
SARS-CoV-2 Variants Identification: Overview of Molecular Existing Methods

 , , , and
, , , and
Abstract
:1. Introduction
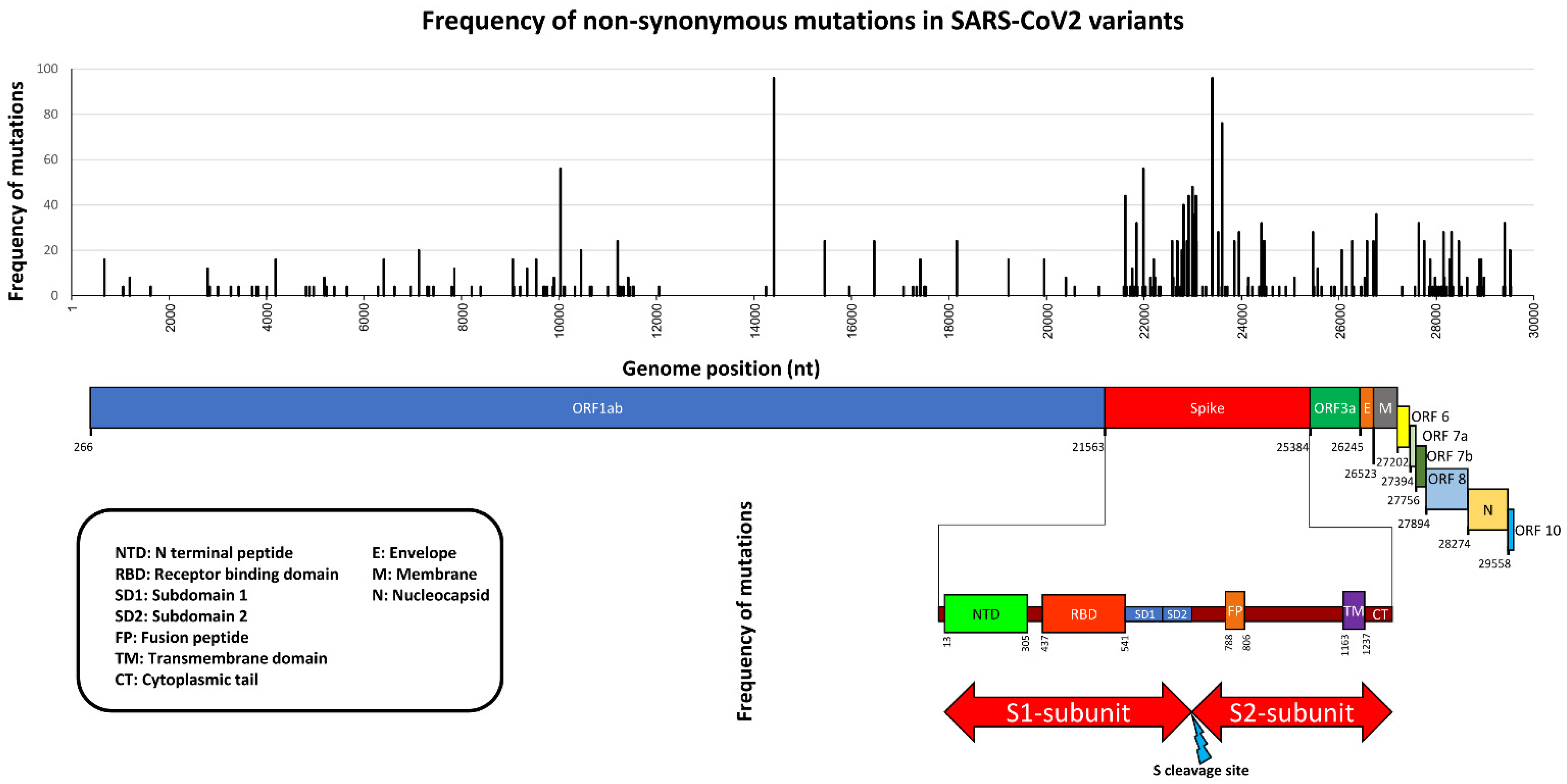
2. Variants Description
3. Detection and Characterization of SARS-CoV-2 Variants by Sequencing
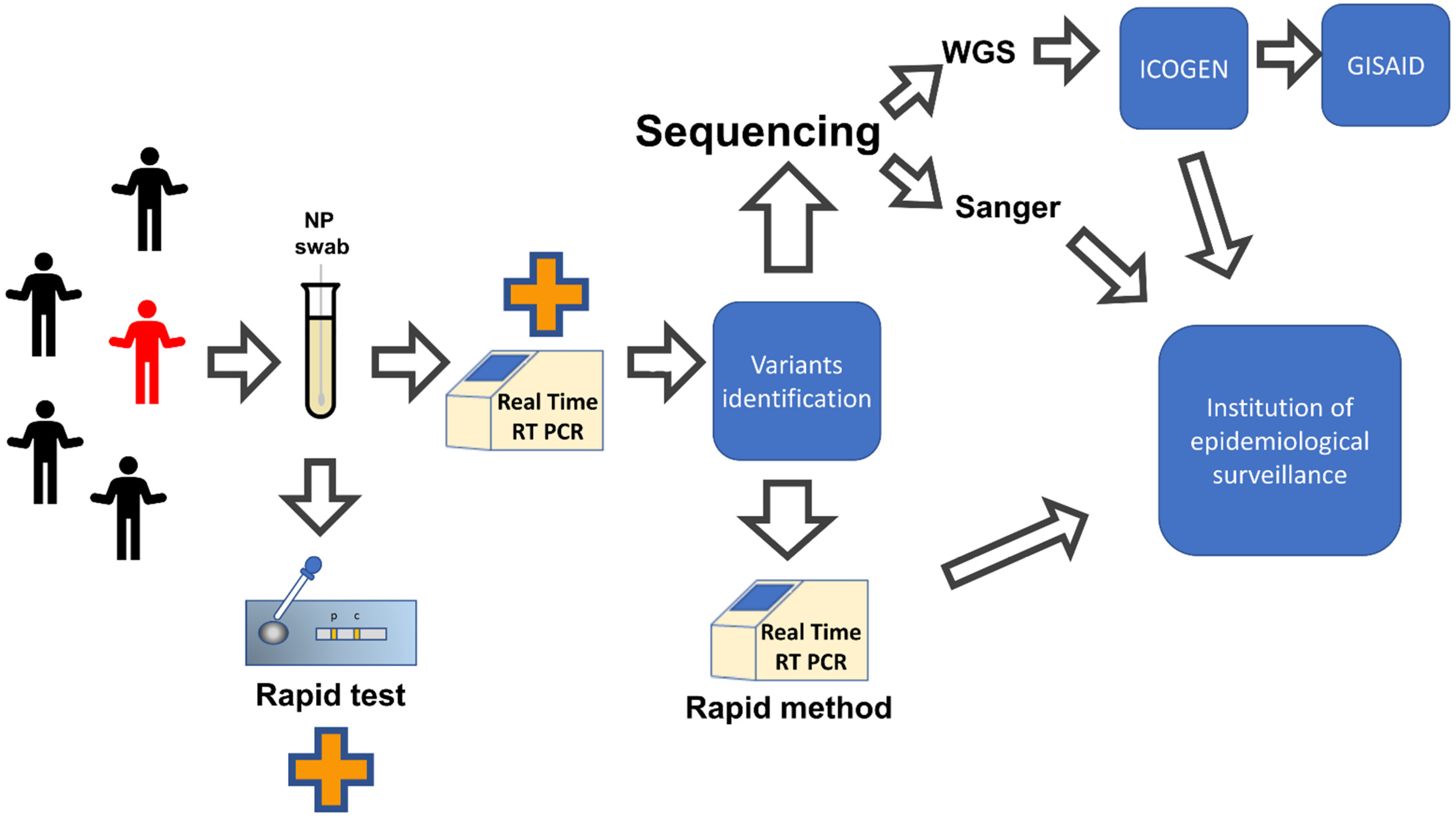
4. Rapid Methods for Early Detection of SARS-CoV-2 Variants
4.1. Main Commercially Available Methods for SARS-CoV-2 Variants Identification
4.2. In House Rapid Methods for SARS-CoV-2 Variants Identification

{kind=link}
{kind=link}
| Method | Amino Acid Mutation Detected | Variants Identified | Ref |
|---|---|---|---|
| Real-Time RT-PCR | Δ156–157 | B.1.617.2 | [117] |
| RT-FRET-PCR | T478K | B.1.617.2 | [118] |
| One-step RT-qPCR | N501Y | B.1.1.7 B.1.351 P.1 BA.1/2/4/5 (no discrimination) | [119] |
| Multiplex RT-qPCR | D3L and Δ242–244 | B.1.1.7 or B.1.351 | [120] |
| Allele-specific polymerase chain reaction (ASP) | E484 K, L452R and P681R | B.1.1.7, B.1.351, B.1.617.2 | [121] |
| Multiplex RT-qPCR | Δ69–70, K417T, K417N, L452R, E484K, E484Q, N501Y, P681H, and P681R | B.1.1.7, B.1.351, P.1, B.1.617.2, BA.1 | [122] |
| CoVarScan Multiplex fragment analysis (Fluorescently labeled RT-PCR amplicons analyzed by capillary electrophoresis) | 8 defined hotspot regions: 5 recurrently deleted regions (RDRs; S:RDR1, S:RDR2, S: RDR3–4, ORF1A, and ORF8) and 3 SNPs (S:N501Y, S:L452R, and S:E484K) | B.1.1.7, B.1.351, P.1, B.1.617.2, BA.1 | [123] |
| RT-PCR/MALDI-TOF | five targets: N1, N2, N3, ORF1A, ORF1AB. | B.1.1.7 (tested december 2020 to april 2021) | [124] |
| Multiplex CRISPR-based diagnostics and microfluidics | D614G, Δ69–70, N501Y, A570D, P681H/R, D80A, K417N, K417T, L18F, E484K/A, H655Y, P26S, Δ156–157, T478K, L452R/Q, S477N | B.1.1.7, B.1.351, P.1, B.1.617.2, BA.1 | [125] |
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 26 August 2022).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.; Tian, J.; Pei, Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. The next phase of SARS-CoV-2 surveillance: Real-time molecular epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, E.; Shafiee, F.; Shahzamani, K.; Ranjbar, M.M.; Alibakhshi, A.; Ahangarzadeh, S.; Beikmohammadi, L.; Shariati, L.; Hooshmandi, S.; Ataei, B.; et al. Novel and emerging mutations of SARS-CoV-2: Biomedical implications. Biomed. Pharmacother. 2021, 139, 111599, Erratum in Biomed. Pharmacother. 2021, 140, 111723. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Andino, R. Closing the gap: The challenges in converging theoretical, computational, experimental and real-life studies in virus evolution. Curr. Opin. Virol. 2012, 2, 515–518. [Google Scholar] [CrossRef]
- Belshaw, R.; Gardner, A.; Rambaut, A.; Pybus, O.G. Pacing a small cage: Mutation and RNA viruses. Trends Ecol. Evol. 2008, 23, 188–193. [Google Scholar] [CrossRef]
- Scrima, M.; Cossu, A.M.; D’Andrea, E.L.; Bocchetti, M.; Abruzzese, Y.; Iannarone, C.; Miarelli1, C.; Grisolia, P.; Melisi, F.; Genua, L.; et al. Genomic Characterization of the Emerging SARS-CoV-2 Lineage in Two Districts of Campania (Italy) Using Next-Generation Sequencing. Front. Virol. 2022, 2, 814114. [Google Scholar] [CrossRef]
- Bansal, K.; Kumar, S. Mutational cascade of SARS-CoV-2 leading to evolution and emergence of omicron variant. Virus Res. 2022, 315, 198765. [Google Scholar] [CrossRef]
