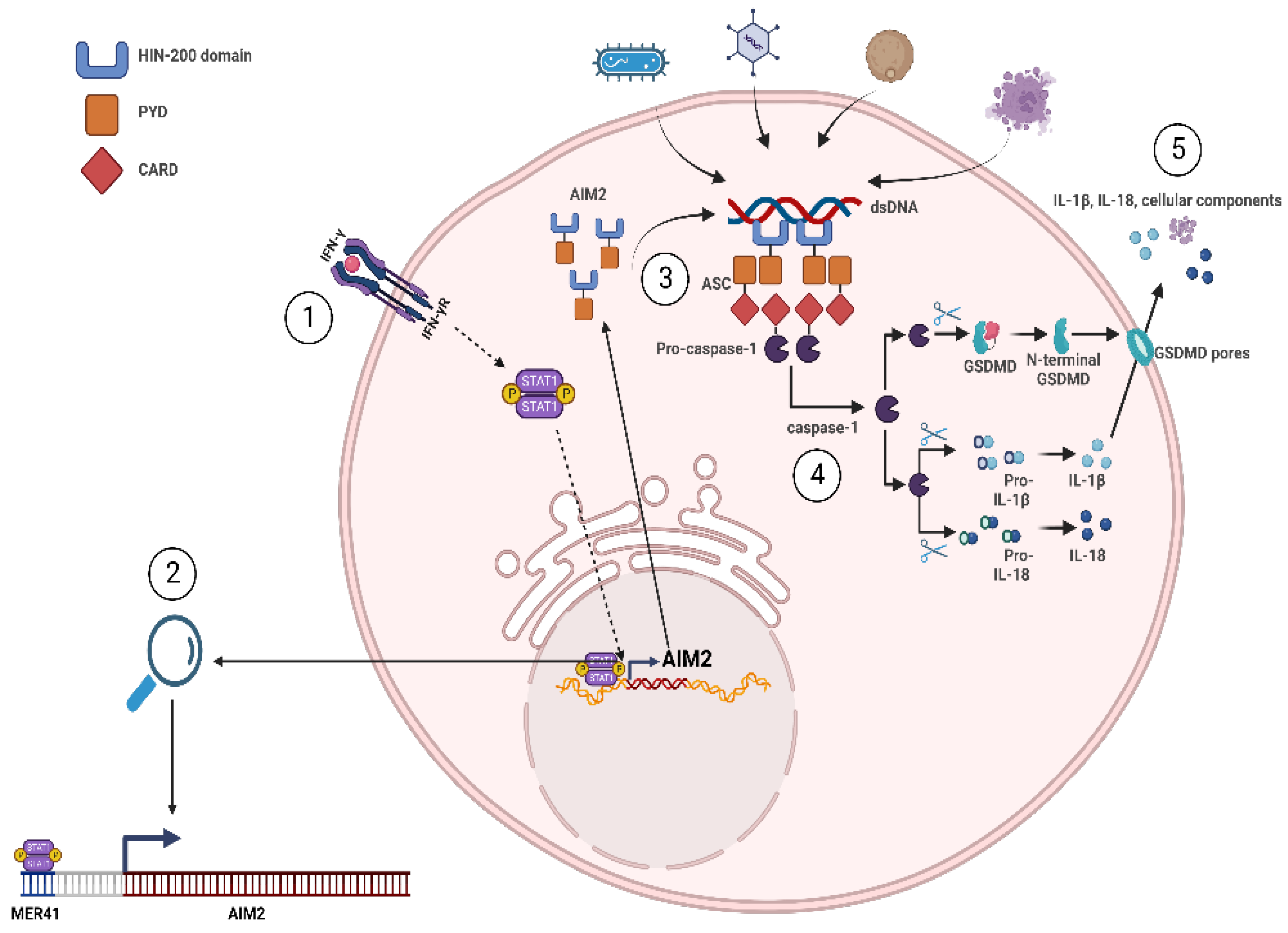
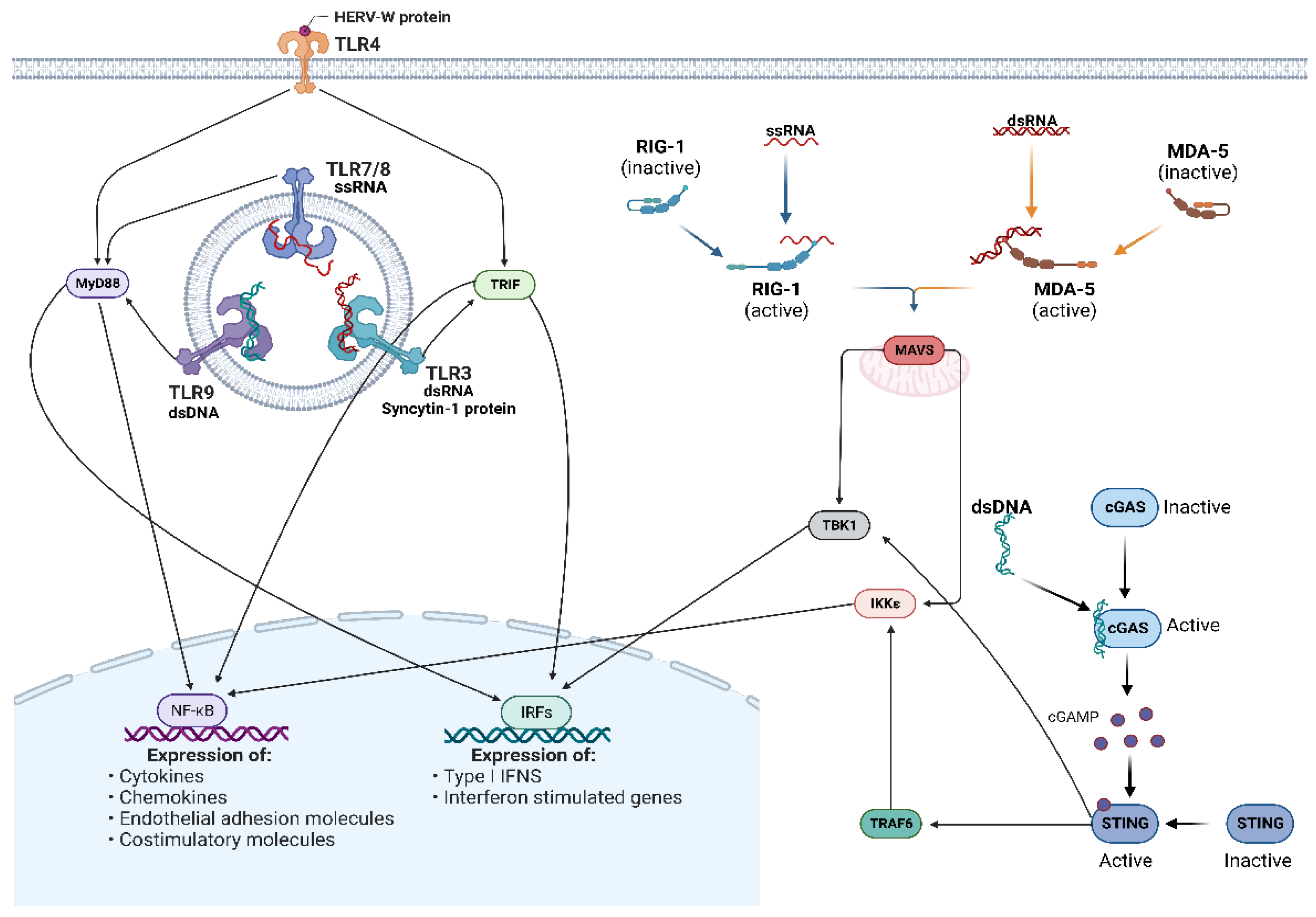
5. HERVs as Activators of the Innate Immune Response
5.1. Toll-Like Receptors
5.2. RIG-I-Like Receptors and MAVS Signaling
5.3. cGAS/STING Pathway
6. HERVs in a Clinical Setting
6.1. HERVs and Chronic Inflammation/Autoimmunity: Multiple Sclerosis
6.2. Rheumatoid Arthritis
6.3. Systemic Lupus Erythematosus
6.4. Pulmonary Arterial Hypertension
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
USU Disclaimer
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Petropoulos, C. Retroviral Taxonomy, Protein Structures, Sequences, and Genetic Maps; Cold Spring Harbor: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Greenwood, A.D.; Ishida, Y.; O’Brien, S.P.; Roca, A.L.; Eiden, M.V. Transmission, Evolution, and Endogenization: Lessons Learned from Recent Retroviral Invasions. Microbiol. Mol. Biol. Rev. 2018, 82, e00044-17. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, G.; Cui, J. Origin and Deep Evolution of Human Endogenous Retroviruses in Pan-Primates. Viruses 2022, 14, 1370. [Google Scholar] [CrossRef]
- Durnaoglu, S.; Lee, S.K.; Ahnn, J. Syncytin, envelope protein of human endogenous retrovirus (HERV): No longer ‘fossil’ in human genome. Anim. Cells Syst. 2021, 25, 358–368. [Google Scholar] [CrossRef]
- Dupressoir, A.; Lavialle, C.; Heidmann, T. From ancestral infectious retroviruses to bona fide cellular genes: Role of the captured syncytins in placentation. Placenta 2012, 33, 663–671. [Google Scholar] [CrossRef]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, E.D. Perpetually mobile footprints of ancient infections in human genome. FEBS Lett. 1998, 428, 1–6. [Google Scholar] [CrossRef] [Green Version]
- de Parseval, N.; Heidmann, T. Human endogenous retroviruses: From infectious elements to human genes. Cytogenet. Genome Res. 2005, 110, 318–332. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Boller, K.; Schonfeld, K.; Lischer, S.; Fischer, N.; Hoffmann, A.; Kurth, R.; Tonjes, R.R. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J. Gen. Virol. 2008, 89, 567–572. [Google Scholar] [CrossRef]
- Faff, O.; Murray, A.B.; Schmidt, J.; Leib-Mosch, C.; Erfle, V.; Hehlmann, R. Retrovirus-like particles from the human T47D cell line are related to mouse mammary tumour virus and are of human endogenous origin. J. Gen. Virol. 1992, 73 Pt 5, 1087–1097. [Google Scholar] [CrossRef]
- Hughes, J.F.; Coffin, J.M. Human endogenous retroviral elements as indicators of ectopic recombination events in the primate genome. Genetics 2005, 171, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Larsson, E. Human endogenous retroviruses. BioEssays 1988, 9, 191–196. [Google Scholar]
- Hanke, K.; Hohn, O.; Bannert, N. HERV-K(HML-2), a seemingly silent subtenant—But still waters run deep. APMIS 2016, 124, 67–87. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.N.; Carnegie, P.R.; Martin, J.; Davari Ejtehadi, H.; Hooley, P.; Roden, D.; Rowland-Jones, S.; Warren, P.; Astley, J.; Murray, P.G. Demystified. Human endogenous retroviruses. Mol. Pathol. 2003, 56, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Turner, G.; Barbulescu, M.; Su, M.; Jensen-Seaman, M.I.; Kidd, K.K.; Lenz, J. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 2001, 11, 1531–1535. [Google Scholar]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Rah, H.; Green, T.; Lee, Y.K.; Lim, D.; Nemzek, J.; Wahl, W.; Greenhalgh, D.; Cho, K. Divergent and dynamic activity of endogenous retroviruses in burn patients and their inflammatory potential. Exp. Mol. Pathol. 2014, 96, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Belshaw, R.; Katzourakis, A.; Paces, J.; Burt, A.; Tristem, M. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol. Biol. Evol. 2005, 22, 814–817. [Google Scholar] [CrossRef] [Green Version]
