
Genomics Analysis to Identify Multiple Genetic Determinants That Drive the Global Transmission of the Pandemic ST95 Lineage of Extraintestinal Pathogenic Escherichia coli (ExPEC)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Collection for Comparative Genomics Analysis
2.2. Pan-Genome Analysis and Phylogenetic Analysis
2.3. In Silico Characterization of ST95 Genomes
2.4. Time-Scale Phylogenetic Analysis
2.5. Virulence Genes, Antimicrobial-Resistant Genes and Plasmid Replicon Analysis
2.6. Plasmid Analysis
2.7. Detection of Pathogenicity Islands (PAIs) in ST95
2.8. Development and Validation of a Multiplex PCR Assay to Rapidly Distinguish ST95 APEC/ExPEC Isolates
3. Results
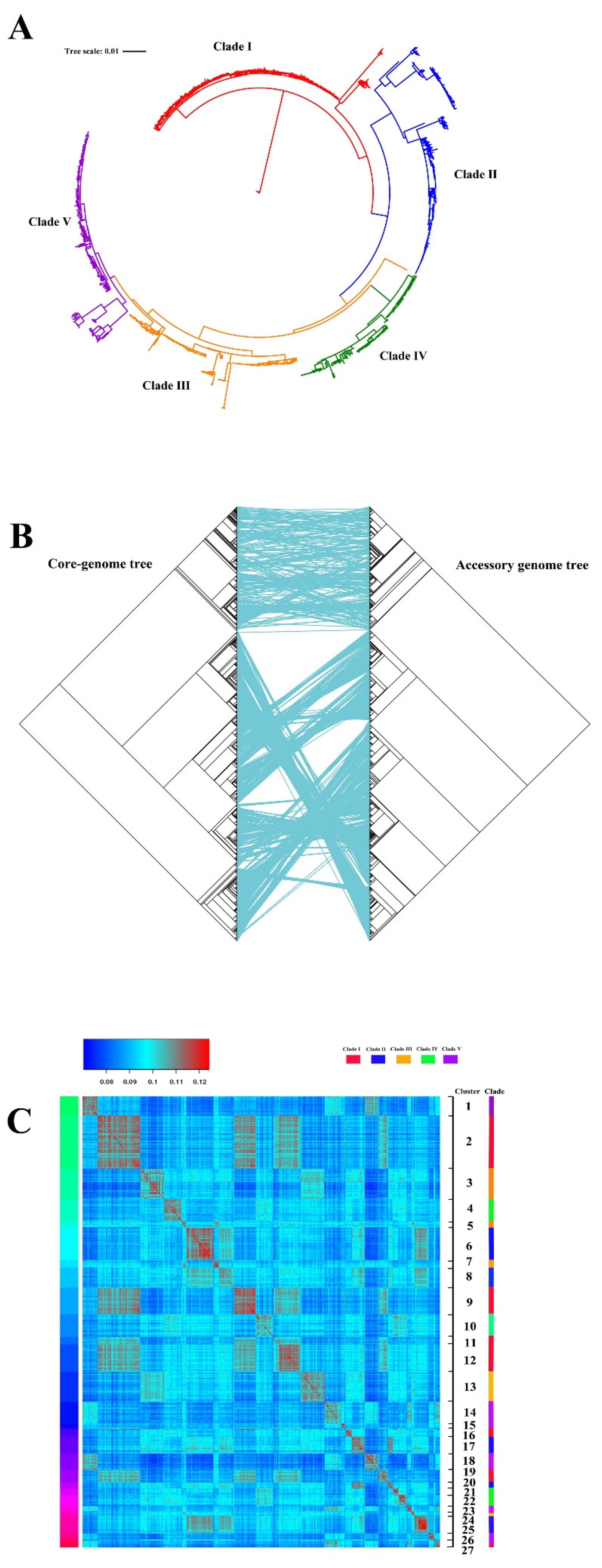
3.1. Pan-Genome Analysis and Phylogeny of ST95 ExPEC
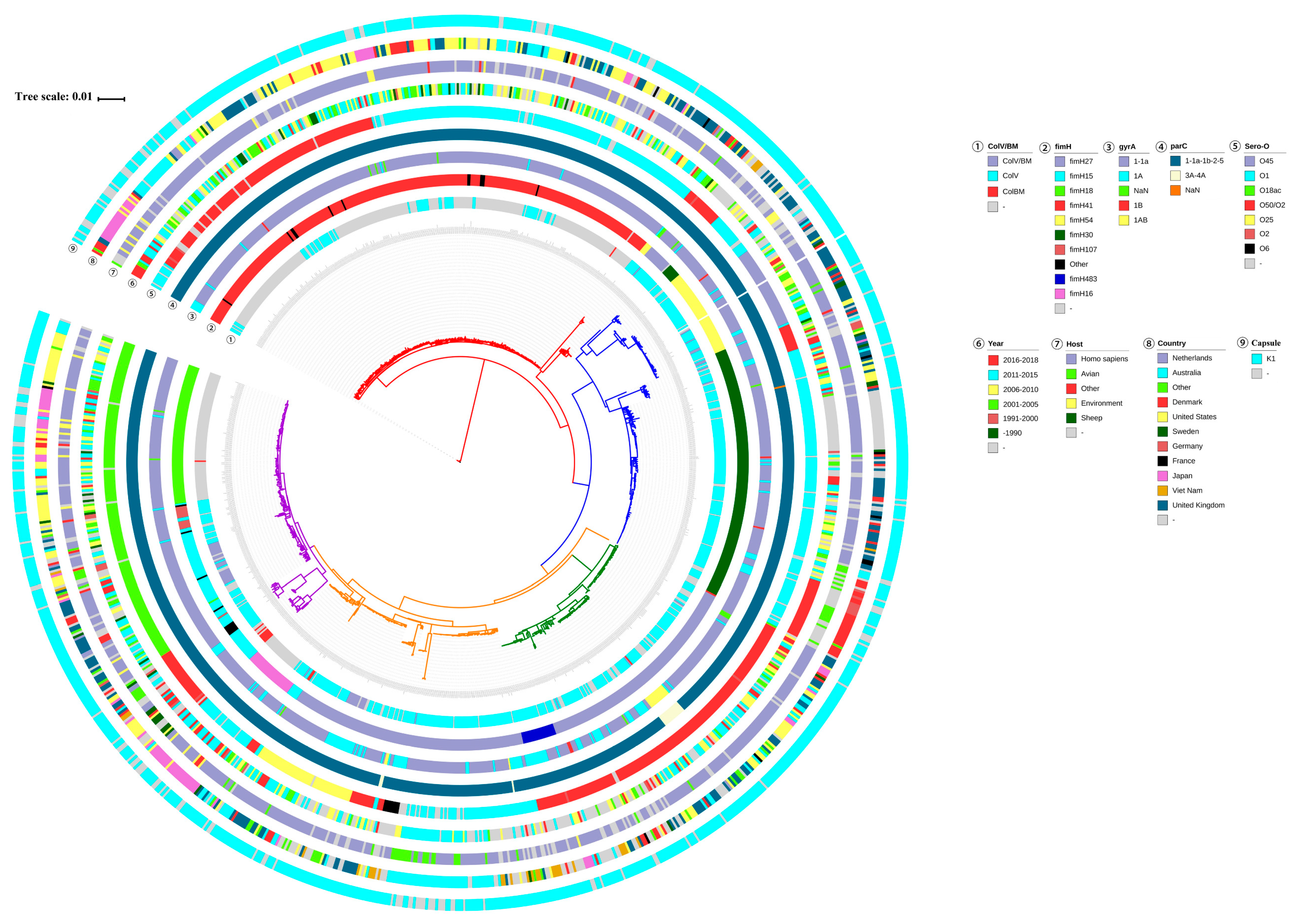
3.2. Genotype Characterization of ST95 ExPEC
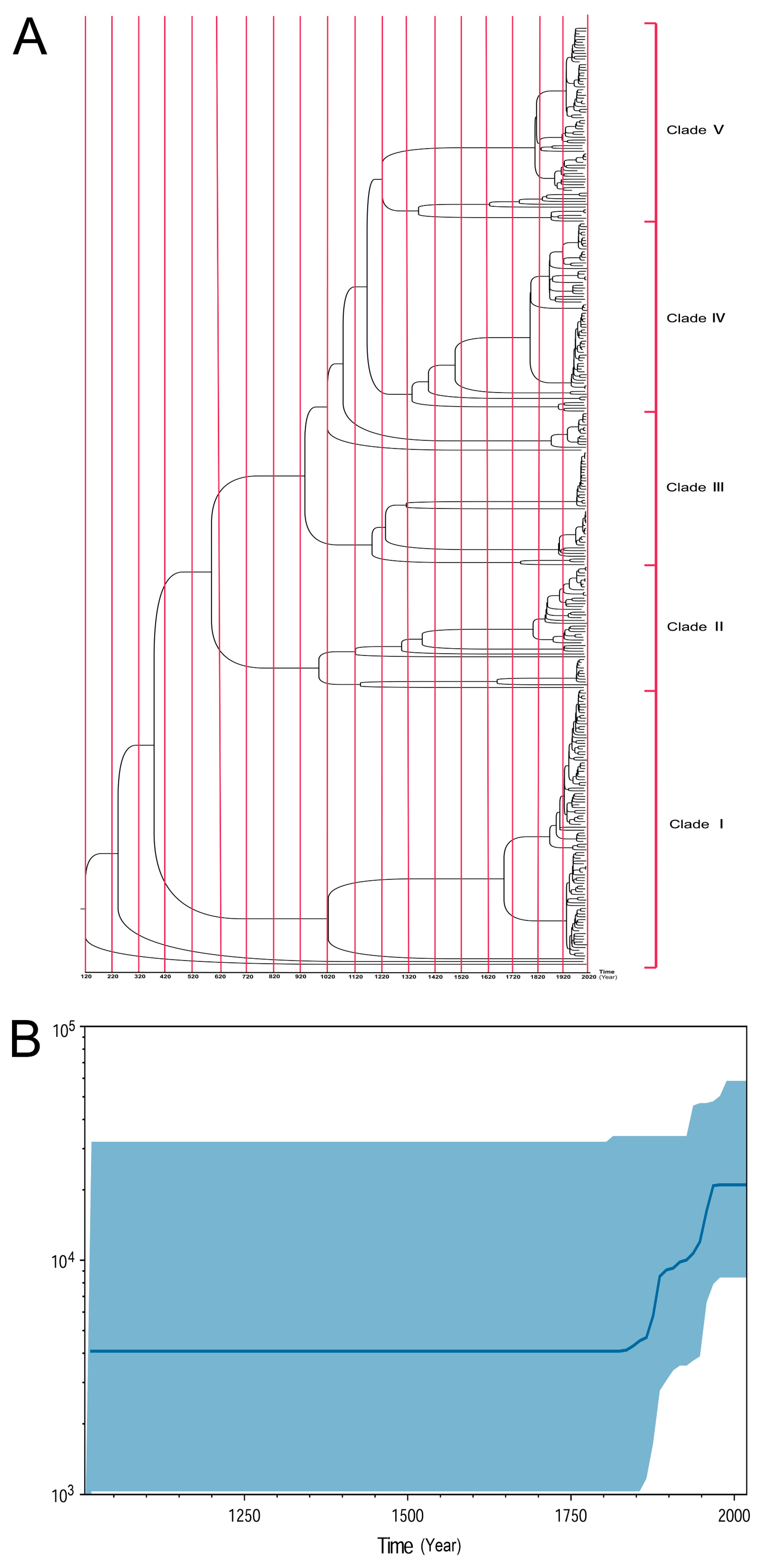
3.3. Time-Scale Emergence of ST95 ExPEC
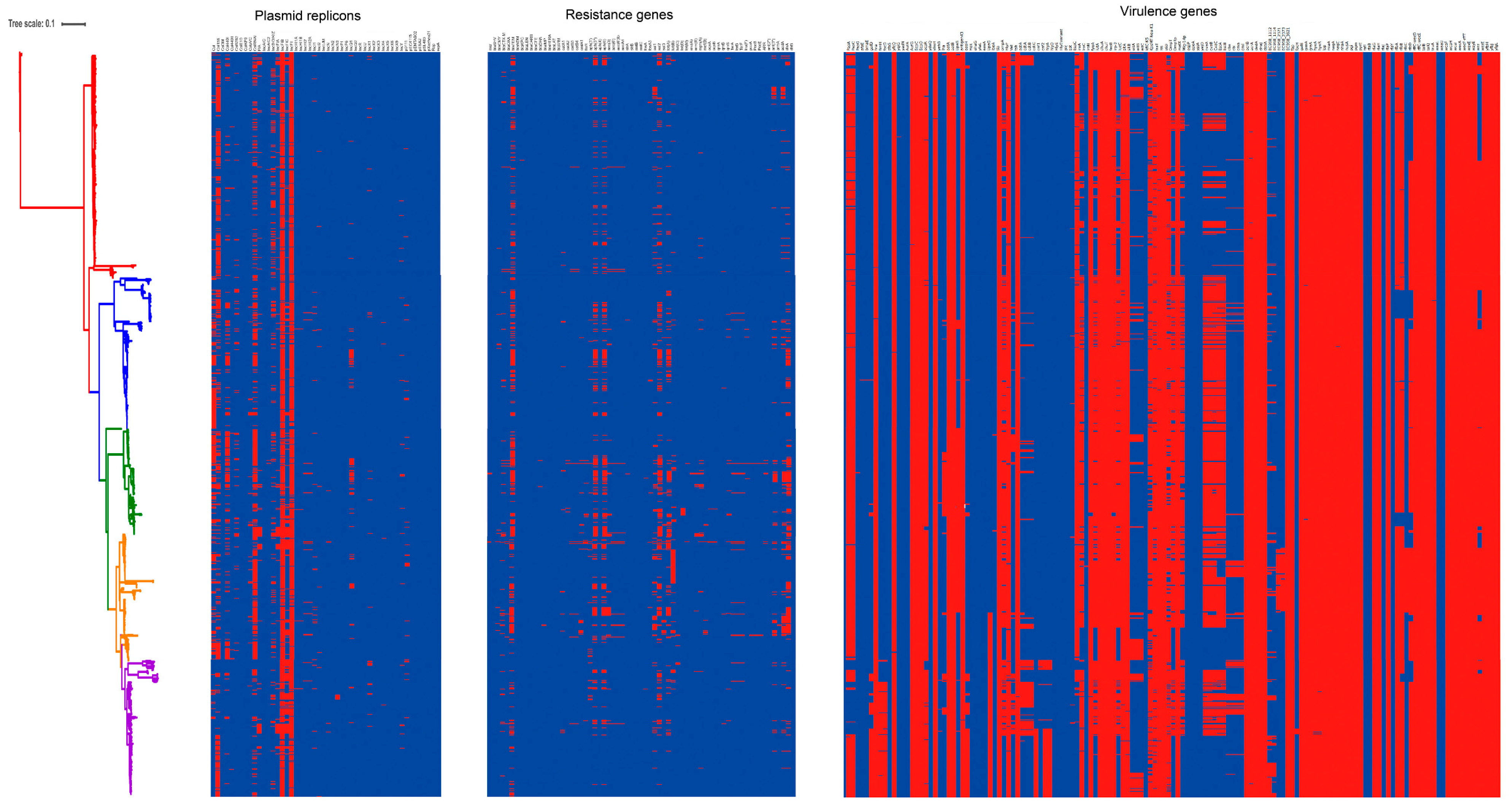
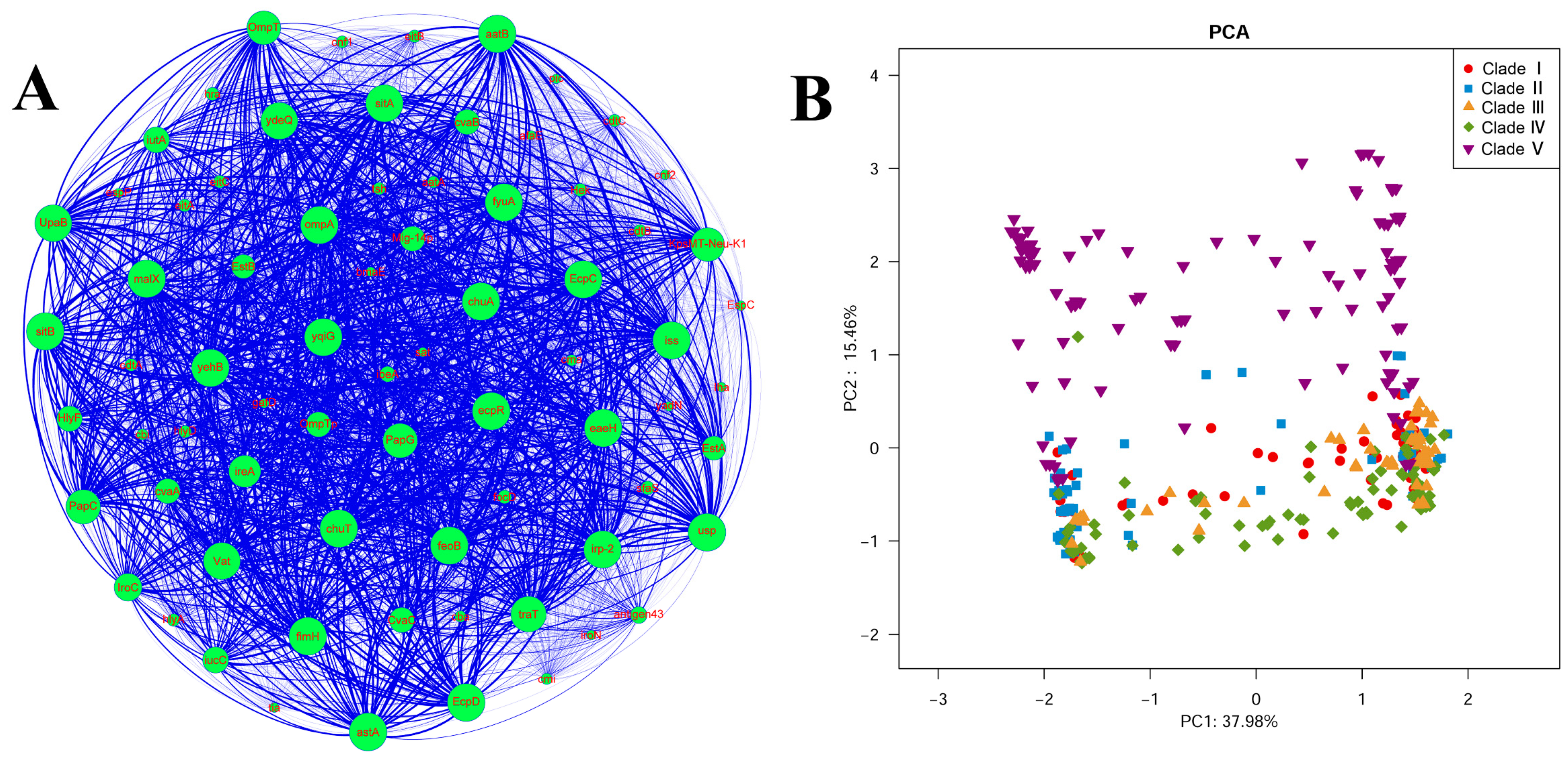
3.4. Carriage of Virulence Genes in ST95 ExPEC
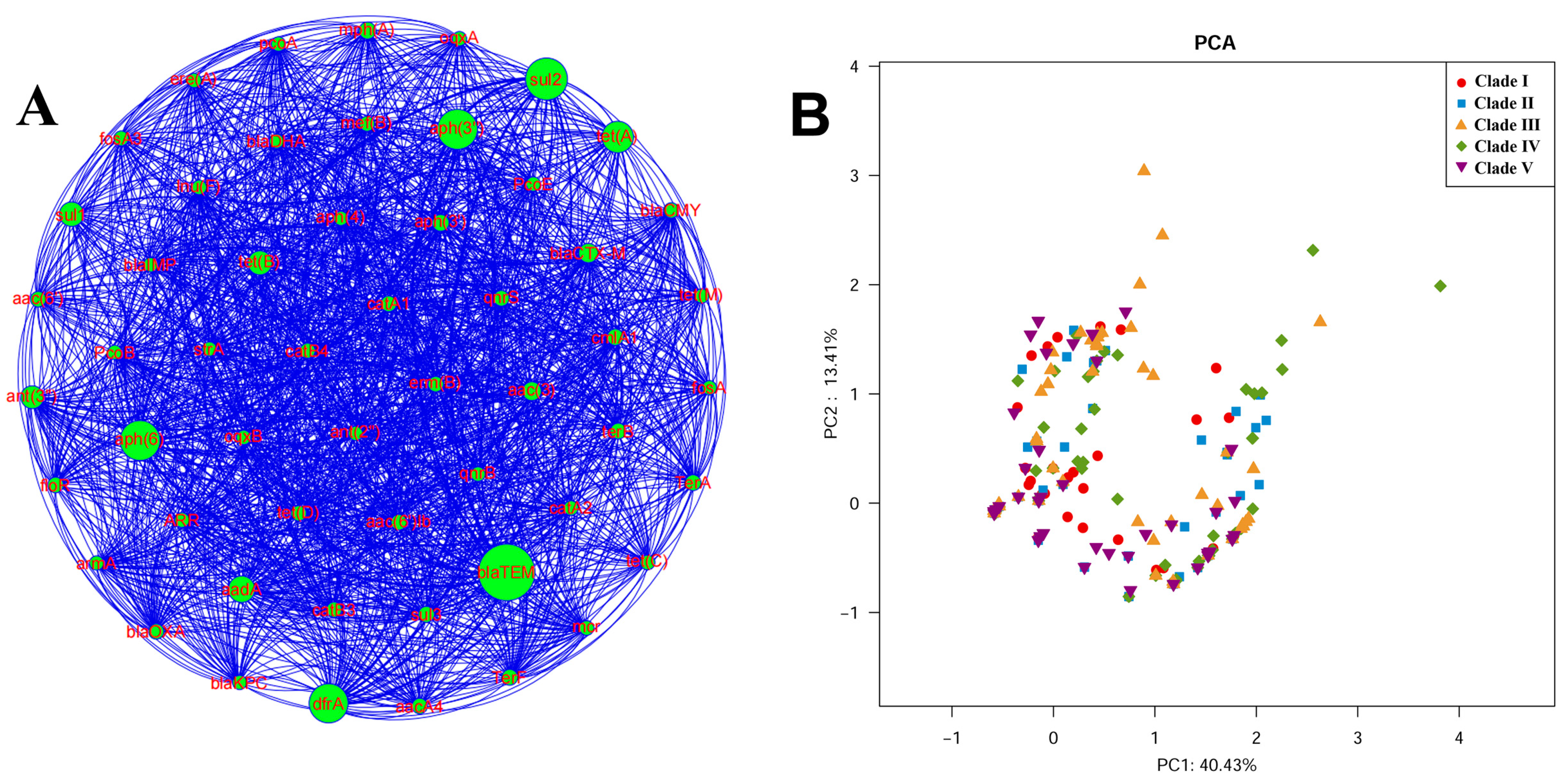
3.5. Carriage of Resistance Genes in ST95 ExPEC
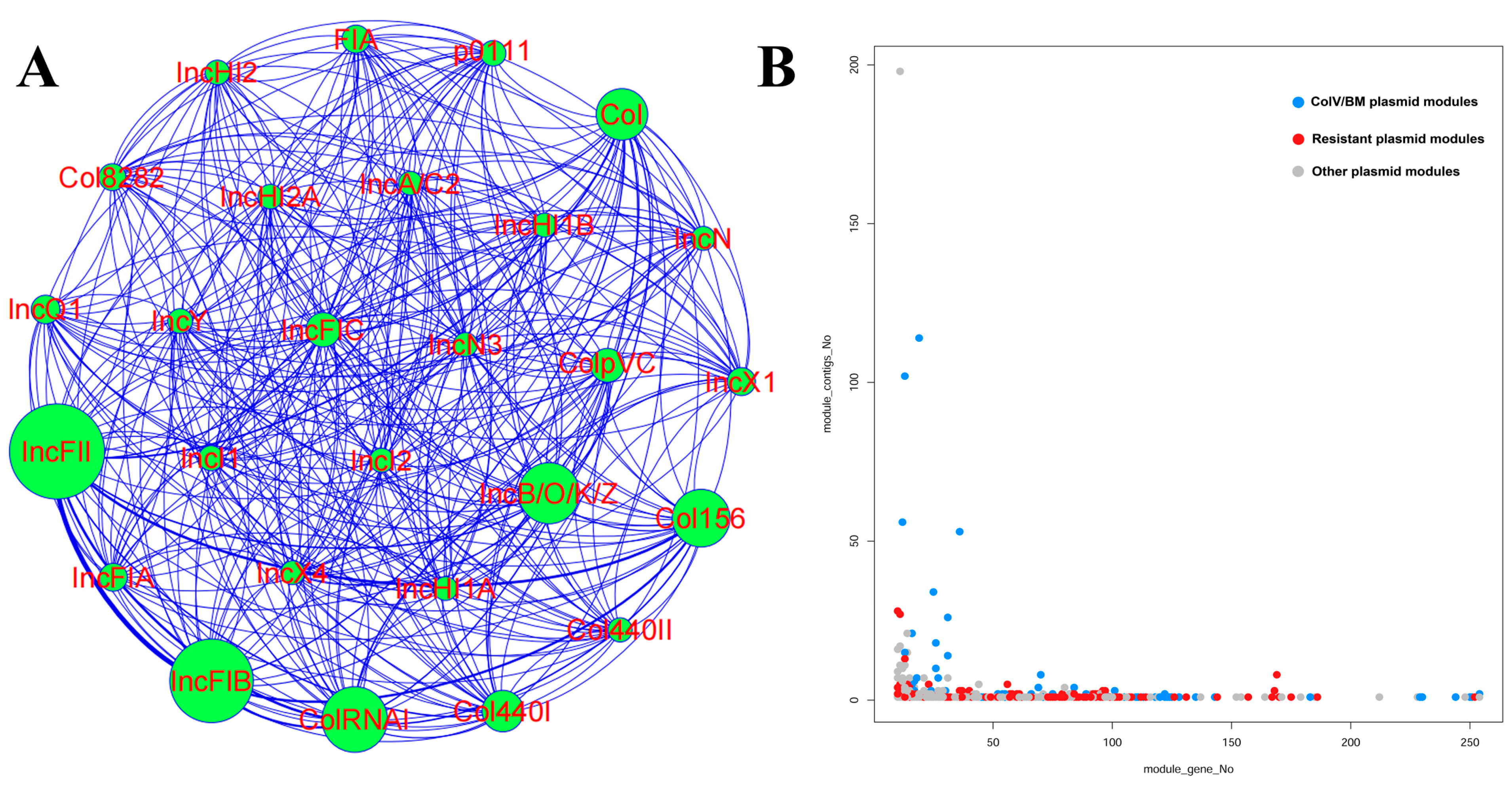
3.6. The Diversity of Plasmid Replicons in ST95 ExPEC
3.7. The Diversity of Mapped Plasmids within the ST95 Pan-Genome
3.8. The Distribution of Genomic Islands among ST95 ExPEC Strains
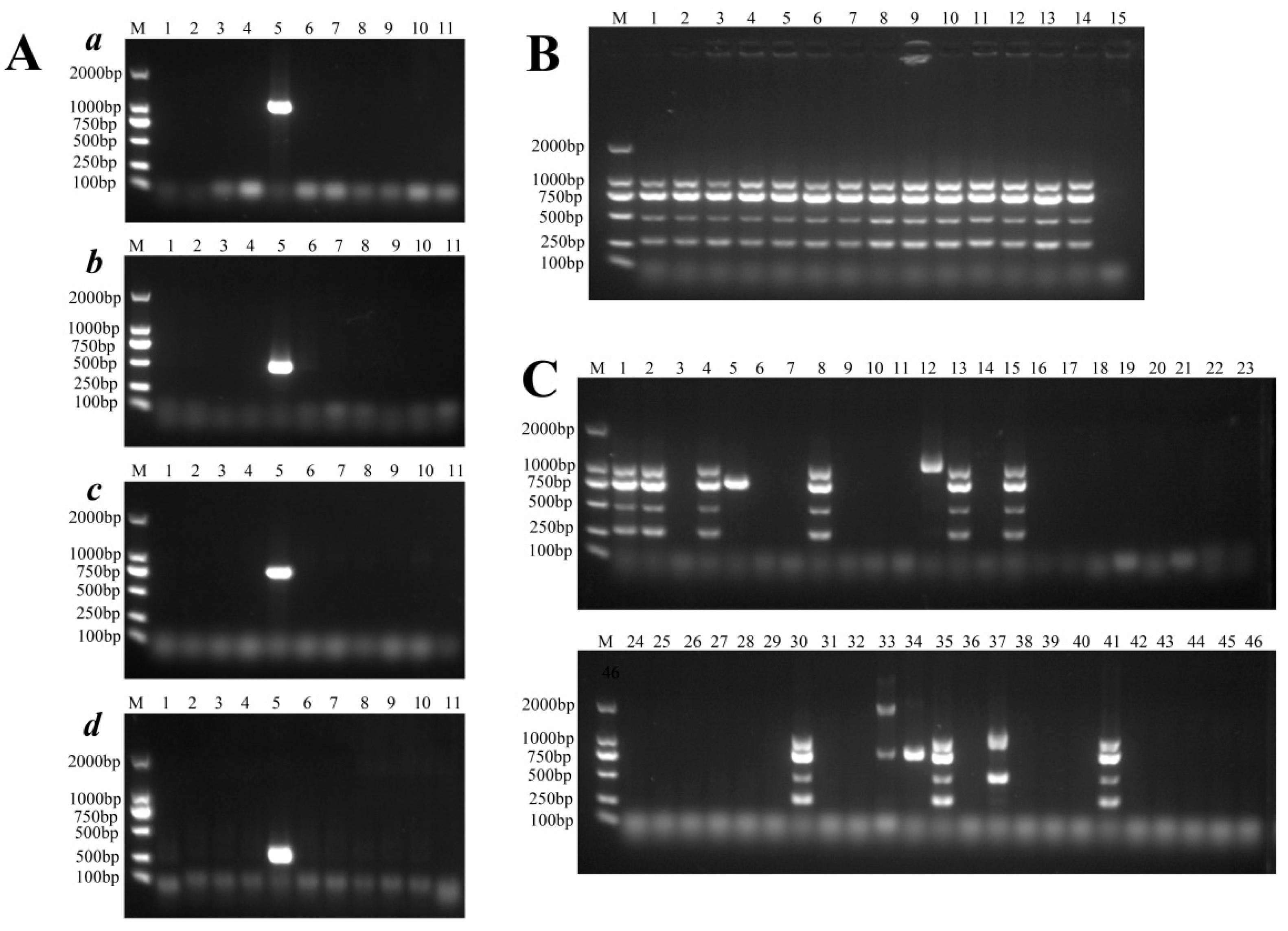
3.9. Rapid and Specific Detection of ST95 APEC/ExPEC Isolates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Johnson, J.R.; Russo, T.A. Extraintestinal pathogenic Escherichia coli: “The other bad E coli”. J. Lab. Clin. Med. 2002, 139, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The Clermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, X.; Ji, Y.; Tang, F.; Sun, Y.; Jiang, M.; Hu, W.; Wu, Y.; Xue, F.; Ren, J.; Zhu, W.; et al. Population structure and antimicrobial resistance traits of avian-origin mcr-1-positive Escherichia coli in Eastern China, 2015 to 2017. Transbound. Emerg. Dis. 2019, 66, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, X.; Zhou, Z.; Jiang, M.; Wang, Z.; Sun, Y.; Tang, F.; Xue, F.; Ren, J.; Dai, J. Chicken-source Escherichia coli within phylogroup F shares virulence genotypes and is closely related to extraintestinal pathogenic E. coli causing human infections. Transbound. Emerg. Dis. 2021, 68, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Ewers, C.; Li, G.W.; Wilking, H.; Kiessling, S.; Alt, K.; Antao, E.M.; Laturnus, C.; Diehl, I.; Glodde, S.; Homeier, T.; et al. Avian pathogenic, uropathogenic, and newborn meningitis-causing Escherichia coli: How closely related are they? Int. J. Med. Microbiol. 2007, 297, 163–176. [Google Scholar] [CrossRef]