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. NextStrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission dynamics and evolutionary history of 2019-nCoV. J. Med. Virol. 2020, 92, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Pereson, M.J.; Mojsiejczuk, L.; Martínez, A.P.; Flichman, D.M.; Garcia, G.H.; Di Lello, F.A. Phylogenetic analysis of SARS-CoV-2 in the first few months since its emergence. J. Med. Virol. 2021, 93, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, Z.; He, F. The phylogenetic relationship within SARS-CoV-2s: An expanding basal clade. Mol. Phylogenet. Evol. 2021, 157, 107017. [Google Scholar] [CrossRef]
- Motayo, B.O.; Oluwasemowo, O.O.; Olusola, B.A.; Akinduti, P.A.; Arege, O.T.; Obafemi, Y.D.; Faneye, A.O.; Isibor, P.O.; Aworunse, O.S.; Oranusi, S.U. Evolution and genetic diversity of SARS-CoV-2 in Africa using whole genome sequences. Int. J. Infect. Dis. 2021, 103, 282–287. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 2–4. [Google Scholar] [CrossRef]
- Alkhatib, M.; Salpini, R.; Carioti, L.; Ambrosio, F.A.; D’Anna, S.; Duca, L.; Costa, G.; Bellocchi, M.C.; Piermatteo, L.; Artese, A.; et al. Update on SARS-CoV-2 Omicron Variant of Concern and Its Peculiar Mutational Profile. Microbiol. Spectr. 2022, 10, e02732-21. [Google Scholar] [CrossRef]
- Lo Presti, A.; Di Martino, A.; Faggioni, G.; Giordani, F.; Fillo, S.; Anselmo, A.; Fain, V.V.; Fortunato, A.; Petralito, G.; Molinari, F.; et al. Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020. Viruses 2022, 14, 472. [Google Scholar] [CrossRef]
- Dolci, M.; Signorini, L.; Cason, C.; Campisciano, G.; Kunderfranco, P.; Pariani, E.; Galli, C.; Petix, V.; Ferrante, P.; Delbueet, S.; et al. Circulation of SARS-CoV-2 Variants among Children from November 2020 to January 2022 in Trieste (Italy). Microorganisms 2022, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Rueca, M.; Giombini, E.; Messina, F.; Bartolini, B.; Di Caro, A.; Capobianchi, M.R.; Gruber, C.E. The Easy-to-Use SARS-CoV-2 Assembler for Genome Sequencing: Development Study. JMIR Bioinf. Biotechnol. 2022, 3, e31536. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Spinella, F.; di Domenico, E.G.; Pontone, M.; Cavallo, I.; Orlandi, G.; Iannazzo, S.; Ricciuto, G.M.; Isg Virology Covid Team; Pellini, R.; et al. Evidence of a SARS-CoV-2 double Spike mutation D614G/S939F potentially affecting immune response of infected subjects. Comput. Struct. Biotechnol. J. 2022, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Micheli, V.; Bracchitta, F.; Rizzo, A.; Mancon, A.; Mileto, D.; Lombardi, A.; Stefanelli, P.; Gismondo, M.R. First identification of the new SARS-CoV-2 Omicron variant (B.1.1.529) in Italy. Clin. Infect. Dis. 2022, 75, 522–524. [Google Scholar] [CrossRef]
- Colavita, F.; Meschi, S.; Gruber, C.E.M.; Rueca, M.; Vairo, F.; Matusali, G.; Lapa, D.; Giombini, E.; De Carli, G.; Spazianteet, M.; et al. Virological and Serological Characterisation of SARS-CoV-2 Infections Diagnosed After mRNA BNT162b2 Vaccination Between December 2020 and March 2021. Front. Med. 2022, 8, 815870. [Google Scholar] [CrossRef]
- Istituto Superiore di Sanità. IRIDA-ARIES Platform. Available online: https://irida.iss.it/irida21-aries/login (accessed on 27 July 2022).