- Mayer, J.; Blomberg, J.; Seal, R.L. A revised nomenclature for transcribed human endogenous retroviral loci. Mob. DNA 2011, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Gifford, R.J.; Blomberg, J.; Coffin, J.M.; Fan, H.; Heidmann, T.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W.E. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 2018, 15, 59. [Google Scholar] [CrossRef]
- Lavie, L.; Medstrand, P.; Schempp, W.; Meese, E.; Mayer, J. Human endogenous retrovirus family HERV-K(HML-5): Status, evolution, and reconstruction of an ancient betaretrovirus in the human genome. J. Virol. 2004, 78, 8788–8798. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.A.; Prassolov, V.; Garazha, A.V. Friends-Enemies: Endogenous Retroviruses Are Major Transcriptional Regulators of Human DNA. Front. Chem. 2017, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Hurst, T.P.; Magiorkinis, G. Epigenetic Control of Human Endogenous Retrovirus Expression: Focus on Regulation of Long-Terminal Repeats (LTRs). Viruses 2017, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Seifarth, W.; Frank, O.; Zeilfelder, U.; Spiess, B.; Greenwood, A.D.; Hehlmann, R.; Leib-Mosch, C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J. Virol. 2005, 79, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Flockerzi, A.; Ruggieri, A.; Frank, O.; Sauter, M.; Maldener, E.; Kopper, B.; Wullich, B.; Seifarth, W.; Muller-Lantzsch, N.; Leib-Mosch, C.; et al. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genom. 2008, 9, 354. [Google Scholar] [CrossRef] [Green Version]
- Burn, A.; Roy, F.; Freeman, M.; Coffin, J.M. Widespread expression of the ancient HERV-K (HML-2) provirus group in normal human tissues. PLoS Biol. 2022, 20, e3001826. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Choy, M.-K.; Movassagh, M.; Goh, H.-G.; Bennett, M.R.; Down, T.A.; Foo, R.S.Y. Genome-wide conserved consensus transcription factor binding motifs are hyper-methylated. BMC Genom. 2010, 11, 519. [Google Scholar] [CrossRef] [Green Version]
- Tate, P.H.; Bird, A.P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993, 3, 226–231. [Google Scholar]
- Nan, X.; Ng, H.-H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Szpakowski, S.; Sun, X.; Lage, J.M.; Dyer, A.; Rubinstein, J.; Kowalski, D.; Sasaki, C.; Costa, J.; Lizardi, P.M. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 2009, 448, 151–167. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar]
- Daskalakis, M.; Brocks, D.; Sheng, Y.H.; Islam, M.S.; Ressnerova, A.; Assenov, Y.; Milde, T.; Oehme, I.; Witt, O.; Goyal, A.; et al. Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle 2018, 17, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, S.; Lin, S.; Guo, Y.; Deng, W.; Zhang, Y.; Xue, Y. WERAM: A database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 2017, 45, D264–D270. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Wang, Y.; Gan, L.; Bai, Y.; Han, W.; Wang, E.; Wang, J. Importance of electrostatic interactions in the association of intrinsically disordered histone chaperone Chz1 and histone H2A.Z-H2B. PLoS Comput. Biol. 2012, 8, e1002608. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kouzarides, T.; Schreiber, S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8695–8700. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Thompson, P.J.; Goyal, P.; Jones, S.J.M.; Singh, P.B.; Karimi, M.M.; Lorincz, M.C. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 2013, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA Methylation and SETDB1/H3K9me3 Regulate Predominantly Distinct Sets of Genes, Retroelements, and Chimeric Transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar]