- Ron, E.Z. Host specificity of septicemic Escherichia coli: Human and avian pathogens. Curr. Opin. Microbiol. 2006, 9, 28–32. [Google Scholar] [CrossRef]
- Russo, T.A.; Johnson, J.R. Medical and economic impact of extraintestinal infections due to Escherichia coli: Focus on an increasingly important endemic problem. Microbes. Infect. 2003, 5, 449–456. [Google Scholar] [CrossRef]
- Manges, A.R.; Geum, H.M.; Guo, A.; Edens, T.J.; Fibke, C.D.; Pitout, J.D.D. Global Extraintestinal Pathogenic Escherichia coli (ExPEC) Lineages. Clin. Microbiol. Rev. 2019, 32, e00135-18. [Google Scholar] [CrossRef]
- Horner, C.; Fawley, W.; Morris, K.; Parnell, P.; Denton, M.; Wilcox, M. Escherichia coli bacteraemia: 2 years of prospective regional surveillance (2010–2012). J. Antimicrob. Chemother. 2014, 69, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Riley, L.W. Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin. Microbiol. Infect. 2014, 20, 380–390. [Google Scholar] [CrossRef]
- Lau, S.H.; Reddy, S.; Cheesbrough, J.; Bolton, F.J.; Willshaw, G.; Cheasty, T.; Fox, A.J.; Upton, M. Major uropathogenic Escherichia coli strain isolated in the northwest of England identified by multilocus sequence typing. J. Clin. Microbiol. 2008, 46, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, B.M.; Zowawi, H.M.; Harris, P.N.A.; Roberts, L.; Ibrahim, E.; Shaikh, N.; Deshmukh, A.; Sid Ahmed, M.A.; Al Maslamani, M.; Cottrell, K.; et al. Discovery of mcr-1-Mediated Colistin Resistance in a Highly Virulent Escherichia coli Lineage. mSphere 2018, 3, e00486-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams-Sapper, S.; Diep, B.A.; Perdreau-Remington, F.; Riley, L.W. Clonal composition and community clustering of drug-susceptible and -resistant Escherichia coli isolates from bloodstream infections. Antimicrob. Agents Chemother. 2013, 57, 490–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires-dos-Santos, T.; Bisgaard, M.; Christensen, H. Genetic diversity and virulence profiles of Escherichia coli causing salpingitis and peritonitis in broiler breeders. Vet. Microbiol. 2013, 162, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.R.; Prussing, C.; Boerlin, P.; Daignault, D.; Dutil, L.; Reid-Smith, R.J.; Zhanel, G.G.; Manges, A.R. Chicken as Reservoir for Extraintestinal Pathogenic Escherichia coli in Humans, Canada. Emerg. Infect. Dis. 2012, 18, 415–421. [Google Scholar] [CrossRef]