- WHO. Genomic Sequencing of SARS-CoV-2. 2021, p. 94. Available online: https://www.who.int/publications/i/item/9789240018440 (accessed on 27 July 2022).
- Yu, W.B.; Tang, G.D.; Zhang, L.; Corlett, R.T. Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2 / HCoV-19) using whole genomic data. Zool. Res. 2020, 41, 247–257. [Google Scholar] [CrossRef]
- Bohn, M.K.; Loh, T.P.; Wang, C.; Mueller, R.; Koch, D.; Sethi, S.; Rawlinson, W.D.; Clementi, M.; Erasmus, R.; Leportier, M.; et al. IFCC Interim Guidelines on Serological Testing of Antibodies against SARS-CoV-2. Clin. Chem. Lab. Med. 2020, 58, 2001–2008. [Google Scholar] [CrossRef]
- Artesi, M.; Bontems, S.; Göbbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.-P.; Bours, V.; et al. A Recurrent Mutation at Position 26340 of SARS-CoV-2 Is Associated with Failure of the E Gene Quantitative Reverse Transcription-PCR Utilized in a Commercial Dual-Target Diagnostic Assay. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef]
- Mavian, C.; Marini, S.; Prosperi, M.; Salemi, M. Erratum: A snapshot of SARS-CoV-2 genome availability up to April 2020 and its implications: Data analysis (JMIR Public Health and Surveillance (2020) 6:2 (e19170). JMIR Public Health Surveill. 2020, 6, 22853. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Peñarrubia, L.; Ruiz, M.; Porco, R.; Rao, S.N.; Juanola-Falgarona, M.; Manissero, D.; López-Fontanals, M.; Pareja, J. Multiple assays in a real-time RT-PCR SARS-CoV-2 panel can mitigate the risk of loss of sensitivity by new genomic variants during the COVID-19 outbreak. Int. J. Infect. Dis. 2020, 97, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, I.; Ijaz, M.; Ahmad, S.; Ahmed, T.; Bari, A.; Abro, A.; Allemailem, K.S.; Almatroudi, A.; Qamar, M.T.U. SARS-CoV-2: An Update on Genomics, Risk Assessment, Potential Therapeutics and Vaccine Development. Int. J. Environ. Res. Public Health 2021, 18, 1626. [Google Scholar] [CrossRef] [PubMed]
- Haidere, M.F.; Ratan, Z.A.; Nowroz, S.; Zaman, S.B.; Jung, Y.J.; Hosseinzadeh, H.; Cho, J.Y. COVID-19 Vaccine: Critical Questions with Complicated Answers. Biomol. Ther. 2021, 29, 1. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.A.J.; Richter, A.; Casey, A.; Osman, H.; Mirza, J.D.; Stockton, J.; Quick, J.; Ratcliffe, L.; Sparks, N.; Cumley, N.; et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evol. 2022, 8, veac050. [Google Scholar] [CrossRef]
- Turcinovic, J.; Schaeffer, B.; Taylor, B.P.; Bouton, T.C.; Odom-Mabey, A.R.; Weber, S.E.; Lodi, S.; Ragan, E.J.; Connor, J.H.; Jacobson, K.R.; et al. Understanding early pandemic SARS-CoV-2 transmission in a medical center by incorporating public sequencing databases to mitigate bias. J. Infect. Dis. 2022, jiac348. [Google Scholar] [CrossRef]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic epidemiology of the SARS-CoV-2 epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef]
- Mencacci, A.; Gili, A.; Camilloni, B.; Bicchieraro, G.; Spaccapelo, R.; Bietta, C.; Stracci, F. Immediate reinfection with Omicron variant after clearance of a previous SARS-CoV-2 infection. J. Infect. Public Health 2022, 15, 983–985. [Google Scholar] [CrossRef]
- Grimaldi, A.; Panariello, F.; Annunziata, P.; Giuliano, T.; Daniele, M.; Pierri, B.; Colantuono, C.; Salvi, M.; Bouché, V.; Manfredi, A.; et al. Improved SARS-CoV-2 sequencing surveillance allows the identification of new variants and signatures in infected patients. Genome Med. 2022, 14, 90. [Google Scholar] [CrossRef]
- Martin, G.E.; Taiaroa, G.; Taouk, M.L.; Savic, I.; O’Keefe, J.; Quach, R.; Prestedge, J.; Krysiak, M.; Caly, L.; Williamson, D.A. Maintaining genomic surveillance using whole-genome sequencing of SARS-CoV-2 from rapid antigen test devices. Lancet Infect. Dis. 2022. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Cheng, Y.X.; Xu, L.; Li, J.Y.; Tao, C.Y.; Ji, C.Y.; Han, N.; Yang, R.; Wu, H.; Li, Y.; et al. Genomic evidence for divergent co-infections of co-circulating SARS-CoV-2 lineages. Comput. Struct. Biotechnol. J. 2022, 20, 4015–4024. [Google Scholar] [CrossRef]
- Sharif, N.; Alzahrani, K.J.; Ahmed, S.N.; Khan, A.; Banjer, H.J.; Alzahrani, F.M.; Parvez, A.K.; Dey, S.K. Genomic surveillance, evolution and global transmission of SARS-CoV-2 during 2019–2022. PLoS ONE 2022, 17, e0271074. [Google Scholar] [CrossRef]
- Helary, M.; Salmona, M.; Walle, É.M.; Feghoul, L.; Mahjoub, N.; Schnepf, N.; Maylin, S.; Chaix, M.-L.; Le Goff, J.; Delaugerre, C. Virological aspects, diagnostic tools and variants of SARS-CoV-2 [Aspects virologiques, diagnostic et variants du SARS-CoV-2]. Rev. Prat. 2022, 72, 494–500. [Google Scholar]
- Isnaini, N.; Mardian, Y.; Lokida, D.; Budiono, F.; Butar-Butar, D.P.; Arlinda, D.; Salim, G.; Kosasih, H.; Wulan, W.N.; Perodin, J.; et al. Mild reinfection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Delta variant: First case report from Indonesia. Front. Med 2022, 9, 906469. [Google Scholar] [CrossRef] [PubMed]
- Andeweg, S.P.; Vennema, H.; Veldhuijzen, I.; Smorenburg, N.; Schmitz, D.; Zwagemaker, F.; van Gageldonk-Lafeber, A.B.; Hahné, S.J.M.; Reusken, C.; Knol, M.J.; et al. Elevated risk of infection with SARS-CoV-2 Beta, Gamma, and Delta variant compared to Alpha variant in vaccinated individuals. Sci. Transl. Med. 2022, eabn4338. [Google Scholar] [CrossRef] [PubMed]
- Acuña-Castillo, C.; Vidal, M.; Inostroza-Molina, A.; Vallejos-Vidal, E.; Luraschi, R.; Figueroa, M.; Barrera-Avalos, C.; Vera, R.; Vargas, S.; Valdes, D.; et al. First Identification of Reinfection by a Genetically Different Variant of SARS-CoV-2 in a Homeless Person from the Metropolitan Area of Santiago, Chile. J. Environ. Public Health 2022, 2022, 3859071. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.D.; Sharma, A.; Lee, H.J.; Yadav, D.K. SARS-CoV-2: Recent Variants and Clinical Efficacy of Antibody-Based Therapy. Front. Cell. Infect. Microbiol. 2022, 12, 839170. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef]
- Gisaid. Gisaid. Available online: https://www.gisaid.org/ (accessed on 27 July 2022).