- Liu, M.; Thomas, S.L.; DeWitt, A.K.; Zhou, W.; Madaj, Z.B.; Ohtani, H.; Baylin, S.B.; Liang, G.; Jones, P.A. Dual Inhibition of DNA and Histone Methyltransferases Increases Viral Mimicry in Ovarian Cancer Cells. Cancer Res. 2018, 78, 5754–5766. [Google Scholar] [CrossRef] [Green Version]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Takemoto, K.; Shinkai, Y. A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun. 2018, 9, 1683. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef] [Green Version]
- Takikita, S.; Muro, R.; Takai, T.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Oda, H.; Kitajima, M.; Oshima, K.; Hattori, M.; et al. A Histone Methyltransferase ESET Is Critical for T Cell Development. J. Immunol. 2016, 197, 2269–2279. [Google Scholar] [CrossRef] [Green Version]
- Curty, G.; Iñiguez, L.P.; Nixon, D.F.; Soares, M.A.; de Mulder Rougvie, M. Hallmarks of retroelement expression in T-cells treated with HDAC inhibitors. Front. Virol. 2021, 1, 756635. [Google Scholar]
- White, C.H.; Beliakova-Bethell, N.; Lada, S.M.; Breen, M.S.; Hurst, T.P.; Spina, C.A.; Richman, D.D.; Frater, J.; Magiorkinis, G.; Woelk, C.H. Transcriptional Modulation of Human Endogenous Retroviruses in Primary CD4+ T Cells Following Vorinostat Treatment. Front. Immunol. 2018, 9, 603. [Google Scholar] [CrossRef]
- Montesion, M.; Williams, Z.H.; Subramanian, R.P.; Kuperwasser, C.; Coffin, J.M. Promoter expression of HERV-K (HML-2) provirus-derived sequences is related to LTR sequence variation and polymorphic transcription factor binding sites. Retrovirology 2018, 15, 57. [Google Scholar] [CrossRef]
- Fuchs, N.V.; Kraft, M.; Tondera, C.; Hanschmann, K.M.; Lower, J.; Lower, R. Expression of the human endogenous retrovirus (HERV) group HML-2/HERV-K does not depend on canonical promoter elements but is regulated by transcription factors Sp1 and Sp3. J. Virol. 2011, 85, 3436–3448. [Google Scholar] [CrossRef]
- Lavie, L.; Kitova, M.; Maldener, E.; Meese, E.; Mayer, J. CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J. Virol. 2005, 79, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Durnaoglu, S.; Lee, S.K.; Ahnn, J. Human Endogenous Retroviruses as Gene Expression Regulators: Insights from Animal Models into Human Diseases. Mol. Cells 2021, 44, 861–878. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Gogvadze, E.; Stukacheva, E.; Buzdin, A.; Sverdlov, E. Human-specific modulation of transcriptional activity provided by endogenous retroviral insertions. J. Virol. 2009, 83, 6098–6105. [Google Scholar] [CrossRef] [Green Version]