- Mora, A.; Viso, S.; López, C.; Alonso, M.P.; García-Garrote, F.; Dabhi, G.; Mamani, R.; Herrera, A.; Marzoa, J.; Blanco, M.; et al. Poultry as reservoir for extraintestinal pathogenic Escherichia coli O45:K1:H7-B2-ST95 in humans. Vet. Microbiol. 2013, 167, 506–512. [Google Scholar] [CrossRef]
- Nandanwar, N.; Janssen, T.; Kuhl, M.; Ahmed, N.; Ewers, C.; Wieler, L.H. Extraintestinal pathogenic Escherichia coli (ExPEC) of human and avian origin belonging to sequence type complex 95 (STC95) portray indistinguishable virulence features. Int. J. Med. Microbiol. 2014, 304, 835–842. [Google Scholar] [CrossRef]
- Jørgensen, S.L.; Stegger, M.; Kudirkiene, E.; Lilje, B.; Poulsen, L.L.; Ronco, T.; Pires Dos Santos, T.; Kiil, K.; Bisgaard, M.; Pedersen, K.; et al. Diversity and Population Overlap between Avian and Human Escherichia coli Belonging to Sequence Type 95. mSphere 2019, 4, e00333-18. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.K.Z.; Jiang, J.W.; Pan, Z.H.; Hu, L.; Wang, S.H.; Wang, H.J.; Leung, F.C.; Dai, J.J.; Fan, H.J. Comparative Genomic Analysis Shows that Avian Pathogenic Escherichia coli Isolate IMT5155 (O2:K1:H5; ST Complex 95, ST140) Shares Close Relationship with ST95 APEC O1:K1 and Human ExPEC O18:K1 Strains. PLoS ONE 2014, 9, e112048. [Google Scholar]
- Tivendale, K.A.; Logue, C.M.; Kariyawasam, S.; Jordan, D.; Hussein, A.; Li, G.; Wannemuehler, Y.; Nolan, L.K. Avian-pathogenic Escherichia coli strains are similar to neonatal meningitis E. coli strains and are able to cause meningitis in the rat model of human disease. Infect. Immun. 2010, 78, 3412–3419. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.X.; Gao, S.; Huan, H.X.; Xu, X.J.; Zhu, X.P.; Yang, W.; Gao, Q.Q.; Liu, X.F. Comparison of virulence factors and expression of specific genes between uropathogenic Escherichia coli and avian pathogenic E-coli in a murine urinary tract infection model and a chicken challenge model. Microbiology 2009, 155, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Pallen, M.J. Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 2015, 13, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Pessia, A.; Grad, Y.; Cobey, S.; Puranen, J.S.; Corander, J. K-Pax2: Bayesian identification of cluster-defining amino acid positions in large sequence datasets. Microb. Genom. 2015, 1, e000025. [Google Scholar] [CrossRef]