- Nextstrain-Real Time RT Tracking of Pathogen Evolution. Available online: https://nextstrain.org (accessed on 27 July 2022).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Available online: https://cov-lineages.org/ (accessed on 27 July 2022).
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef]
- Duarte, C.M.; Ketcheson, D.I.; Eguíluz, V.M.; Agustí, S.; Fernández-Gracia, J.; Jamil, T.; Laiolo, E.; Gojobori, T.; Alam, I. Rapid evolution of SARS-CoV-2 challenges human defenses. Sci. Rep. 2022, 12, 6457. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- European Centre for Diseases Prevention and Control (ECDC). SARS-CoV-2 Variants of Concern as of 25 Aug 2022. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 25 August 2022).
- Helix. Viral Surveillance. Available online: https://www.helix.com/pages/helix-covid-19-surveillance-dashboard (accessed on 25 August 2022).
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. MBio 2022, 13, e02979-21. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 25 August 2022).
- He, X.; Hong, W.; Pan, X.; Lu, G.; Wei, X. SARS-CoV-2 Omicron variant: Characteristics and prevention. MedComm 2021, 2, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and Sub-Variants (BA.1, BA.2 and BA.3) of SARS-CoV-2 Spike Infectivity and Pathogenicity: A Comparative Sequence and Structural-based Computational Assessment. bioRxiv. 2022. bioRxiv: 3:2022.02.11.480029. Available online: https://www.biorxiv.org/content/10.1101/2022.02.11.480029v1%0Ahttps://www.biorxiv.org/content/10.1101/2022.02.11.480029v1.abstract (accessed on 25 August 2022).
- Desingu, P.A.; Nagarajan, K.; Dhama, K. Emergence of Omicron third lineage BA.3 and its importance. J. Med. Virol. 2022, 94, 1808–1810. [Google Scholar] [CrossRef]
- WHO. Enhancing response to Omicron SARS-CoV-2 Variant: Technical Brief and Priority Actions for Member States. Available online: https://www.who.int/docs/default-source/coronaviruse/2022-01-21-global-technical-brief-and-priority-action-on-omicron-sars-cov-2-variant.pdf (accessed on 28 January 2022).
- Mohapatra, R.K.; Kandi, V.; Verma, S.; Dhama, K. Challenges of the Omicron (B.1.1.529) Variant and Its Lineages: A Global Perspective. ChemBioChem 2022, 23, e202200059. [Google Scholar] [CrossRef]
- Phan, T.; Boes, S.; McCullough, M.; Gribschaw, J.; Marsh, J.W.; Harrison, L.H.; Wells, A. First detection of SARS-CoV-2 Omicron BA.4 variant in Western Pennsylvania, United States. J. Med. Virol. 2022, 10, 4593–4594. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.A.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 1–6. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Statement on Omicron Sublineage BA.2 by WHO. Available online: https://www.who.int/news/item/22-02-2022-statement-on-omicron-sublineage-ba.2 (accessed on 22 February 2022).
- Yu, J.; Collier, A.Y.; Rowe, M.; Mardas, F.; Ventura, J.D.; Wan, H.; Miller, J.; Powers, O.; Chung, B.; Siamatu, M.; et al. Neutralization of the SARS-CoV-2 Omicron BA.1 and BA.2 Variants. N. Engl. J. Med. 2022, 386, 1579–1580. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K.; et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell 2022, 185, 2103–2115.e19. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, R.K.; Kandi, V.; Tuli, H.S.; Chakraborty, C.; Dhama, K. The recombinant variants of SARS-CoV-2: Concerns continues amid COVID-19 pandemic. J. Med. Virol. 2022, 94, 3506–3508. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: What do we know about the delta omicron recombinant variant? BMJ 2022, 376, o792. [Google Scholar] [CrossRef] [PubMed]
- WHO. Coronavirus disease (COVID-19)-Weekly Epidemiological Update 1. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200817-weekly-epi-update-1.pdf?sfvrsn=b6d49a76_4 (accessed on 19 August 2020).
- UKHSA. SARS-CoV-2 Variants of Concern and Variants under Investigation in England- Technical Briefing 31. Sage. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1040076/Technical_Briefing_31.pdf (accessed on 10 December 2021).
- VanInsberghe, D.; Neish, A.S.; Lowen, A.C.; Koelle, K. Recombinant SARS-CoV-2 Genomes Are Currently Circulating at Low Levels. In bioRxiv Prepr. Serv. Biol.; 2021; pp. 1–20. Available online: http://www.ncbi.nlm.nih.gov/pubmed/33758853%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC7987012 (accessed on 25 August 2022).
- Duerr, R.; Dimartino, D.; Marier, C.; Zappile, P.; Wang, G.; Plitnick, J.; Griesemer, S.B.; Lasek-Nesselquist, E.; Dittmann, M.; Ortigoza, M.B.; et al. Delta-Omicron recombinant SARS-CoV-2 in a transplant patient treated with Sotrovimab. bioRxiv. 2022. bioRxiv: 2022.04.06.487325. Available online: https://www.biorxiv.org/content/10.1101/2022.04.06.487325v1%0Ahttps://www.biorxiv.org/content/10.1101/2022.04.06.487325v1 (accessed on 25 August 2022).
- Guidance for Representative and Targeted Genomic SARS-CoV-2 Monitoring by ECDC. Available online: https://www.ecdc.europa.eu/en/publications-data/guidance-representative-and-targeted-genomic-sars-cov-2-monitoring (accessed on 25 August 2022).
