
Identification of Genetic Variants Associated with Severe Myocardial Bridging through Whole-Exome Sequencing
 , ,
, ,
Abstract
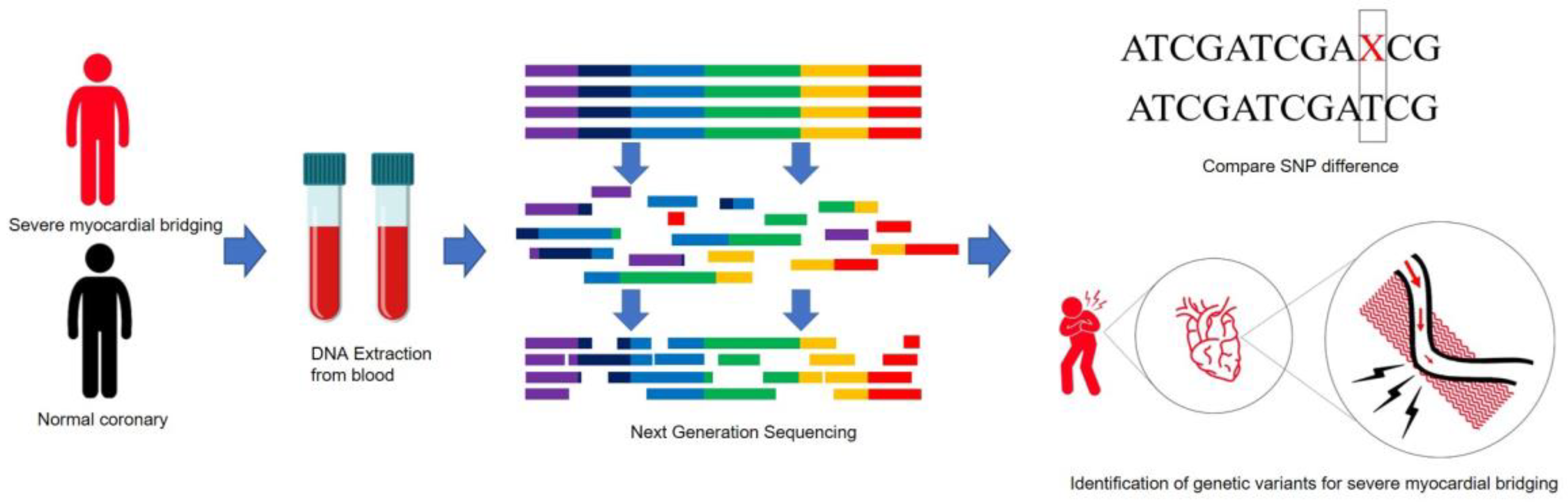
:1. Introduction
2. Results
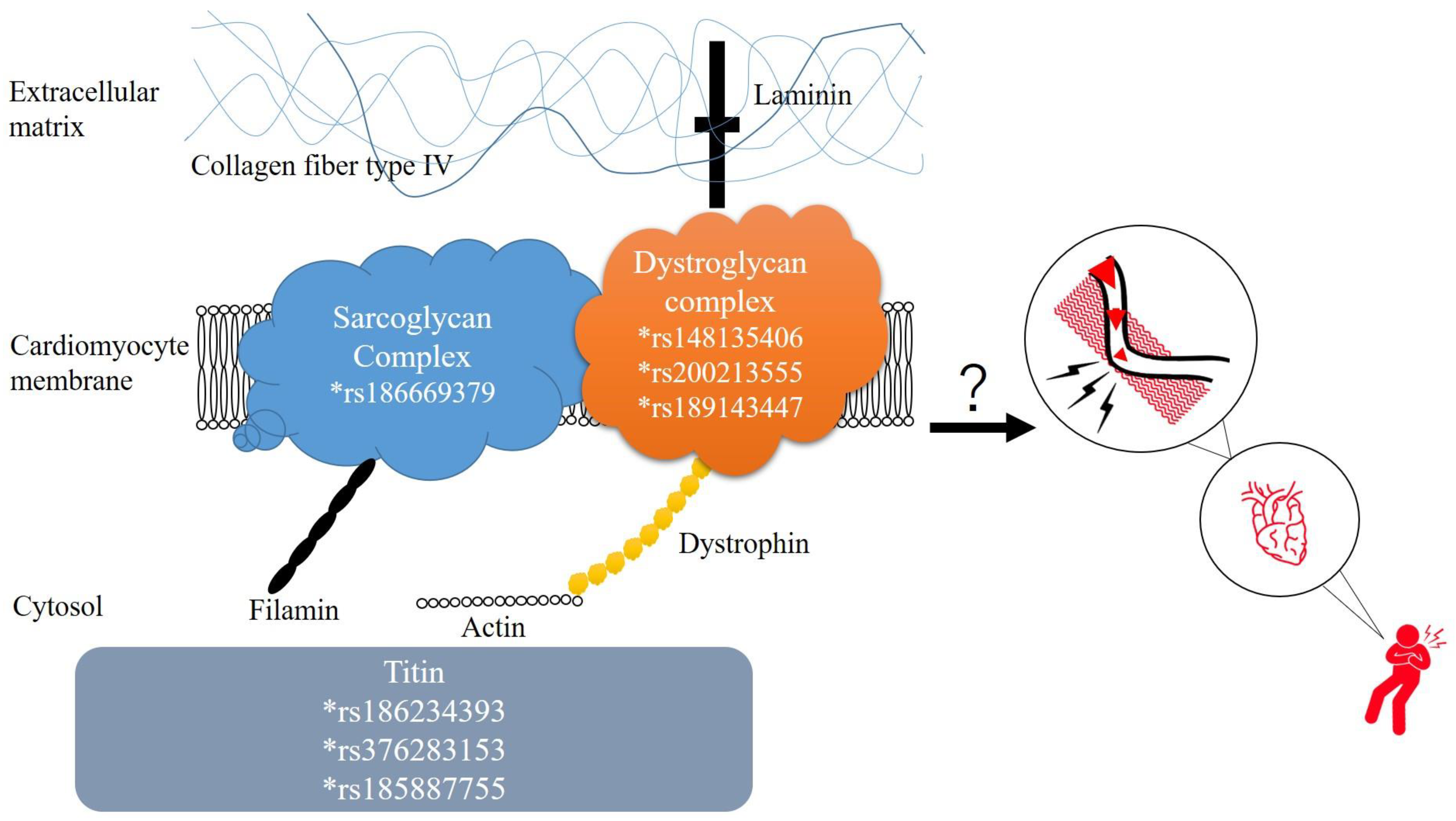
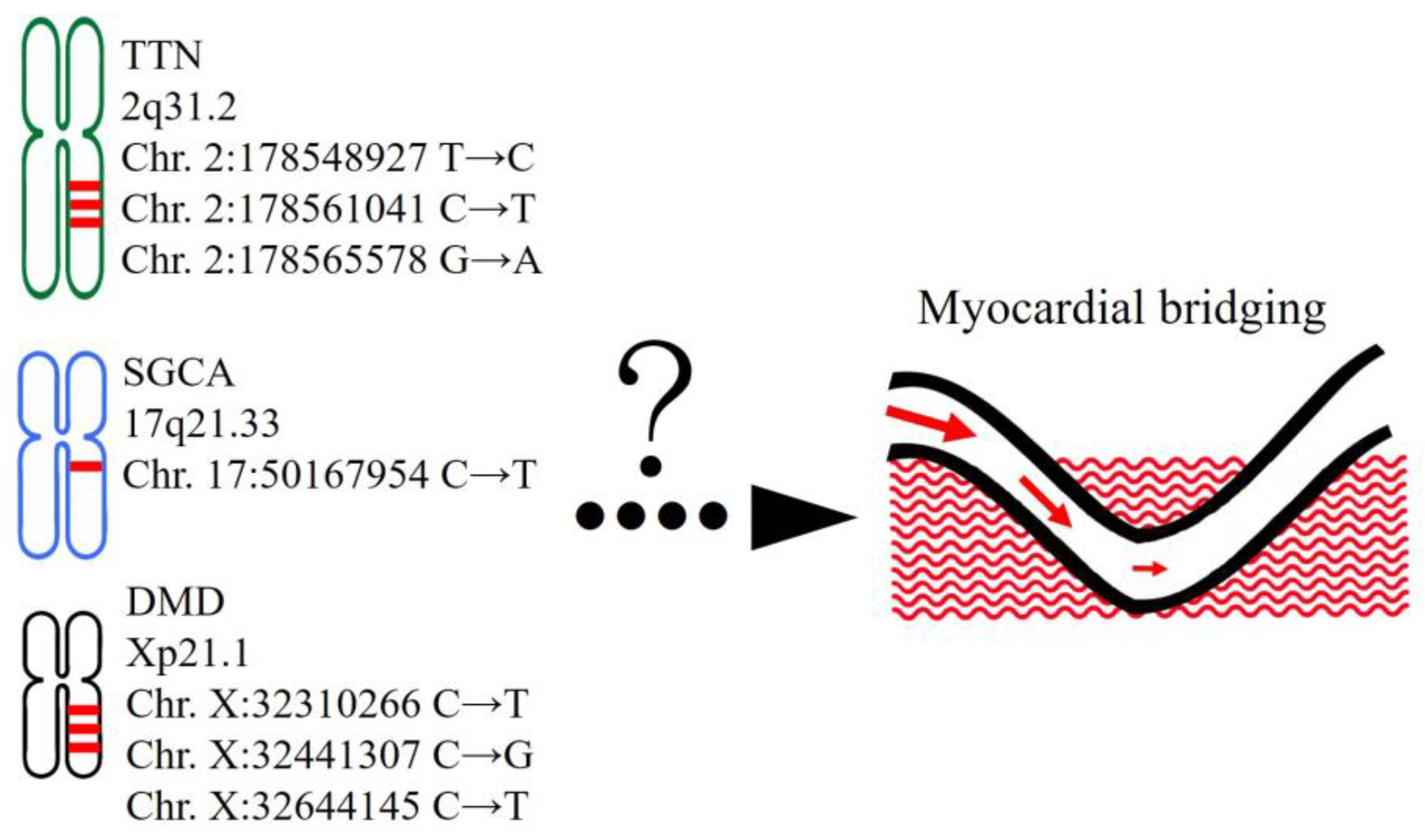
2.1. Variants Detected in Patients with SMB
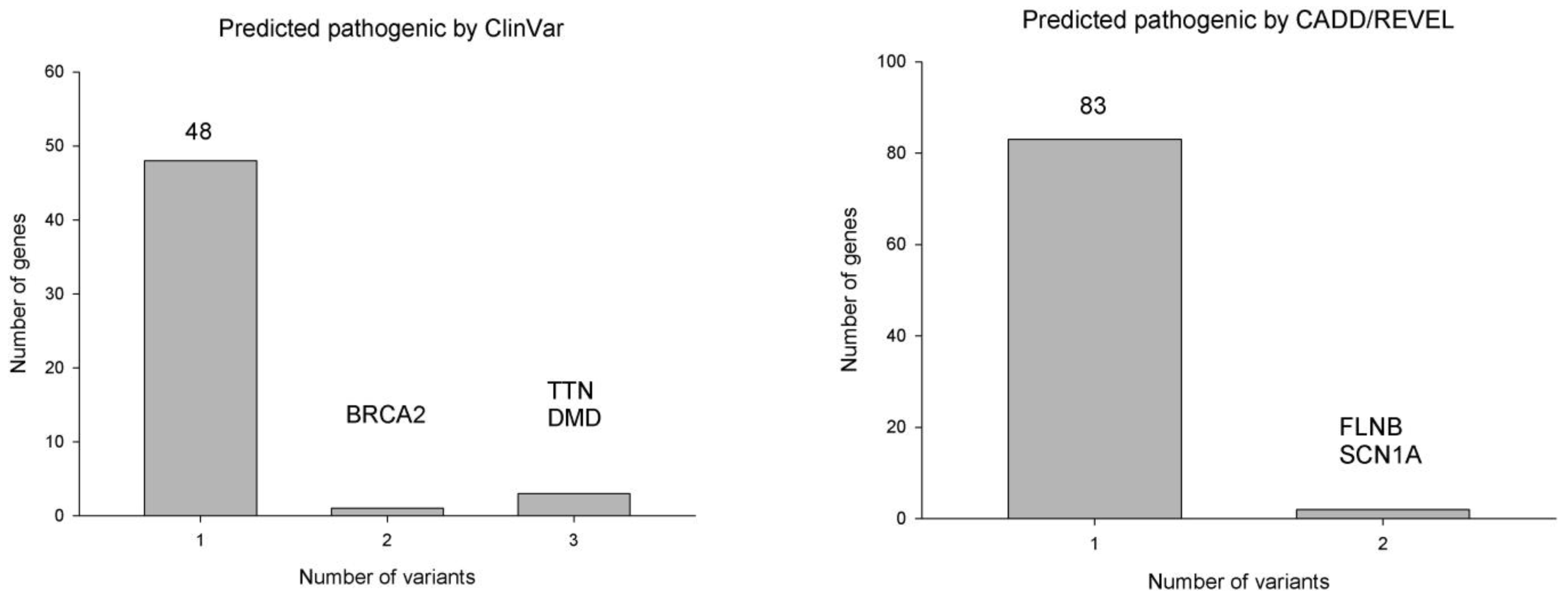
2.2. Genes with Multiple Rare Pathogenic Variants in Patients with SMB
2.3. Rare Pathogenic Variants Identified by Both ClinVar and CADD/REVEL
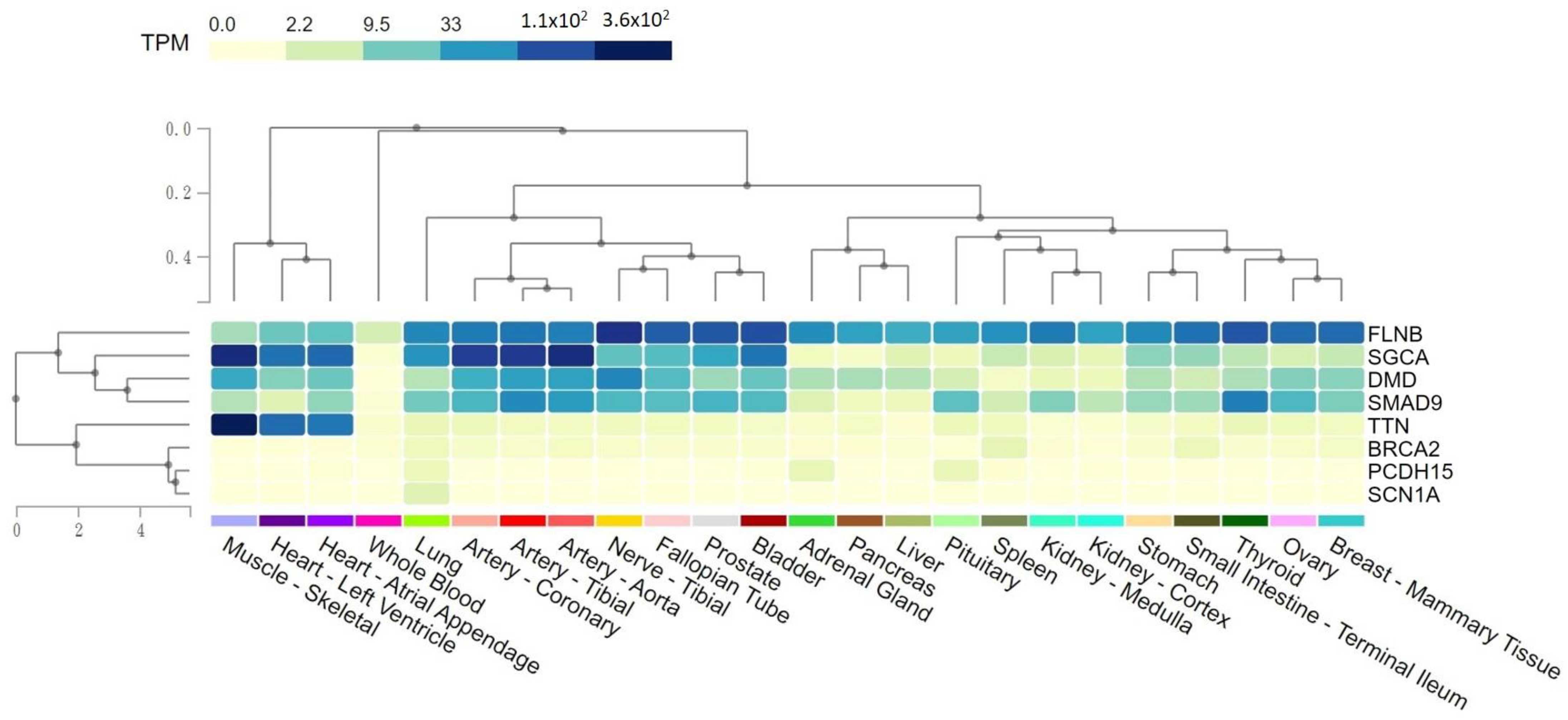
2.4. Tissue Expression Levels of Rare Variants Potentially Pathogenic for SMB
2.5. Functional Annotation of Rare Variants for SMB
3. Discussion
4. Materials and Methods
4.1. Participants
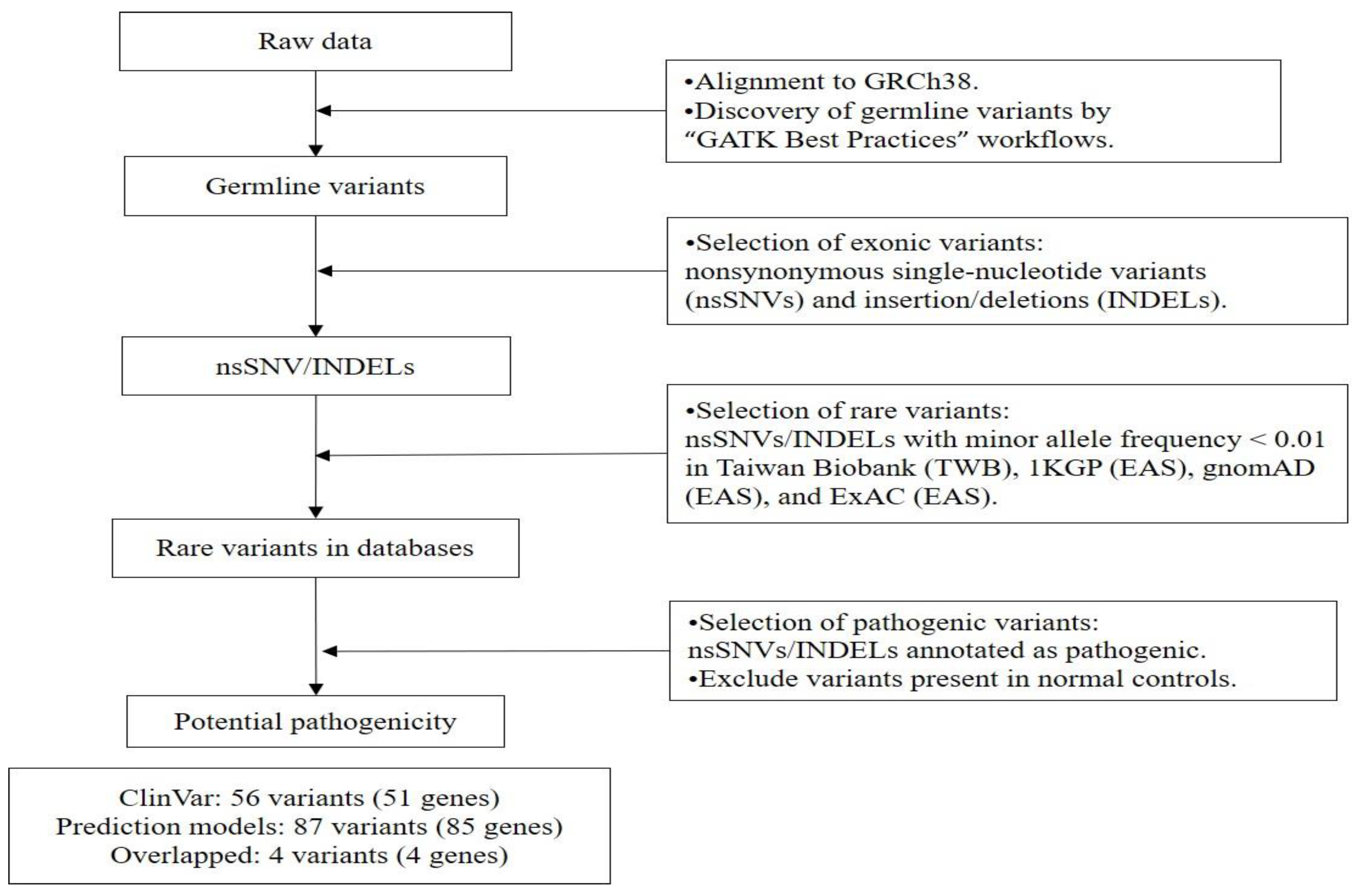
4.2. Sample Preparation, Whole-Exome Sequencing, and Bioinformatics Analysis
4.3. Pathogenicity Prediction
4.4. Functional Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corban, M.T.; Hung, O.Y.; Eshtehardi, P.; Rasoul-Arzrumly, E.; McDaniel, M.; Mekonnen, G.; Timmins, L.H.; Lutz, J.; Guyton, R.A.; Samady, H. Myocardial bridging: Contemporary understanding of pathophysiology with implications for diagnostic and therapeutic strategies. J. Am. Coll. Cardiol. 2014, 63, 2346–2355. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kawawa, Y.; Kohda, E.; Shimada, K.; Ishii, T. Significance of the anatomical properties of a myocardial bridge in coronary heart disease. Circ. J. Off. J. Jpn. Circ. Soc. 2011, 75, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Schnittger, I.; Nagy, A.; Zanley, E.; Xu, Y.; Song, Y.; Nieman, K.; Tremmel, J.A.; Dey, D.; Boyd, J.; et al. Relationship Between Coronary Atheroma, Epicardial Adipose Tissue Inflammation, and Adipocyte Differentiation Across the Human Myocardial Bridge. J. Am. Heart Assoc. 2021, 10, e021003. [Google Scholar] [CrossRef] [PubMed]