- Joensen, K.G.; Tetzschner, A.M.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and Easy In Silico Serotyping of Escherichia coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef] [Green Version]
- Roer, L.; Tchesnokova, V.; Allesoe, R.; Muradova, M.; Chattopadhyay, S.; Ahrenfeldt, J.; Thomsen, M.C.F.; Lund, O.; Hansen, F.; Hammerum, A.M.; et al. Development of a Web Tool for Escherichia coli Subtyping Based on fimH Alleles. J. Clin. Microbiol. 2017, 55, 2538–2543. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Tchesnokova, V.; Johnston, B.; Clabots, C.; Roberts, P.L.; Billig, M.; Riddell, K.; Rogers, P.; Qin, X.; Butler-Wu, S.; et al. Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J. Infect. Dis. 2013, 207, 919–928. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Uhlemann, A.C.; McAdam, P.R.; Sullivan, S.B.; Knox, J.R.; Khiabanian, H.; Rabadan, R.; Davies, P.R.; Fitzgerald, J.R.; Lowy, F.D. Evolutionary Dynamics of Pandemic Methicillin-Sensitive Staphylococcus aureus ST398 and Its International Spread via Routes of Human Migration. mBio 2017, 8, e01375-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemoth. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.; Jiang, M.; Wen, Z.; Wang, Z.; Wang, M.; Xu, Y.; Zhuge, X.; Dai, J. Complete genomic analysis of ST117 lineage extraintestinal pathogenic Escherichia coli (ExPEC) to reveal multiple genetic determinants to drive its global transmission: ST117 E. coli as an emerging multidrug-resistant foodborne ExPEC with zoonotic potential. Transbound. Emerg. Dis. 2022. [Google Scholar] [CrossRef]
- Yoon, S.H.; Park, Y.K.; Lee, S.; Choi, D.; Oh, T.K.; Hur, C.G.; Kim, J.F. Towards pathogenomics: A web-based resource for pathogenicity islands. Nucleic Acids Res. 2007, 35, D395–D400. [Google Scholar] [CrossRef] [Green Version]
- Clermont, O.; Dixit, O.V.A.; Vangchhia, B.; Condamine, B.; Dion, S.; Bridier-Nahmias, A.; Denamur, E.; Gordon, D. Characterization and rapid identification of phylogroup G in Escherichia coli, a lineage with high virulence and antibiotic resistance potential. Environ. Microbiol. 2019, 21, 3107–3117. [Google Scholar] [CrossRef]