- European Centre for Disease Prevention and Control, Europe WHORO for. Methods for the Detection and Characterisation of SARS-CoV-2 Variants–First Update What Is New in This Update: Key Messages. 13 December 2021. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Methods-for-the-detection-char-SARS-CoV-2-variants_2nd%20update_final.pdf (accessed on 25 August 2022).
- Van der Moeren, N.; Selhorst, P.; Ha, M.; Heireman, L.; Van Gaal, P.J.; Breems, D.; Meysman, P.; Laukens, K.; Verstrepen, W.; Van Gasse, N.; et al. Viral Evolution and Immunology of SARS-CoV-2 in a Persistent Infection after Treatment with Rituximab. Viruses 2022, 14, 752. [Google Scholar] [CrossRef]
- Martínez-Chinchilla, C.; Vazquez-Montero, L.; Palazón-Carrión, N.; Fernández-Román, I.M.; López-Barba, J.; de la Cruz-Merino, L.; Rodríguez-Baño, J.; Palacios-Baena, Z.R. Persistence of SARS-CoV-2 Infection in Severely Immunocompromised Patients With Complete Remission B-Cell Lymphoma and Anti-CD20 Monoclonal Antibody Therapy: A Case Report of Two Cases. Front. Immunol. 2022, 13, 860891. [Google Scholar] [CrossRef]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 exhibits intra-host genomic plasticity and low-frequency polymorphic quasispecies. J. Clin. Virol. 2020, 131, 104585. [Google Scholar] [CrossRef]
- Rueca, M.; Bartolini, B.; Gruber, C.E.M.; Piralla, A.; Baldanti, F.; Giombini, E.; Messina, F.; Marchioni, L.; Ippolito, G.; Di Caro, A.; et al. Compartmentalized Replication of SARS-CoV-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis. Microorganisms 2020, 8, 1302. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 954–956. [Google Scholar] [CrossRef]
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef]
- Tshiabuila, D.; Giandhari, J.; Pillay, S.; Ramphal, U.; Ramphal, Y.; Maharaj, A.; Anyaneji, U.J.; Naidoo, Y.; Tegally, H.; San, E.J.; et al. Comparison of SARS-CoV-2 sequencing using the ONT GridION and the Illumina MiSeq. BMC Genom. 2022, 23, 319. [Google Scholar] [CrossRef] [PubMed]
- Fissel, J.A.; Mestas, J.; Chen, P.Y.; Flores-Vazquez, J.; Truong, T.T.; Bootwalla, M.; Maglinte, D.T.; Gai, X.; Bard, J.D. Implementation of a Streamlined SARS-CoV-2 Whole-Genome Sequencing Assay for Expeditious Surveillance during the Emergence of the Omicron Variant. J. Clin. Microbiol. 2022, 60, e0256921. [Google Scholar] [CrossRef]
- Plitnick, J.; Griesemer, S.; Lasek-Nesselquist, E.; Singh, N.; Lamson, D.M.; St George, K. Whole-Genome Sequencing of SARS-CoV-2: Assessment of the Ion Torrent AmpliSeq Panel and Comparison with the Illumina MiSeq ARTIC Protocol. J. Clin. Microbiol. 2021, 59, e0064921. [Google Scholar] [CrossRef]
- Jacot, D.; Pillonel, T.; Greub, G.; Bertelli, C. Assessment of SARS-CoV-2 Genome Sequencing: Quality Criteria and Low-Frequency Variants. J. Clin. Microbiol. 2021, 59, e0094421. [Google Scholar] [CrossRef] [PubMed]
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 708. [Google Scholar] [CrossRef]
- Dragen by Illumina. Available online: https://emea.illumina.com/products/by-type/informatics-products/dragen-bio-it-platform.html#dragen (accessed on 25 August 2022).
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Hölzer, M. poreCov-An Easy to Use, Fast, and Robust Workflow for SARS-CoV-2 Genome Reconstruction via Nanopore Sequencing. Front. Genet. 2021, 12, 1397. [Google Scholar] [CrossRef]
- Tilloy, V.; Cuzin, P.; Leroi, L.; Guérin, E.; Durand, P.; Alain, S. ASPICov: An automated pipeline for identification of SARS-Cov2 nucleotidic variants. PLoS ONE 2022, 17, e0262953. [Google Scholar] [CrossRef]
- Lo, C.-C.; Shakya, M.; Connor, R.; Davenport, K.; Flynn, M.; Gutiérrez, A.M.Y.; Hu, B.; Li, P.-E.; Jackson, E.P.; Xu, Y.; et al. EDGE COVID-19: A web platform to generate submission-ready genomes from SARS-CoV-2 sequencing efforts. Bioinformatics 2022, 38, 2700–2704. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer1, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- de Sabato, L.; Vaccari, G.; Knijn, A.; Ianiro, G.; Bartolo, I.D.; Morabito, S. SARS-CoV-2 RECoVERY: A multi-platform open-source bioinformatic pipeline for the automatic construction and analysis of SARS-CoV-2 genomes from NGS sequencing data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Salles, T.S.; Cavalcanti, A.C.; da Costa, F.B.; Dias, V.Z.; de Souza, L.M.; de Meneses, M.D.F.; da Silva, J.A.S.; Amaral, C.D.; Felix, J.R.; Pereira, D.A.; et al. Genomic surveillance of SARS-CoV-2 Spike gene by sanger sequencing. PLoS ONE 2022, 17, e0262170. [Google Scholar] [CrossRef] [PubMed]
- Illumina COVIDSeq Test by Illumina. Available online: https://emea.illumina.com/products/by-type/ivd-products/covidseq.html (accessed on 26 August 2022).
- Ampliseq for Illumina SARS-CoV-2 Research Panel by Illumina. Available online: https://emea.illumina.com/products/by-brand/ampliseq/community-panels/sars-cov-2.html (accessed on 26 August 2022).
- Respiratory Virus Oligo Panel by Illumina. Available online: https://emea.illumina.com/products/by-type/sequencing-kits/library-prep-kits/respiratory-virus-oligo-panel.html (accessed on 26 August 2022).
- Respiratory Pathogen ID/AMR Enrichment Panel Kit by Illumina. Available online: https://emea.illumina.com/products/by-type/sequencing-kits/library-prep-kits/respiratory-pathogen-id-panel.html (accessed on 26 August 2022).
- Ion AmpliSeq SARS-CoV-2 Insight Research Assay by LifeTechnologies. Available online: https://www.thermofisher.com/it/en/home/life-science/sequencing/dna-sequencing/microbial-sequencing/microbial-identification-ion-torrent-next-generation-sequencing/viral-typing/coronavirus-research.html (accessed on 26 August 2022).
- ARTIC Network by ARTIC Network. Available online: https://artic.network/ncov-2019 (accessed on 26 August 2022).
- ARTIC Protocol by ARTIC Network. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-bp2l6n26rgqe/v1?version_warning=no (accessed on 26 August 2022).