- Suntsova, M.; Gogvadze, E.V.; Salozhin, S.; Gaifullin, N.; Eroshkin, F.; Dmitriev, S.E.; Martynova, N.; Kulikov, K.; Malakhova, G.; Tukhbatova, G.; et al. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc. Natl. Acad. Sci. USA 2013, 110, 19472–19477. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.L.; Zhao, Z.K.; Zhu, F. The role of human endogenous retroviral long terminal repeat sequences in human cancer (Review). Int. J. Mol. Med. 2013, 32, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Garazha, A.; Ivanova, A.; Suntsova, M.; Malakhova, G.; Roumiantsev, S.; Zhavoronkov, A.; Buzdin, A. New bioinformatic tool for quick identification of functionally relevant endogenous retroviral inserts in human genome. Cell Cycle 2015, 14, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Wang, B.; Tian, Y.; Yin, Q. AIM2 Inflammasome Assembly and Signaling. Adv. Exp. Med. Biol. 2019, 1172, 143–155. [Google Scholar] [CrossRef]
- Matyszewski, M.; Morrone, S.R.; Sohn, J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl. Acad. Sci. USA 2018, 115, E1963–E1972. [Google Scholar] [CrossRef] [Green Version]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Ruangkiattikul, N.; Nerlich, A.; Abdissa, K.; Lienenklaus, S.; Suwandi, A.; Janze, N.; Laarmann, K.; Spanier, J.; Kalinke, U.; Weiss, S.; et al. cGAS-STING-TBK1-IRF3/7 induced interferon-beta contributes to the clearing of non tuberculous mycobacterial infection in mice. Virulence 2017, 8, 1303–1315. [Google Scholar] [CrossRef] [Green Version]
- Perron, H.; Germi, R.; Bernard, C.; Garcia-Montojo, M.; Deluen, C.; Farinelli, L.; Faucard, R.; Veas, F.; Stefas, I.; Fabriek, B.O.; et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult. Scler. 2012, 18, 1721–1736. [Google Scholar] [CrossRef]
- Sun, S.; Qi, W.; Rehrauer, H.; Ronner, M.; Hariharan, A.; Wipplinger, M.; Meiller, C.; Stahel, R.; Fruh, M.; Cerciello, F.; et al. Viral Mimicry Response Is Associated With Clinical Outcome in Pleural Mesothelioma. JTO Clin. Res. Rep. 2022, 3, 100430. [Google Scholar] [CrossRef]
- Attermann, A.S.; Bjerregaard, A.M.; Saini, S.K.; Gronbaek, K.; Hadrup, S.R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 2018, 29, 2183–2191. [Google Scholar] [CrossRef]
- Mikhalkevich, N.; O’Carroll, I.P.; Tkavc, R.; Lund, K.; Sukumar, G.; Dalgard, C.L.; Johnson, K.R.; Li, W.; Wang, T.; Nath, A.; et al. Response of human macrophages to gamma radiation is mediated via expression of endogenous retroviruses. PLoS Pathog. 2021, 17, e1009305. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Wang, P.; Li, S.; Zeng, J.; Tu, X.; Yan, Q.; Xiao, Z.; Pan, M.; Zhu, F. Syncytin-1, an endogenous retroviral protein, triggers the activation of CRP via TLR3 signal cascade in glial cells. Brain Behav. Immun. 2018, 67, 324–334. [Google Scholar] [CrossRef]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J. Immunol. 2006, 176, 7636–7644. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Lubben, W.; Slomka, H.; Gebler, J.; Konert, M.; Cai, C.; Neubrandt, L.; Prazeres da Costa, O.; Paul, S.; Dehnert, S.; et al. Nucleic acid-sensing Toll-like receptors are essential for the control of endogenous retrovirus viremia and ERV-induced tumors. Immunity 2012, 37, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Lima-Junior, D.S.; Krishnamurthy, S.R.; Bouladoux, N.; Collins, N.; Han, S.J.; Chen, E.Y.; Constantinides, M.G.; Link, V.M.; Lim, A.I.; Enamorado, M.; et al. Endogenous retroviruses promote homeostatic and inflammatory responses to the microbiota. Cell 2021, 184, 3794–3811.e19. [Google Scholar] [CrossRef]
- Hidmark, A.; von Saint Paul, A.; Dalpke, A.H. Cutting edge: TLR13 is a receptor for bacterial RNA. J. Immunol. 2012, 189, 2717–2721. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Nie, L.; Cai, S.Y.; Shao, J.Z.; Chen, J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front. Immunol. 2018, 9, 1523. [Google Scholar] [CrossRef] [Green Version]