- Rodriguez-Siek, K.E.; Giddings, C.W.; Doetkott, C.; Johnson, T.J.; Fakhr, M.K.; Nolan, L.K. Comparison of Escherichia coli isolates implicated in human urinary tract infection and avian colibacillosis. Microbiology 2005, 151 Pt 6, 2097–2110. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Siek, K.E.; Giddings, C.W.; Doetkott, C.; Johnson, T.J.; Nolan, L.K. Characterizing the APEC pathotype. Vet. Res. 2005, 36, 241–256. [Google Scholar] [CrossRef] [Green Version]
- Dias, R.C.; Moreira, B.M.; Riley, L.W. Use of fimH single-nucleotide polymorphisms for strain typing of clinical isolates of Escherichia coli for epidemiologic investigation. J. Clin. Microbiol. 2010, 48, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Tartof, S.Y.; Solberg, O.D.; Riley, L.W. Genotypic analyses of uropathogenic Escherichia coli based on fimH single nucleotide polymorphisms (SNPs). J. Med. Microbiol. 2007, 56 Pt 10, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoesser, N.; Sheppard, A.E.; Peirano, G.; Anson, L.W.; Pankhurst, L.; Sebra, R.; Phan, H.T.T.; Kasarskis, A.; Mathers, A.J.; Peto, T.E.A.; et al. Genomic epidemiology of global Klebsiella pneumoniae carbapenemase (KPC)-producing Escherichia coli. Sci. Rep. 2017, 7, 5917. [Google Scholar] [CrossRef] [Green Version]
- Stephens, C.M.; Adams-Sapper, S.; Sekhon, M.; Johnson, J.R.; Riley, L.W. Genomic Analysis of Factors Associated with Low Prevalence of Antibiotic Resistance in Extraintestinal Pathogenic Escherichia coli Sequence Type 95 Strains. mSphere 2017, 2, e00390-16. [Google Scholar] [CrossRef] [Green Version]
- Fricke, W.F.; McDermott, P.F.; Mammel, M.K.; Zhao, S.; Johnson, T.J.; Rasko, D.A.; Fedorka-Cray, P.J.; Pedroso, A.; Whichard, J.M.; Leclerc, J.E.; et al. Antimicrobial resistance-conferring plasmids with similarity to virulence plasmids from avian pathogenic Escherichia coli strains in Salmonella enterica serovar Kentucky isolates from poultry. Appl. Environ. Microbiol. 2009, 75, 5963–5971. [Google Scholar] [CrossRef] [Green Version]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [Green Version]
- Parreira, V.R.; Gyles, C.L. A novel pathogenicity island integrated adjacent to the thrW tRNA gene of avian pathogenic Escherichia coli encodes a vacuolating autotransporter toxin. Infect. Immun. 2003, 71, 5087–5096. [Google Scholar] [CrossRef] [Green Version]