- Sberna, G.; Fabeni, L.; Berno, G.; Carletti, F.; Specchiarello, E.; Colavita, F.; Meschi, S.; Matusali, G.; Garbuglia, A.R.; Bordi, L.; et al. Rapid and qualitative identification of SARS-CoV-2 mutations associated with Variants of Concern (VOC) using a multiplex RT-PCR assay coupled with melting analysis. Int. J. Infect. Dis. 2022, 122, 401–404. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1201971222003678 (accessed on 25 August 2022). [CrossRef] [PubMed]
- Nielsen, M.C.; Machado, R.R.G.; Mitchell, B.M.; McConnell, A.J.; Saada, N.I.; Weaver, S.C.; Ren, P. A Comparison of Seegene Technologies Novaplex SARS-CoV-2 Variants I, II, and IV Assays with Spike Gene Sequencing for Detection of Known Severe Acute Respiratory Syndrome Coronavirus 2 Variants. J. Mol. Diagn. 2022, 24, 455–461. [Google Scholar] [CrossRef]
- Implications of the Emergence and Spread of the SARS-CoV-2 B.1.1. 529 Variant of Concern (Omicron) for the EU/EEA by ECDC. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Implications-emergence-spread-SARS-CoV-2%20B.1.1.529-variant-concern-Omicron-for-the-EU-EEA-Nov2021.pdf (accessed on 26 November 2022).
- De Pace, V.; Bruzzone, B.; Orsi, A.; Ricucci, V.; Domnich, A.; Guarona, G.; Randazzo, N.; Stefanelli, F.; Battolla, E.; Dusi, P.A.; et al. Comparative Analysis of Five Multiplex RT-PCR Assays in the Screening of SARS-CoV-2 Variants. Microorganisms 2022, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Alejo-Cancho, I.; Gual-de-Torrella, A.; Gallego, M.; Urrutikoetxea-Gutierrez, M.; Lejarraga, C.; López de Goikoetxea, M.J. Misidentification of the SARS-CoV-2 Mu variant using commercial mutation screening assays. Arch. Virol. 2022, 167, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- ABL Advanced Biological Laboratories. Ultragene Assay SARS-CoV-2 Multi Variants Deletions V1 (CE-IVD). Available online: https://www.ablsa.com/laboratory-applications/ultragene-combo2screen-3/ (accessed on 25 August 2022).
- S.r.l. C. COVID-19 Ultra Variant Catcher Kit. Available online: https://www.clonit.it/en/company/news/covid-19-ultra-variant-catcher-kit/ (accessed on 25 August 2022).
- Diasorin. Simplexa® SARS-CoV-2 Variants Direct (RUO). Available online: https://molecular.diasorin.com/us/kit/simplexa-sars-cov-2-variants-direct/ (accessed on 25 August 2022).
- Elitech. Respiratory Pathogen Infections SARS-CoV-2 Variants ELITe MGB® Kit. Available online: https://www.elitechgroup.com/product/sars-cov-2-variants-elite-mgb-kit (accessed on 25 August 2022).
- ThermoFisher. TaqPathTM COVID-19 CE-IVD RT-PCR Kit Instructions for Use. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0019215_TaqPathCOVID-19_CE-IVD_RT-PCR%20Kit_IFU.pdf (accessed on 25 August 2022).
- Seegene. NovaplexTM SARS-CoV-2 Variants Assays*. Available online: https://seegenetech.com/novaplex-sars-cov-2-variants-ii-assay/#:~:text=TheNovaplexTMSARS-CoV,viralnucleicacidsandvariants (accessed on 25 August 2022).
- Hamill, V.; Noll, L.; Lu, N.; Tsui, W.N.T.; Porter, E.P.; Gray, M.; Sebhatu, T.; Goerl, K.; Brown, S.; Palinski, R.; et al. Molecular detection of SARS-CoV-2 strains and differentiation of Delta variant strains. Transbound. Emerg. Dis. 2022, 1–11. [Google Scholar] [CrossRef]
- Barua, S.; Bai, J.; John, P.; Hanzlicek, G.; Noll, L.; Johnson, C.; Yin, J.-H.; Wang, C. Identi fi cation of the SARS-CoV-2 Delta variant C22995A using a high- resolution melting curve RT-FRET-PCR. Emerg. Microbes Infect. 2021, 11, 11–14. [Google Scholar]
- Chan, C.T.M.; Leung, J.S.L.; Lee, L.K.; Lo, H.W.H.; Wong, E.Y.K.; Wong, D.S.H.; Ng, T.T.L.; Lao, H.-Y.; Lu, K.K.; Jim, S.H.-C.; et al. A low-cost TaqMan minor groove binder probe-based one-step RT-qPCR assay for rapid identification of N501Y variants of SARS-CoV-2. J. Virol. Methods 2022, 299, 114333. [Google Scholar] [CrossRef]
- Erster, O.; Mendelson, E.; Levy, V.; Kabat, A.; Mannasse, B.; Asraf, H.; Azar, R.; Ali, Y.; Shirazi, R.; Bucris, E.; et al. Rapid and High-Throughput Reverse Transcriptase Quantitative PCR (RT-qPCR) Assay for Identification and Differentiation between SARS-CoV-2 Variants B.1.1.7 and B.1.351. Microbiol. Spectr. 2021, 9, 1–9. [Google Scholar] [CrossRef]
- Brito-Mutunayagam, S.; Maloney, D.; McAllister, G.; Dewar, R.; McHugh, M.; Templeton, K. Rapid detection of SARS-CoV-2 variants using allele-specific PCR. J. Virol. Methods 2022, 303, 114497. [Google Scholar] [CrossRef]
- Chung, H.-Y.; Jian, M.-J.; Chang, C.-K.; Lin, J.-C.; Yeh, K.-M.; Chen, C.-W.; Hsieh, S.-S.; Hung, K.-S.; Tang, S.-H.; Perng, C.-L.; et al. Emergency SARS-CoV-2 Variants of Concern: Novel Multiplex Real-Time RT-PCR Assay for Rapid Detection and Surveillance. Microbiol. Spectr. 2022, 10, e0251321. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.E.; Zheng, H.; Yao, H.; Cantarel, B. Multiplex Fragment Analysis for Flexible Detection of All SARS-CoV-2 Variants of Concern. Clin. Chem. 2022, 11, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.M.; Banu, R.; Gonzalez-Reiche, A.S.; van de Guchte, A.; Khan, Z.; Shrestha, P.; Cao, L.; Chen, F.; Shi, H.; Hanna, A.; et al. Robust clinical detection of SARS-CoV-2 variants by RT-PCR/MALDI-TOF multitarget approach. J. Med. Virol. 2022, 94, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Welch, N.L.; Zhu, M.; Hua, C.; Weller, J.; Mirhashemi, M.E.; Nguyen, T.G.; Mantena, S.; Bauer, M.R.; Shaw, B.M.; Ackerman, C.M.; et al. Multiplexed CRISPR-based microfluidic platform for clinical testing of respiratory viruses and identification of SARS-CoV-2 variants. Nat. Med. 2022, 28, 1083–1094. [Google Scholar] [CrossRef]