- Teragawa, H.; Oshita, C.; Ueda, T. The Myocardial Bridge: Potential Influences on the Coronary Artery Vasculature. Clin. Med. Insights Cardiol. 2019, 13, 1179546819846493. [Google Scholar] [CrossRef]
- Bruce, C.; Ubhi, N.; McKeegan, P.; Sanders, K. Systematic Review and Meta-Analysis of Cardiovascular Consequences of Myocardial Bridging in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2023, 188, 110–119. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, S.; Cui, H.; Tang, B.; Wang, S. Associations of myocardial bridging with adverse cardiac events: A meta-analysis of published observational cohort studies involving 4556 individuals. Ann. Transl. Med. 2020, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Sternheim, D.; Power, D.A.; Samtani, R.; Kini, A.; Fuster, V.; Sharma, S. Myocardial Bridging: Diagnosis, Functional Assessment, and Management: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 2196–2212. [Google Scholar] [CrossRef] [PubMed]
- Waterbury, T.M.; Tarantini, G.; Vogel, B.; Mehran, R.; Gersh, B.J.; Gulati, R. Non-atherosclerotic causes of acute coronary syndromes. Nat. Rev. Cardiol. 2020, 17, 229–241. [Google Scholar] [CrossRef]
- Kim, S.S.; Jeong, M.H.; Kim, H.K.; Kim, M.C.; Cho, K.H.; Lee, M.G.; Ko, J.S.; Park, K.H.; Sim, D.S.; Yoon, N.S.; et al. Long-term clinical course of patients with isolated myocardial bridge. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 538–543. [Google Scholar] [CrossRef]