- Kariyawasam, S.; Johnson, T.J.; Nolan, L.K. The pap operon of avian pathogenic Escherichia coli strain O1: K1 is located on a novel pathogenicity island. Infect. Immun. 2006, 74, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Dobrindt, U.; Blum-Oehler, G.; Nagy, G.; Schneider, G.; Johann, A.; Gottschalk, G.; Hacker, J. Genetic structure and distribution of four pathogenicity islands (PAI I(536) to PAI IV(536)) of uropathogenic Escherichia coli strain. Infect. Immun. 2002, 70, 6365–6372. [Google Scholar] [CrossRef] [Green Version]
- Lalioui, L.; Le Bouguenec, C. afa-8 gene cluster is carried by a pathogenicity island inserted into the tRNA(Phe) of human and bovine pathogenic Escherichia coli isolates (vol 69, pg 937, 2001). Infect. Immun. 2002, 70, 3308. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.H.; Chen, Y.H.; Kong, G.; Chen, S.H.; Besemer, J.; Borodovsky, M.; Jong, A. A novel genetic island of meningitic Escherichia coli K1 containing the ibeA invasion gene (GimA): Functional annotation and carbon-source-regulated invasion of human brain microvascular endothelial cells. Funct. Integr. Genom. 2001, 1, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Klemm, E.; Dougan, G. Advances in Understanding Bacterial Pathogenesis Gained from Whole-Genome Sequencing and Phylogenetics. Cell Host. Microbe 2016, 19, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.M.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, R.; Rubin, J.; Thys, E.; Friedman, C.R.; Riley, L.W. Persistent Pandemic Lineages of Uropathogenic Escherichia coli in a College Community from 1999 to 2017. J. Clin. Microbiol. 2018, 56, e01834-17. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.M.; Geyik, S.; Clermont, O.; O’Brien, C.L.; Huang, S.; Abayasekara, C.; Rajesh, A.; Kennedy, K.; Collignon, P.; Pavli, P.; et al. Fine-Scale Structure Analysis Shows Epidemic Patterns of Clonal Complex 95, a Cosmopolitan Escherichia coli Lineage Responsible for Extraintestinal Infection. mSphere 2017, 2, e00168-17. [Google Scholar] [CrossRef] [Green Version]
- Bidet, P.; Mahjoub-Messai, F.; Blanco, J.; Blanco, J.; Dehem, M.; Aujard, Y.; Bingen, E.; Bonacorsi, S. Combined Multilocus Sequence Typing and O Serogrouping Distinguishes Escherichia coli Subtypes Associated with Infant Urosepsis and/or Meningitis. J. Infect. Dis. 2007, 196, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, S.J.; Chattopadhyay, S.; Aprikian, P.; Obata-Yasuoka, M.; Yarova-Yarovaya, Y.; Stapleton, A.; Ba-Thein, W.; Dykhuizen, D.; Johnson, J.R.; Sokurenko, E.V. Clonal analysis reveals high rate of structural mutations in fimbrial adhesins of extraintestinal pathogenic Escherichia coli. Mol. Microbiol. 2006, 59, 975–988. [Google Scholar] [CrossRef] [Green Version]