- Behzadi, P.; Garcia-Perdomo, H.A.; Karpinski, T.M. Toll-Like Receptors: General Molecular and Structural Biology. J. Immunol. Res 2021, 2021, 9914854. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Discrimination between Self and Non-Self-Nucleic Acids by the Innate Immune System. Int. Rev. Cell Mol. Biol. 2019, 344, 1–30. [Google Scholar] [CrossRef]
- Domansky, A.N.; Kopantzev, E.P.; Snezhkov, E.V.; Lebedev, Y.B.; Leib-Mosch, C.; Sverdlov, E.D. Solitary HERV-K LTRs possess bi-directional promoter activity and contain a negative regulatory element in the U5 region. FEBS Lett. 2000, 472, 191–195. [Google Scholar] [CrossRef]
- Qiu, X.; Xiao, Y.; Wu, J.; Gan, L.; Huang, Y.; Wang, J. C-Reactive Protein and Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2019, 10, 384. [Google Scholar] [CrossRef]
- Luan, Y.Y.; Yao, Y.M. The Clinical Significance and Potential Role of C-Reactive Protein in Chronic Inflammatory and Neurodegenerative Diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Li, S.; Hu, Y.; Yu, H.; Luo, F.; Zhang, Q.; Zhu, F. Implication of the env gene of the human endogenous retrovirus W family in the expression of BDNF and DRD3 and development of recent-onset schizophrenia. Schizophr. Bull. 2011, 37, 988–1000. [Google Scholar] [CrossRef]
- Christensen, T. HERVs in Neuropathogenesis. J. Neuroimmune Pharmacol. 2010, 5, 326–335. [Google Scholar] [CrossRef]
- Maeshima, N.; Fernandez, R.C. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Shinya, K.; Ito, M.; Makino, A.; Tanaka, M.; Miyake, K.; Eisfeld, A.J.; Kawaoka, Y. The TLR4-TRIF pathway protects against H5N1 influenza virus infection. J. Virol. 2012, 86, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Georgel, P.; Jiang, Z.; Kunz, S.; Janssen, E.; Mols, J.; Hoebe, K.; Bahram, S.; Oldstone, M.B.; Beutler, B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 2007, 362, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Rolland, A.; Jouvin-Marche, E.; Saresella, M.; Ferrante, P.; Cavaretta, R.; Creange, A.; Marche, P.; Perron, H. Correlation between disease severity and in vitro cytokine production mediated by MSRV (multiple sclerosis associated retroviral element) envelope protein in patients with multiple sclerosis. J. Neuroimmunol. 2005, 160, 195–203. [Google Scholar] [CrossRef]
- Perron, H.; Garson, J.A.; Bedin, F.; Beseme, F.; Paranhos-Baccala, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.W.; Voisset, C.; Blond, J.L.; et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588. [Google Scholar] [CrossRef]
- Jin, X.; Li, X.; Guan, F.; Zhang, J. Human Endogenous Retroviruses and Toll-Like Receptors. Viral Immunol. 2022. [Google Scholar] [CrossRef]
- Gottle, P.; Schichel, K.; Reiche, L.; Werner, L.; Zink, A.; Prigione, A.; Kury, P. TLR4 Associated Signaling Disrupters as a New Means to Overcome HERV-W Envelope-Mediated Myelination Deficits. Front. Cell. Neurosci. 2021, 15, 777542. [Google Scholar] [CrossRef]
- Kremer, D.; Schichel, T.; Forster, M.; Tzekova, N.; Bernard, C.; van der Valk, P.; van Horssen, J.; Hartung, H.P.; Perron, H.; Kury, P. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation. Ann. Neurol. 2013, 74, 721–732. [Google Scholar] [CrossRef]
- Duperray, A.; Barbe, D.; Raguenez, G.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Perron, H.; Marche, P.N. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int. Immunol. 2015, 27, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, X.; Huang, J.; Li, H.; Yan, Q.; Zhu, F. Human endogenous retrovirus W family envelope protein (HERV-W env) facilitates the production of TNF-alpha and IL-10 by inhibiting MyD88s in glial cells. Arch. Virol. 2021, 166, 1035–1045. [Google Scholar] [CrossRef]