| Lineage -> | Alpha | Beta (B.1.351) | Gamma (P.1) | Zeta | Eta | Kappa (B.1.617.1) | Delta (B.1.617.2) | Delta (AY.1) | Delta (AY.2) | Delta (AY.4) | Delta (AY.4.2) | Delta (AY.43) | Lambda | Mu | Omicron | Omicron (BA.2) | Omicron (BA.4/BA.5) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (B.1.1.7) | (P.2) | B.1.525) | (C.37) | (B.1.621) | (BA.1) | |||||||||||||
| Gene | Spike Regions/Domains | Mutations | ||||||||||||||||
| Spike (1273 aa) | NTD (aa13-304) | L18F | L18F | |||||||||||||||
| T19R | T19R | T19R | T19R | T19R | T19R | T19I | T19I | |||||||||||
| T20N | ||||||||||||||||||
| LPP24-26del | LPP24-26del | |||||||||||||||||
| A24S | A24S | |||||||||||||||||
| P26S | ||||||||||||||||||
| Q52R | ||||||||||||||||||
| A67V | A67V | |||||||||||||||||
| HV69-70del | HV69-70del | V70F | HV69-70del | HV69-70del | ||||||||||||||
| G75V | ||||||||||||||||||
| T76I | ||||||||||||||||||
| D80A | ||||||||||||||||||
| T95I | T95I | T95I | T95I | T95I | ||||||||||||||
| D138Y | ||||||||||||||||||
| G142D | G142D | G142D | G142D | G142D | G142D | G142D | G142D | G142D | ||||||||||
| VYY143-145del | ||||||||||||||||||
| ins144-145VN | ||||||||||||||||||
| Y144del | Y144del | |||||||||||||||||
| Y145H | ||||||||||||||||||
| Y145N | ||||||||||||||||||
| E154K | ||||||||||||||||||
| FR157-158del | FR157-158del | FR157-158del | FR157-158del | FR157-158del | FR157-158del | |||||||||||||
| R190S | ||||||||||||||||||
| N211del | ||||||||||||||||||
| L212I | ||||||||||||||||||
| V213G | V213G | |||||||||||||||||
| ins215-217EPE | ||||||||||||||||||
| D215G | ||||||||||||||||||
| A222V | A222V | |||||||||||||||||
| LAL242-244del | ||||||||||||||||||
| RSYLTPG246-252del | ||||||||||||||||||
| R246I | ||||||||||||||||||
| W258L | ||||||||||||||||||
| RBD (aa319-541) | G339D | G339D | G339D | |||||||||||||||
| S371L | S371F | S371F | ||||||||||||||||
| S373P | S373P | S373P | ||||||||||||||||
| S375F | S375F | S375F | ||||||||||||||||
| T376A | T376A | |||||||||||||||||
| D405N | D405N | |||||||||||||||||
| R408S | R408S | |||||||||||||||||
| K417N | K417T | K417N | K417N | K417N | K417N | K417N | ||||||||||||
| N440K | N440K | N440K | ||||||||||||||||
| G446S | ||||||||||||||||||
| L452R | L452R | L452R | L452R | L452R | L452R | L452R | L452Q | L452R | ||||||||||
| S477N | S477N | S477N | ||||||||||||||||
| T478K | T478K | T478K | T478K | T478K | T478K | T478K | T478K | T478K | ||||||||||
| E484K | E484K | E484K | E484K | E484Q | E484K | |||||||||||||
| E484A | E484A | E484A | ||||||||||||||||
| F486V | ||||||||||||||||||
| F490S | ||||||||||||||||||
| Q493R | Q493R | |||||||||||||||||
| G496S | ||||||||||||||||||
| Q498R | Q498R | Q498R | ||||||||||||||||
| N501Y | N501Y | N501Y | N501Y | N501Y | N501Y | N501Y | ||||||||||||
| Y505H | Y505H | Y505H | ||||||||||||||||
| CTS1/S2 (aa543-1273) | T547K | |||||||||||||||||
| A570D | ||||||||||||||||||
| D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | D614G | ||
| H655Y | H655Y | H655Y | H655Y | |||||||||||||||
| Q677H | ||||||||||||||||||
| N679K | N679K | N679K | ||||||||||||||||
| P681H | P681R | P681R | P681R | P681R | P681R | P681R | P681R | P681H | P681H | P681H | P681H | |||||||
| A701V | ||||||||||||||||||
| T716I | ||||||||||||||||||
| N764K | N764K | N764K | ||||||||||||||||
| D796Y | D796Y | D796Y | ||||||||||||||||
| N856K | ||||||||||||||||||
| T859N | ||||||||||||||||||
| F888L | ||||||||||||||||||
| D950N | D950N | D950N | D950N | D950N | D950N | |||||||||||||
| Q954H | Q954H | Q954H | ||||||||||||||||
| N969K | N969K | N969K | ||||||||||||||||
| L981F | ||||||||||||||||||
| S982A | ||||||||||||||||||
| T1027I | ||||||||||||||||||
| Q1071H | ||||||||||||||||||
| D1118H | ||||||||||||||||||
| V1176F | V1176F | |||||||||||||||||
| Tecnology | NGS Platform | Kit for Library Preparation | Methodology | Characteristics and Data Output | Time for Library Prep, Sequencing and Analysis |
|---|---|---|---|---|---|
| Illumina | NovaSeq 6000; NextSeq 2000; NextSeq 500/550; NextSeq 550Dx (in RUO mode) | Illumina COVIDSeq Test [101] | Amplicon based | The Illumina COVIDSeq Test leverages a modified version of the validated, publicly available ARTIC multiplex PCR protocol, with 98 amplicons designed to amplify SARS-CoV-2 virus-specific sequences | Library prep: 6–9 h Sequencing:12–44 h depending on flowcell and platform |
| All Illumina platforms | AmpliSeq for Illumina SARS-CoV-2 Research Panel [102] | Amplicon based | 247 amplicons in 2 pools; >99% coverage of the Coronavirus genome (~30 kb) and covers all potential serotypes | Library prep: <9 h Sequencing:12–44 h depending on flowcell and platform | |
| MiniSeq; MiSeq, MiSeq; NextSeq 550 | Respiratory Virus Oligo Panel [103] | Enrichment | Targets and characterizes ~40 common respiratory viruses, including SARS-CoV-2. hybrid–capture methods allows for highly sensitive detection; near-complete sequence data of targets | Library prep: <9 h. Sequencing: 12–30 h depending on flowcell and platform | |
| MiniSeq, MiSeq, MiSeq, NextSeq 550 | Respiratory Pathogen ID/AMR Enrichment Panel Kit [104] | Enrichment | >280 respiratory pathogens, including SARS-CoV-2; Report full genome coverage of SARS-CoV-2 | Library prep: <9 h. Sequencing: 12–30 h depending on flowcell and platform | |