- Kallonen, T.; Brodrick, H.J.; Harris, S.R.; Corander, J.; Brown, N.M.; Martin, V.; Peacock, S.J.; Parkhill, J. Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome Res. 2017, 27, 1437–1449. [Google Scholar] [CrossRef] [Green Version]
- Stoesser, N.; Sheppard, A.E.; Pankhurst, L.; De Maio, N.; Moore, C.E.; Sebra, R.; Turner, P.; Anson, L.W.; Kasarskis, A.; Batty, E.M.; et al. Evolutionary History of the Global Emergence of the Escherichia coli Epidemic Clone ST131. mBio 2016, 7, e02162. [Google Scholar] [CrossRef] [Green Version]
- McNally, A.; Oren, Y.; Kelly, D.; Pascoe, B.; Dunn, S.; Sreecharan, T.; Vehkala, M.; Valimaki, N.; Prentice, M.B.; Ashour, A.; et al. Combined Analysis of Variation in Core, Accessory and Regulatory Genome Regions Provides a Super-Resolution View into the Evolution of Bacterial Populations. PLoS Genet. 2016, 12, e1006280. [Google Scholar] [CrossRef] [Green Version]
- Massot, M.; Daubie, A.S.; Clermont, O.; Jaureguy, F.; Couffignal, C.; Dahbi, G.; Mora, A.; Blanco, J.; Branger, C.; Mentre, F.; et al. Phylogenetic, virulence and antibiotic resistance characteristics of commensal strain populations of Escherichia coli from community subjects in the Paris area in 2010 and evolution over 30 years. Microbiology 2016, 162, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Flament-Simon, S.C.; Nicolas-Chanoine, M.H.; Garcia, V.; Duprilot, M.; Mayer, N.; Alonso, M.P.; Garcia-Menino, I.; Blanco, J.E.; Blanco, M.; Blanco, J. Clonal Structure, Virulence Factor-encoding Genes and Antibiotic Resistance of Escherichia coli, Causing Urinary Tract Infections and Other Extraintestinal Infections in Humans in Spain and France during 2016. Antibiotics 2020, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.J.; Wannemuehler, Y.; Doetkott, C.; Johnson, S.J.; Rosenberger, S.C.; Nolan, L.K. Identification of Minimal Predictors of Avian Pathogenic Escherichia coli Virulence for Use as a Rapid Diagnostic Tool. J. Clin. Microbiol. 