- Bender, A.T.; Tzvetkov, E.; Pereira, A.; Wu, Y.; Kasar, S.; Przetak, M.M.; Vlach, J.; Niewold, T.B.; Jensen, M.A.; Okitsu, S.L. TLR7 and TLR8 Differentially Activate the IRF and NF-kappaB Pathways in Specific Cell Types to Promote Inflammation. Immunohorizons 2020, 4, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Jeong, B.H.; Lee, Y.J.; Carp, R.I.; Kim, Y.S. The prevalence of human endogenous retroviruses in cerebrospinal fluids from patients with sporadic Creutzfeldt-Jakob disease. J. Clin. Virol. 2010, 47, 136–142. [Google Scholar] [CrossRef]
- Douville, R.N.; Nath, A. Chapter 22—Human endogenous retroviruses and the nervous system. In Handbook of Clinical Neurology; Tselis, A.C., Booss, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 123, pp. 465–485. [Google Scholar]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.B.; Silva, C.; Holden, J.; Warren, K.G.; Clark, A.W.; Power, C. Monocyte activation and differentiation augment human endogenous retrovirus expression: Implications for inflammatory brain diseases. Ann. Neurol. 2001, 50, 434–442. [Google Scholar] [CrossRef]
- Dembny, P.; Newman, A.G.; Singh, M.; Hinz, M.; Szczepek, M.; Kruger, C.; Adalbert, R.; Dzaye, O.; Trimbuch, T.; Wallach, T.; et al. Human endogenous retrovirus HERV-K(HML-2) RNA causes neurodegeneration through Toll-like receptors. JCI Insight 2020, 5, e131093. [Google Scholar] [CrossRef]
- Forsbach, A.; Nemorin, J.G.; Montino, C.; Muller, C.; Samulowitz, U.; Vicari, A.P.; Jurk, M.; Mutwiri, G.K.; Krieg, A.M.; Lipford, G.B.; et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 2008, 180, 3729–3738. [Google Scholar] [CrossRef] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Rigby, R.E.; Webb, L.M.; Mackenzie, K.J.; Li, Y.; Leitch, A.; Reijns, M.A.; Lundie, R.J.; Revuelta, A.; Davidson, D.J.; Diebold, S.; et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 2014, 33, 542–558. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Bruns, A.M.; Leser, G.P.; Lamb, R.A.; Horvath, C.M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 2014, 55, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Quicke, K.M.; Kim, K.Y.; Horvath, C.M.; Suthar, M.S. RNA Helicase LGP2 Negatively Regulates RIG-I Signaling by Preventing TRIM25-Mediated Caspase Activation and Recruitment Domain Ubiquitination. J. Interferon Cytokine Res. 2019, 39, 669–683. [Google Scholar] [CrossRef]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Xodo, L.E. Endogenous Retroviruses (ERVs): Does RLR (RIG-I-Like Receptors)-MAVS Pathway Directly Control Senescence and Aging as a Consequence of ERV De-Repression? Front. Immunol. 2022, 13, 917998. [Google Scholar] [CrossRef]
- Peisley, A.; Wu, B.; Yao, H.; Walz, T.; Hur, S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 2013, 51, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.; Eberle, F.; Seitz, S.; Mucke, N.; Huber, C.M.; Kiani, N.; Kaderali, L.; Lohmann, V.; Dalpke, A.; Bartenschlager, R. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 2011, 286, 27278–27287. [Google Scholar] [CrossRef]
- Yu, Q.; Qu, K.; Modis, Y. Cryo-EM Structures of MDA5-dsRNA Filaments at Different Stages of ATP Hydrolysis. Mol. Cell 2018, 72, 999–1012.e6. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M., Jr. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19, e45000. [Google Scholar] [CrossRef]