| Ion Torrent | GeneStudio S5/S5 Prime | Ion AmpliSeq SARS-CoV-2 Insight Research Assay [105] | Amplicon based | Two pools with amplicons ranging from 125 bp to 275 bp in length and covers >99% of the SARS-CoV-2 genome, including all serotypes. | Less than 24 h from nucleic acid to data (with overnight Ion Chef System run) |
| Genexus Integrated Sequencer (Automated sequencer) | Ion AmpliSeq SARS-CoV-2 Insight Research Assay [105] | Amplicon based | Two pools with amplicons ranging from 125 bp to 275 bp in length and covers >99% of the SARS-CoV-2 genome, including all serotypes. | Library prep plus sequencing and Analysis: <24 h With <30 min hands-on time. | |
| Oxford Nanopore | MinION; GridION; PromethION. | ARTIC protocol [106,107] | Amplicon based | Two pools with amplicons for cover entire genome of SARS-CoV-2 | Library prep: 6–9 h Sequencing: it depends on the experiment goal and samples multiplexing |
| Company | SARS-CoV-2 Variants Assay’s Name | Regulatory Approval Status | Including RNA Extraction | Turnaround Time | Limit of Detection or Ct Cutoff Value | Mutation Detected | Interpretation of SARS-CoV-2 Variants Based on Detection of Mutations |
|---|---|---|---|---|---|---|---|
| ABL Advanced Biological Laboratories | UltraGene Assay SARS-CoV-2 Multi Variants Deletions V1 | CE - IVD | No | <1.30 h | 1150 TCID50/mL (QuantStudio 5) 115 TCID50/mL (UltraGene qPCR) | 69-70del, Y144del, 242-244del, 3675-3677del | 69-70del + Y144del + 3675-3677del --> B.1.1.7 242-244del + 3675-3677del --> B.1.351 3675-3677del --> P1 Lineage |
| Clonit S.r.l. | COVID-19 Variant Catcher COVID-19 Ultra Variant Catcher | CE - IVD | No | ≈2 h <1.30 h | SARS-CoV-2 Positive RNA Ct < 35 SARS-CoV-2 Positive RNA Ct < 30 | 69-70del, E484K, N501Y L452R, E484K, E484Q, N501Y | 69-70del + N501Y --> B.1.1.7 E484K + N501Y --> B.1.351/P1 Lineage N501Y --> B.1.1.7 N501Y + E484K --> B.1.351/P.1 L452R --> B.1.617.2/B.1.427/B.1.429/B.1.526/B.1.526.1 |
| Diasorin Molecular | Simplexa™ SARS-CoV-2 Variants Direct assay | RUO | Yes | <2 h | 500 copies/mL | N501Y, E484K, E484Q, L452 | N501Y + E484K --> B.1.351/P.1 L452R --> B.1.617.2/B.1.427/B.1.429/B.1.526/B.1.526.1 N501Y --> B.1.1.7 E484K -->B.1.525/P2 L452R + E484Q --> B.1.617.1/B.1.617.3 N501Y + E484Q --> B.1.621 |
| ELITech Group | SARS-CoV-2 Variants ELITe MGB® Kit SARS-CoV-2 Extended ELITe MGB® Kit | RUO | No | INA | INA | E484K, N501Y L452R, E484K, E484Q, N501Y | N501Y --> B.1.1.7 N501Y + E484K --> B.1.351/P.1 E484K -->B.1.525/P2 N501Y --> B.1.1.7 N501Y + E484K --> B.1.351/P.1 L452R --> B.1.617.2/B.1.427/B.1.429/B.1.526/B.1.526.1 |
| Life Technologies | TaqPath™ COVID 19 CE IVD RT PCR Kit | CE - IVD | No | <1.30 h | 10 GCE/reaction | 69-70del | 69-70del --> B.1.1.7/B.1.525/BA.1 |
| Seegene | Allplex™ SARS-CoV-2 Variants I Assay | CE - IVD | No (possibility of automated extraction and PCR setup) | INA | INA | 69-70del, E484K, N501Y | 69-70del + N501Y --> B.1.1.7/B.1.1.529 |
| E484K + N501Y --> B.1.351/P1/P.3 | |||||||
| 69-70del + E484K --> B.1.525 | |||||||
| Allplex™ SARS-CoV-2 Variants II Assay | CE - IVD in progress | INA | L452R, W152C, K417T, K417N | K417N --> B.1.351/B.1.1.529/AY.1/BA.2 | |||
| K417T --> P.1 | |||||||
| L452R + W152C --> B.1.429/ B.1.427 | |||||||
| L452R --> B.1.617.1/ B.1.617.2 | |||||||
| Novaplex™ SARS-CoV-2 Variants Assays I | RUO | 4 h (starting from extraction) | 69-70del, E484K, N501Y | 69-70del + N501Y --> B.1.1.7/B.1.529 | |||
| E484K + N501Y --> B.1.351/P1/P.3 | |||||||
| 69-70del + E484K --> B.1.525 | |||||||
| Novaplex™ SARS-CoV-2 Variants II Assay | L452R, W152C, K417T, K417N | K417N --> B.1.351/B.1.1.529/AY.1/BA.2 | |||||
| K417T --> P.1 | |||||||
| L452R + W152C --> B.1.429/ B.1.427 | |||||||
| L452R --> B.1.617.1/ B.1.617.2 | |||||||
| Novaplex™ SARS-CoV-2 Variants IV Assay | L452R, P681R, K417N | L452R + P681R --> B.1.617.1/ B.1.617.2 | |||||
| K417N --> B.1.351/B.1.1.529/BA.2 | |||||||
| L452R + P681R + K417N --> AY.1 | |||||||
| L425R --> B.1.427/B.1.429 | |||||||
| Novaplex™ SARS-CoV-2 Variants V Assay | L452Q, F490S,P681R, L452R | P681R + L452R --> B.1.617.1/B.1.617.2/AY.1 | |||||
| L425R --> B.1.427/B.1.429 | |||||||
| L452Q+F490S --> C.37 | |||||||
| Novaplex™ SARS-CoV-2 Variants VI Assay | L452Q, F490S, R346K, D950N | D950N --> B.1.617.1/ B.1.617.2/AY.1 | |||||
| L452Q + F490S --> C.37 | |||||||
| R346K + D950N --> B.1.621 | |||||||
| Novaplex™ SARS-CoV-2 Variants VII Assay | 69-70del, E484A, N501Y, RdRp | 69-70del + N501Y + RdRp --> B.1.1.7 | |||||
| N501Y + RdRp --> B.1.351/P.1/P.3 | |||||||
| 69-70del + E484A + N501Y + RdRp--> B.1.1.529 | |||||||
| E484A + N501Y + RdRp --> BA.2 | |||||||
| RdRp --> B.1.617.2/AY1/B.1.427/429/P.2/B.1.526/B.1.617.1/C.37/B.1.621 | |||||||
| 69-70del + RdRp --> B.1.525 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berno, G.; Fabeni, L.; Matusali, G.; Gruber, C.E.M.; Rueca, M.; Giombini, E.; Garbuglia, A.R. SARS-CoV-2 Variants Identification: Overview of Molecular Existing Methods. Pathogens 2022, 11, 1058. https://doi.org/10.3390/pathogens11091058
Berno G, Fabeni L, Matusali G, Gruber CEM, Rueca M, Giombini E, Garbuglia AR. SARS-CoV-2 Variants Identification: Overview of Molecular Existing Methods. Pathogens. 2022; 11(9):1058. https://doi.org/10.3390/pathogens11091058
Chicago/Turabian StyleBerno, Giulia, Lavinia Fabeni, Giulia Matusali, Cesare Ernesto Maria Gruber, Martina Rueca, Emanuela Giombini, and Anna Rosa Garbuglia. 2022. "SARS-CoV-2 Variants Identification: Overview of Molecular Existing Methods" Pathogens 11, no. 9: 1058. https://doi.org/10.3390/pathogens11091058