2008, 46, 3987–3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.J.; Siek, K.E.; Johnson, S.J.; Nolan, L.K. DNA sequence of a ColV plasmid and prevalence of selected plasmid-encoded virulence genes among avian Escherichia coli strains. J. Bacteriol. 2006, 188, 745–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, Y.; Pitout, J.D.D.; Peirano, G.; DeVinney, R.; Noguchi, T.; Yamamoto, M.; Gomi, R.; Matsuda, T.; Nakano, S.; Nagao, M.; et al. Rapid Identification of Different Escherichia coli Sequence Type 131 Clades. Antimicrob. Agents Chemother. 2017, 61, e00179-17. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Johnston, B.D.; Gordon, D.M. Rapid and Specific Detection of the Escherichia coli Sequence Type 648 Complex within Phylogroup F. J. Clin. Microbiol. 2017, 55, 1116–1121. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Primer Sequence | Length (nt) | Tm (°C) | % GC | Amplicon Size (bp) |
|---|---|---|---|---|---|
| p-sfmH _For CTCCAGAAAACAATACCACACC | 22 | 50.5 | 45.5 | 996bp | |
| p-sfmH _Rev ATTCCACTCGCAATATAGCCAG | 22 | 52.9 | 45.5 | ||
| group4861-F TACAACAGTTTCCTCCAGCC | 20 | 49.2 | 50 | 474bp | |
| group4861-R CTTTAACCATGACATCCCAG | 20 | 46.9 | 45 | ||
| group 8607-F TCTTGTTCGTTGTTTTTATCGTCGG | 25 | 58.3 | 40% | 775bp | |
| group 8607-R TTTTATCACGCCAGGTGAAGAGTGA | 25 | 58.8 | 44% | ||
| group9754-F ACAAAATGGCTAAAAAGGAGTGATG | 25 | 54.9 | 36% | 256bp | |
| group9754-R CCACCGACTTGTAATTCCTCTAACA | 25 | 55.5 | 44% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, F.; Cheng, J.; Jiang, M.; Wang, Z.; Wen, Z.; Wang, M.; Ren, J.; Zhuge, X. Genomics Analysis to Identify Multiple Genetic Determinants That Drive the Global Transmission of the Pandemic ST95 Lineage of Extraintestinal Pathogenic Escherichia coli (ExPEC). Pathogens 2022, 11, 1489. https://doi.org/10.3390/pathogens11121489
Xia F, Cheng J, Jiang M, Wang Z, Wen Z, Wang M, Ren J, Zhuge X. Genomics Analysis to Identify Multiple Genetic Determinants That Drive the Global Transmission of the Pandemic ST95 Lineage of Extraintestinal Pathogenic Escherichia coli (ExPEC). Pathogens. 2022; 11(12):1489. https://doi.org/10.3390/pathogens11121489
Chicago/Turabian StyleXia, Fufang, Jinlong Cheng, Min Jiang, Zhongxing Wang, Zhe Wen, Min Wang, Jianluan Ren, and Xiangkai Zhuge. 2022. "Genomics Analysis to Identify Multiple Genetic Determinants That Drive the Global Transmission of the Pandemic ST95 Lineage of Extraintestinal Pathogenic Escherichia coli (ExPEC)" Pathogens 11, no. 12: 1489. https://doi.org/10.3390/pathogens11121489