- Sun, S.; Frontini, F.; Qi, W.; Hariharan, A.; Ronner, M.; Wipplinger, M.; Blanquart, C.; Rehrauer, H.; Fonteneau, J.F.; Felley-Bosco, E. Endogenous retrovirus expression activates type-I interferon signaling in an experimental mouse model of mesothelioma development. Cancer Lett. 2021, 507, 26–38. [Google Scholar] [CrossRef]
- Mameli, G.; Madeddu, G.; Mei, A.; Uleri, E.; Poddighe, L.; Delogu, L.G.; Maida, I.; Babudieri, S.; Serra, C.; Manetti, R.; et al. Activation of MSRV-type endogenous retroviruses during infectious mononucleosis and Epstein-Barr virus latency: The missing link with multiple sclerosis? PLoS ONE 2013, 8, e78474. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Rangel, S.C.; da Silva, M.D.; da Silva, A.L.; de Melo Batista dos Santos, J.; Neves, L.M.; Pedrosa, A.; Rodrigues, F.M.; dos Santos Trettel, C.; Furtado, G.E.; de Barros, M.P.; et al. Human endogenous retroviruses and the inflammatory response: A vicious circle associated with health and illness. Front. Immunol. 2022, 13, 1057791. [Google Scholar] [CrossRef]
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991. [Google Scholar] [CrossRef]
- Mameli, G.; Serra, C.; Astone, V.; Castellazzi, M.; Poddighe, L.; Fainardi, E.; Neri, W.; Granieri, E.; Dolei, A. Inhibition of multiple-sclerosis-associated retrovirus as biomarker of interferon therapy. J. Neurovirol. 2008, 14, 73–77. [Google Scholar] [CrossRef]
- Nali, L.H.; Olival, G.S.; Montenegro, H.; da Silva, I.T.; Dias-Neto, E.; Naya, H.; Spangenberg, L.; Penalva-de-Oliveira, A.C.; Romano, C.M. Human endogenous retrovirus and multiple sclerosis: A review and transcriptome findings. Mult. Scler. Relat. Disord. 2022, 57, 103383. [Google Scholar] [CrossRef]
- Hartung, H.P.; Derfuss, T.; Cree, B.A.; Sormani, M.P.; Selmaj, K.; Stutters, J.; Prados, F.; MacManus, D.; Schneble, H.M.; Lambert, E.; et al. Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study. Mult. Scler. 2022, 28, 429–440. [Google Scholar] [CrossRef]
- Freimanis, G.; Hooley, P.; Ejtehadi, H.D.; Ali, H.A.; Veitch, A.; Rylance, P.B.; Alawi, A.; Axford, J.; Nevill, A.; Murray, P.G.; et al. A role for human endogenous retrovirus-K (HML-2) in rheumatoid arthritis: Investigating mechanisms of pathogenesis. Clin. Exp. Immunol. 2010, 160, 340–347. [Google Scholar] [CrossRef]
- Reynier, F.; Verjat, T.; Turrel, F.; Imbert, P.E.; Marotte, H.; Mougin, B.; Miossec, P. Increase in human endogenous retrovirus HERV-K (HML-2) viral load in active rheumatoid arthritis. Scand. J. Immunol. 2009, 70, 295–299. [Google Scholar] [CrossRef]
- Nelson, P.N.; Roden, D.; Nevill, A.; Freimanis, G.L.; Trela, M.; Ejtehadi, H.D.; Bowman, S.; Axford, J.; Veitch, A.M.; Tugnet, N.; et al. Rheumatoid arthritis is associated with IgG antibodies to human endogenous retrovirus gag matrix: A potential pathogenic mechanism of disease? J. Rheumatol. 2014, 41, 1952–1960. [Google Scholar] [CrossRef]
- Stearrett, N.; Dawson, T.; Rahnavard, A.; Bachali, P.; Bendall, M.L.; Zeng, C.; Caricchio, R.; Perez-Losada, M.; Grammer, A.C.; Lipsky, P.E.; et al. Expression of Human Endogenous Retroviruses in Systemic Lupus Erythematosus: Multiomic Integration with Gene Expression. Front. Immunol. 2021, 12, 661437. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Evolving story of autoantibodies in systemic lupus erythematosus. J. Autoimmun. 2020, 110, 102356. [Google Scholar] [CrossRef]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, M.; Gunn, B.M.; Venkataraman, A.; Kong, Y.; Kang, I.; Rakib, T.; Townsend, M.J.; Costenbader, K.H.; Alter, G.; Iwasaki, A. Antibodies against human endogenous retrovirus K102 envelope activate neutrophils in systemic lupus erythematosus. J. Exp. Med. 2021, 218, e20191766. [Google Scholar] [CrossRef]