- Okada, K.; Hibi, K.; Ogino, Y.; Maejima, N.; Kikuchi, S.; Kirigaya, H.; Kirigaya, J.; Sato, R.; Nakahashi, H.; Minamimoto, Y.; et al. Impact of Myocardial Bridge on Life-Threatening Ventricular Arrhythmia in Patients With Implantable Cardioverter Defibrillator. J. Am. Heart Assoc. 2020, 9, e017455. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Okada, K.; Kitahara, H.; Luikart, H.; Yock, P.G.; Yeung, A.C.; Schnittger, I.; Tremmel, J.A.; Fitzgerald, P.J.; Khush, K.K.; et al. Impact of myocardial bridging on coronary artery plaque formation and long-term mortality after heart transplantation. Int. J. Cardiol. 2023, 379, 24–32. [Google Scholar] [CrossRef]
- Guner, A.; Atmaca, S.; Balaban, I.; Turkmen, I.; Ceneli, D.; Turkvatan, A.; Oner, E.; Surgit, O.; Guler, A.; Uzun, F.; et al. Relationship between myocardial bridging and fatal ventricular arrhythmias in patients with hypertrophic cardiomyopathy: The HCM-MB study. Herz 2023, 48, 399–407. [Google Scholar] [CrossRef]
- Kato, K.; Kitahara, H.; Saito, Y.; Fujimoto, Y.; Sakai, Y.; Ishibashi, I.; Himi, T.; Kobayashi, Y. Impact of myocardial bridging on in-hospital outcome in patients with takotsubo syndrome. J. Cardiol. 2017, 70, 615–619. [Google Scholar] [CrossRef]
- Roberts, W.; Charles, S.M.; Ang, C.; Holda, M.K.; Walocha, J.; Lachman, N.; Tubbs, R.S.; Loukas, M. Myocardial bridges: A meta-analysis. Clin. Anat. 2021, 34, 685–709. [Google Scholar] [CrossRef]
- Hostiuc, S.; Negoi, I.; Rusu, M.C.; Hostiuc, M. Myocardial Bridging: A Meta-Analysis of Prevalence. J. Forensic Sci. 2018, 63, 1176–1185. [Google Scholar] [CrossRef]
- Rogers, I.S.; Tremmel, J.A.; Schnittger, I. Myocardial bridges: Overview of diagnosis and management. Congenit. Heart Dis. 2017, 12, 619–623. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.X.; Wu, Z.J.; Tang, J.; Su, X.X.; Chen, Y.J.; Jin, W. Association between APOE gene and myocardial bridge in Chinese Han population: A case-control study. Int. J. Clin. Exp. Pathol. 2016, 9, 4587–4594. [Google Scholar]
- Maruyama, K.; Kimura, S.; Ohashi, K.; Kuwano, Y. Connectin, an elastic protein of muscle. Identification of “titin” with connectin. J. Biochem. 1981, 89, 701–709. [Google Scholar] [CrossRef]
- Eldemire, R.; Tharp, C.A.; Taylor, M.R.G.; Sbaizero, O.; Mestroni, L. The Sarcomeric Spring Protein Titin: Biophysical Properties, Molecular Mechanisms, and Genetic Mutations Associated with Heart Failure and Cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 121. [Google Scholar] [CrossRef] [PubMed]
- Linke, W.A.; Hamdani, N. Gigantic business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef]
- Trinick, J.; Knight, P.; Whiting, A. Purification and properties of native titin. J. Mol. Biol. 1984, 180, 331–356. [Google Scholar] [CrossRef]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Begay, R.L.; Graw, S.; Sinagra, G.; Merlo, M.; Slavov, D.; Gowan, K.; Jones, K.L.; Barbati, G.; Spezzacatene, A.; Brun, F.; et al. Role of Titin Missense Variants in Dilated Cardiomyopathy. J. Am. Heart Assoc. 2015, 4, e002645. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. Genet. Causes Card. Dis. 2019, 7, 45–91. [Google Scholar] [CrossRef]
- Brown, S.S.; Malinoff, H.L.; Wicha, M.S. Connectin: Cell surface protein that binds both laminin and actin. Proc. Natl. Acad. Sci. USA 1983, 80, 5927–5930. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, D.; Khan, S.; Khera, R.; Bhatt, A.; Garg, S.; Grodin, J.L.; Morlend, R.; Araj, F.G.; Amin, A.A.; Thibodeau, J.T.; et al. Predictors of Death in Adults With Duchenne Muscular Dystrophy-Associated Cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e006340. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Cardona, A.; He, X.; Halnon, N.; Markham, L.; Soslow, J.H.; Puchalski, M.D.; Auerbach, S.R.; et al. Stabilization of Early Duchenne Cardiomyopathy With Aldosterone Inhibition: Results of the Multicenter AIDMD Trial. J. Am. Heart Assoc. 2019, 8, e013501. [Google Scholar] [CrossRef]
- Mirouse, V. Evolution and developmental functions of the dystrophin-associated protein complex: Beyond the idea of a muscle-specific cell adhesion complex. Front. Cell Dev. Biol. 2023, 11, 1182524. [Google Scholar] [CrossRef]
- Ozawa, E.; Mizuno, Y.; Hagiwara, Y.; Sasaoka, T.; Yoshida, M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005, 32, 563–576. [Google Scholar] [CrossRef]
- Alonso-Perez, J.; Gonzalez-Quereda, L.; Bello, L.; Guglieri, M.; Straub, V.; Gallano, P.; Semplicini, C.; Pegoraro, E.; Zangaro, V.; Nascimento, A.; et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain 2020, 143, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Vainzof, M.; Souza, L.S.; Gurgel-Giannetti, J.; Zatz, M. Sarcoglycanopathies: An update. Neuromuscul. Disord. 2021, 31, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Duclos, F.; Apostol, B.; Straub, V.; Lee, J.C.; Allamand, V.; Venzke, D.P.; Sunada, Y.; Moomaw, C.R.; Leveille, C.J.; et al. Characterization of delta-sarcoglycan, a novel component of the oligomeric sarcoglycan complex involved in limb-girdle muscular dystrophy. J. Biol. Chem. 1996, 271, 32321–32329. [Google Scholar] [CrossRef]
- Fayssoil, A.; Renault, G.; Guerchet, N.; Marchiol-Fournigault, C.; Fougerousse, F.; Richard, I. Cardiac Characterization of sgca-Null Mice Using High Resolution Echocardiography. Neurol. Int. 2013, 5, e22. [Google Scholar] [CrossRef] [PubMed]