- Tokuyama, M.; Kong, Y.; Song, E.; Jayewickreme, T.; Kang, I.; Iwasaki, A. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2018, 115, 12565–12572. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Carmona-Rivera, C.; Kaplan, M.J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front. Immunol. 2012, 3, 380. [Google Scholar] [CrossRef] [Green Version]
- Angeletti, A.; Volpi, S.; Bruschi, M.; Lugani, F.; Vaglio, A.; Prunotto, M.; Gattorno, M.; Schena, F.; Verrina, E.; Ravelli, A.; et al. Neutrophil Extracellular Traps-DNase Balance and Autoimmunity. Cells 2021, 10, 2667. [Google Scholar] [CrossRef]
- Baker, V.S.; Imade, G.E.; Molta, N.B.; Tawde, P.; Pam, S.D.; Obadofin, M.O.; Sagay, S.A.; Egah, D.Z.; Iya, D.; Afolabi, B.B.; et al. Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar. J 2008, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.C.; Potoka, K.C.; Champion, H.C.; Mora, A.L.; Gladwin, M.T. Pulmonary arterial hypertension: The clinical syndrome. Circ. Res. 2014, 115, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, S.; Howard, L.S.; Gomberg-Maitland, M.; Hoeper, M.M. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation 2020, 141, 678–693. [Google Scholar] [CrossRef]
- Oldroyd, S.H.; Manek, G.; Sankari, A.; Bhardwaj, A. Pulmonary Hypertension; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Levine, D.J. Pulmonary arterial hypertension: Updates in epidemiology and evaluation of patients. Am. J. Manag. Care 2021, 27, S35–S41. [Google Scholar] [CrossRef]
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmuller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in pulmonary arterial hypertension. Chest 2012, 141, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Miyagawa, K.; Chen, S.Y.; Tamosiuniene, R.; Wang, L.; Sharpe, O.; Samayoa, E.; Harada, D.; Moonen, J.A.J.; Cao, A.; et al. Upregulation of Human Endogenous Retrovirus-K Is Linked to Immunity and Inflammation in Pulmonary Arterial Hypertension. Circulation 2017, 136, 1920–1935. [Google Scholar] [CrossRef]
- Otsuki, S.; Saito, T.; Taylor, S.; Li, D.; Moonen, J.R.; Marciano, D.P.; Harper, R.L.; Cao, A.; Wang, L.; Ariza, M.E.; et al. Monocyte-released HERV-K dUTPase engages TLR4 and MCAM causing endothelial mesenchymal transition. JCI Insight 2021, 6, e146416. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRR | Type of Antigen | Source |
|---|---|---|
| TLR3 | HERV-derived dsRNA; syncytin-1 protein | [36,75,76] |
| TLR4 | HERV-W Env | [77] |
| TLR8 | HERV-K (HML-2) RNA | [78] |
| TLR9 | HERV-derived RNA:DNA heteroduplexes (speculative) | [79,80] |
| MDA5 | HERV-derived dsRNA | [36,75] |
| cGAS | HERV-derived dsDNA | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russ, E.; Iordanskiy, S. Endogenous Retroviruses as Modulators of Innate Immunity. Pathogens 2023, 12, 162. https://doi.org/10.3390/pathogens12020162
Russ E, Iordanskiy S. Endogenous Retroviruses as Modulators of Innate Immunity. Pathogens. 2023; 12(2):162. https://doi.org/10.3390/pathogens12020162
Chicago/Turabian StyleRuss, Eric, and Sergey Iordanskiy. 2023. "Endogenous Retroviruses as Modulators of Innate Immunity" Pathogens 12, no. 2: 162. https://doi.org/10.3390/pathogens12020162