- Coral-Vazquez, R.; Cohn, R.D.; Moore, S.A.; Hill, J.A.; Weiss, R.M.; Davisson, R.L.; Straub, V.; Barresi, R.; Bansal, D.; Hrstka, R.F.; et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: A novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999, 98, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Des Rosiers, C.; Petrof, B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.Y.; Yang, J.H.; Yeh, E.C.; Tsai, M.F.; Kao, H.J.; Lo, C.Z.; Chang, L.P.; Lin, W.J.; Hsieh, F.J.; Belsare, S.; et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom. Med. 2021, 6, 10. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Gene Ontology, C. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr. | Position | Ref. | Alt. | Gene | Type | rs Number | ClinVar | CADD | REVEL | Number of Alt. Alleles in Patients with SMB | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||||||||
| Pathogenic Variants Predicted by ClinVar | |||||||||||||||||
| 2 | 178548927 | T | C | TTN | nsSNV | rs186234393 | CIP | 13.78 | 0.496 | 1 | - | - | - | - | - | - | - |
| 2 | 178561041 | C | T | TTN | nsSNV | rs376283153 | CIP | 18.93 | 0.14 | 1 | - | - | - | - | - | - | - |
| 2 | 178565578 | G | A | TTN | nsSNV | rs185887755 | CIP | 20.8 | 0.533 | - | - | - | - | - | - | 1 | 1 |
| 13 | 32338731 | A | G | BRCA2 | nsSNV | rs117187202 | CIP | 0.395 | 0.146 | - | - | - | - | - | - | 1 | - |
| 13 | 32340191 | T | C | BRCA2 | nsSNV | rs80358811 | CIP | 0.002 | 0.14 | - | - | 1 | - | - | - | - | - |
| X | 32310266 | C | T | DMD | nsSNV | rs148135406 | CIP | 17.97 | 0.147 | - | - | - | - | - | - | 1 | - |
| X | 32441307 | C | G | DMD | nsSNV | rs200213555 | CIP | 25.8 | 0.312 | - | - | - | 1 | - | - | - | - |
| X | 32644145 | C | T | DMD | nsSNV | rs189143447 | CIP | 27.4 | 0.294 | - | - | 1 | - | - | - | - | - |
| Pathogenic Variants Predicted by CADD/REVEL | |||||||||||||||||
| 2 | 165994164 | C | T | SCN1A | nsSNV | rs121918808 | LB | 32 | 0.817 | - | - | - | - | - | - | - | 1 |
| 2 | 166041286 | A | G | SCN1A | nsSNV | rs773695263 | CIP | 25.9 | 0.884 | - | - | - | - | 1 | - | - | - |
| 3 | 58008647 | A | T | FLNB | nsSNV | N/A | N/A | 32 | 0.953 | - | - | 1 | - | - | - | - | - |
| 3 | 58154853 | C | T | FLNB | nsSNV | rs369477886 | N/A | 34 | 0.876 | - | - | - | - | 1 | - | - | - |
| Chr. | Position | Ref. | Alt. | Gene | Type | rs Number | SMB No. |
|---|---|---|---|---|---|---|---|
| 2 | 166041286 | A | G | SCN1A | nsSNV | rs773695263 | 5 |
| 10 | 53857257 | C | T | PCDH15 | nsSNV | rs201137087 | 4 |
| 13 | 36879563 | T | C | SMAD9 | nsSNV | rs397514715 | 6 |
| 17 | 50167954 | C | T | SGCA | nsSNV | rs186669379 | 3 |
| Functional Annotation | Reference Genes in Category | SMB Genes in Category | p Value | FDR |
|---|---|---|---|---|
| Knockout mouse phenotype | ||||
| Abnormal soleus morphology | 21 | 4 | 3.84 × 10−5 | 2.50 × 10−2 |
| Impaired skeletal muscle contractility | 38 | 6 | 1.25 × 10−6 | 8.18 × 10−3 |
| Absent startle reflex | 39 | 5 | 2.91 × 10−5 | 2.22 × 10−2 |
| Decreased skeletal muscle mass | 107 | 8 | 6.75 × 10−6 | 1.85 × 10−2 |
| Abnormal skeletal muscle mass | 121 | 8 | 1.67 × 10−5 | 2.04 × 10−2 |
| Abnormal muscle fiber morphology | 322 | 13 | 8.84 × 10−6 | 1.85 × 10−2 |
| Increased or absent threshold for auditory brainstem response | 310 | 12 | 3.06 × 10−5 | 2.22 × 10−2 |
| Abnormal skeletal muscle morphology | 381 | 14 | 1.14 × 10−5 | 1.85 × 10−2 |
| Abnormal cardiovascular system physiology | 1421 | 29 | 2.56 × 10−5 | 2.22 × 10−2 |
| Abnormal cardiovascular system morphology | 1794 | 34 | 1.88 × 10−5 | 2.04 × 10−2 |
| KEGG pathway | ||||
| Arrhythmogenic right ventricular cardiomyopathy | 72 | 7 | 3.49 × 10−6 | 1.14 × 10−3 |
| GO term categories | ||||
| Detection of mechanical stimulus | 43 | 6 | 6.03 × 10−7 | 2.74 × 10−3 |
| Muscle contraction | 339 | 14 | 1.52 × 10−7 | 1.38 × 10−3 |
| Muscle system process | 423 | 14 | 2.15 × 10−6 | 3.33 × 10−3 |
| Monovalent inorganic cation transport | 513 | 15 | 4.17 × 10−6 | 4.21 × 10−3 |
| Inorganic cation transmembrane transport | 722 | 19 | 9.42 × 10−7 | 2.85 × 10−3 |
| Cation transmembrane transport | 810 | 20 | 1.27 × 10−6 | 2.88 × 10−3 |
| Metal ion transport | 841 | 20 | 2.25 × 10−6 | 3.33 × 10−3 |
| Inorganic ion transmembrane transport | 808 | 19 | 4.91 × 10−6 | 4.47 × 10−3 |
| Cation transport | 1111 | 23 | 3.73 × 10−6 | 4.22 × 10−3 |
| Ion transport | 1608 | 29 | 2.56 × 10−6 | 3.33 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-L.; Ting, J.; Lin, M.-R.; Chang, W.-C.; Shih, C.-M. Identification of Genetic Variants Associated with Severe Myocardial Bridging through Whole-Exome Sequencing. J. Pers. Med. 2023, 13, 1509. https://doi.org/10.3390/jpm13101509
Yang T-L, Ting J, Lin M-R, Chang W-C, Shih C-M. Identification of Genetic Variants Associated with Severe Myocardial Bridging through Whole-Exome Sequencing. Journal of Personalized Medicine. 2023; 13(10):1509. https://doi.org/10.3390/jpm13101509
Chicago/Turabian StyleYang, Tsung-Lin, Jafit Ting, Min-Rou Lin, Wei-Chiao Chang, and Chun-Ming Shih. 2023. "Identification of Genetic Variants Associated with Severe Myocardial Bridging through Whole-Exome Sequencing" Journal of Personalized Medicine 13, no. 10: 1509. https://doi.org/10.3390/jpm13101509