
Pb2+ Uptake by Magnesite: The Competition between Thermodynamic Driving Force and Reaction Kinetics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thermodynamic Background
3. Materials and Methods
3.1. Materials
3.2. Batch Interaction
3.3. Precipitations
3.4. In Situ AFM
3.5. Solid Characterization
4. Results
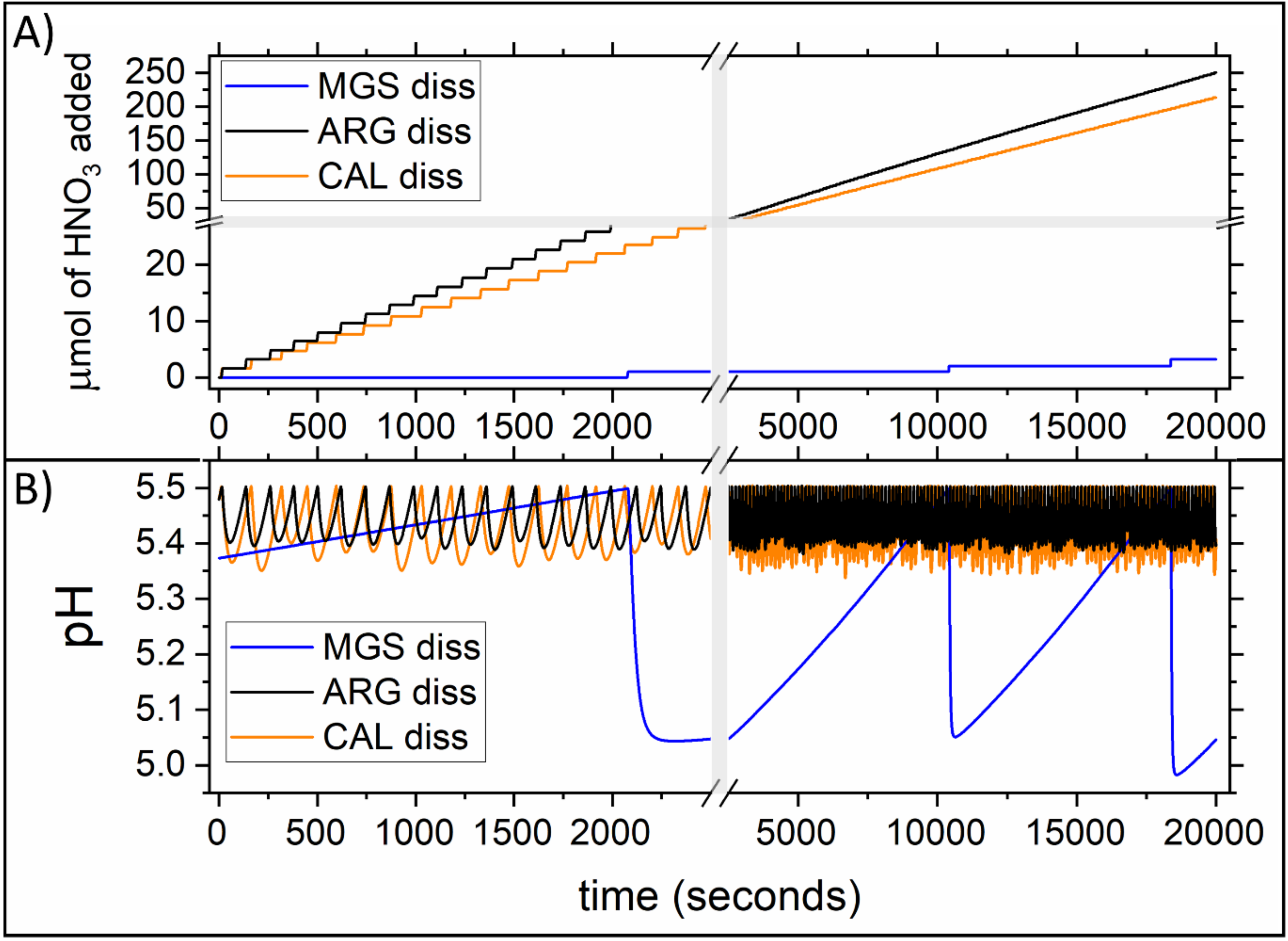
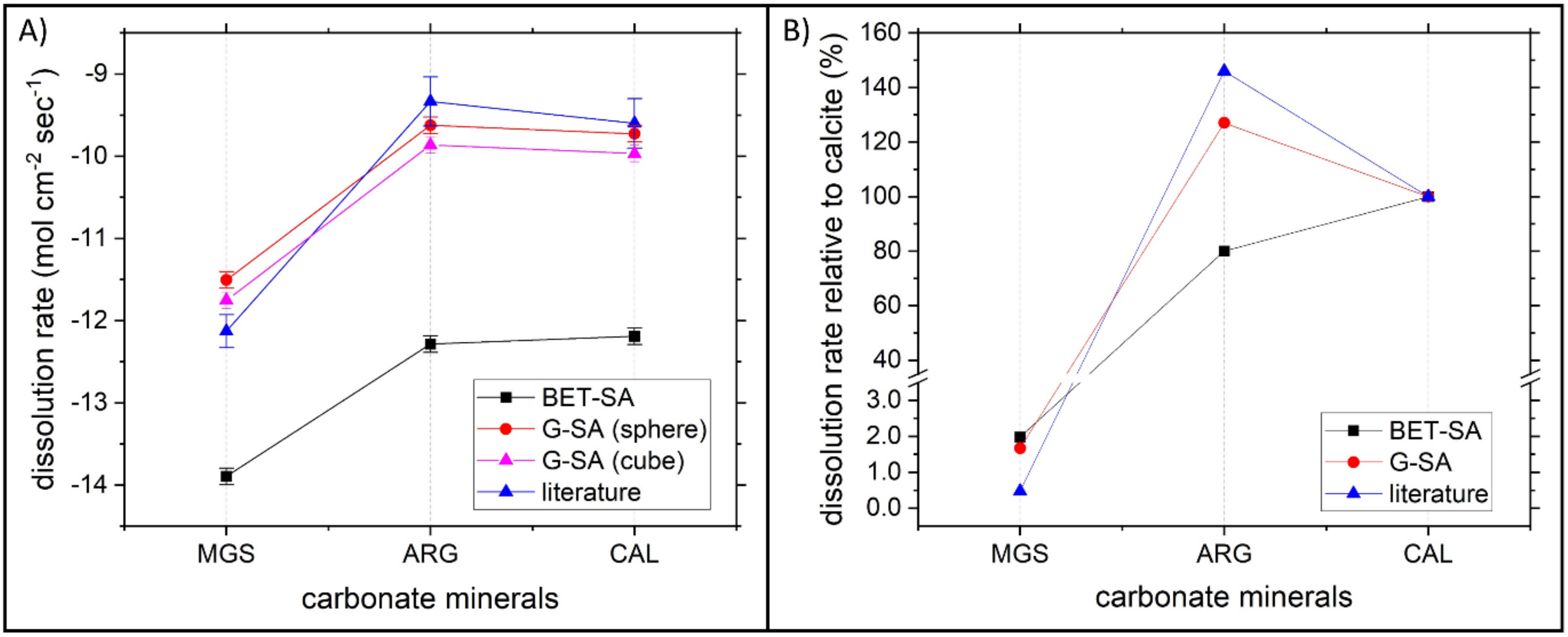
4.1. Comparison among the Dissolution Rate of Magnesite, Aragonite and Calcite
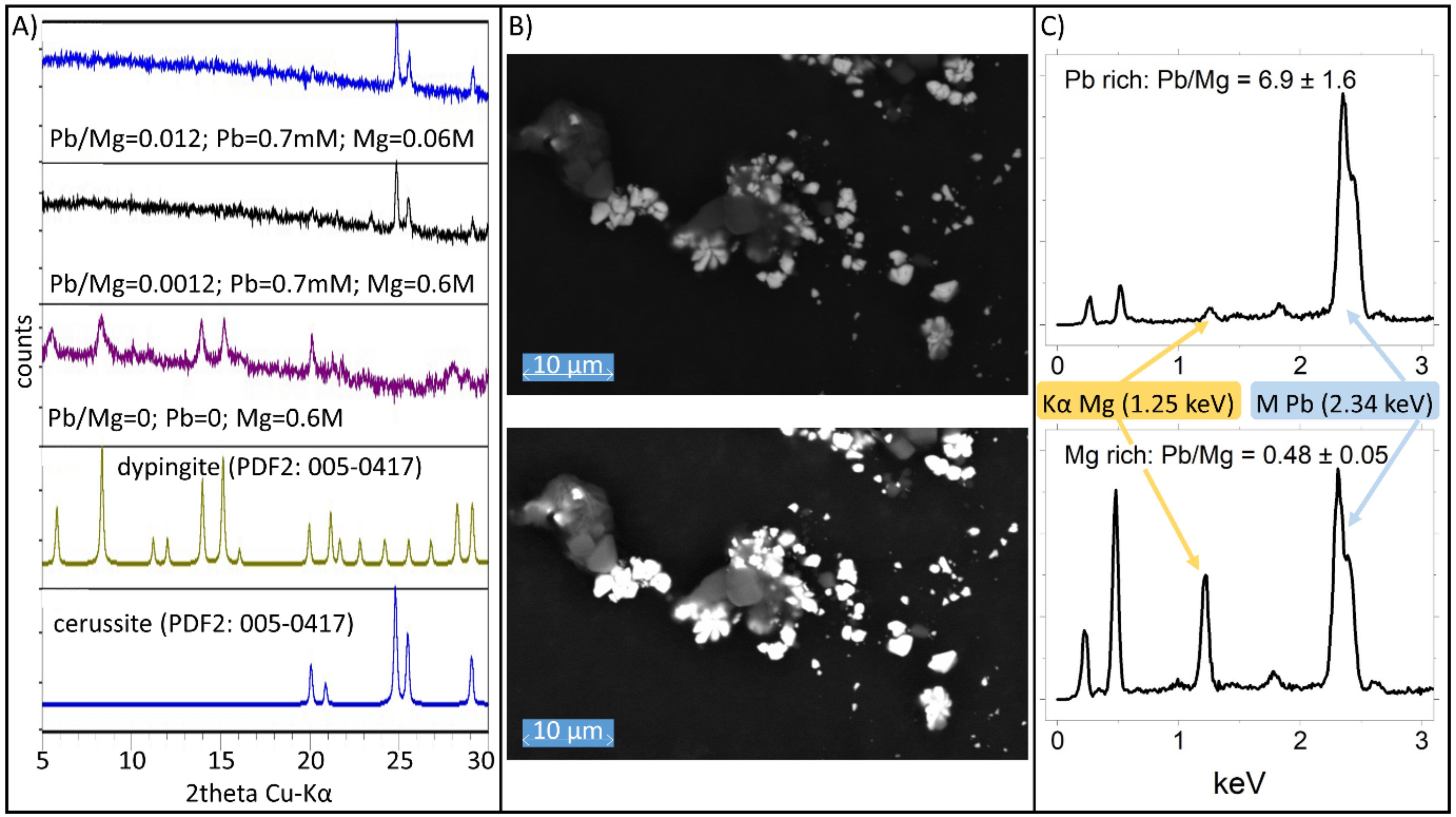
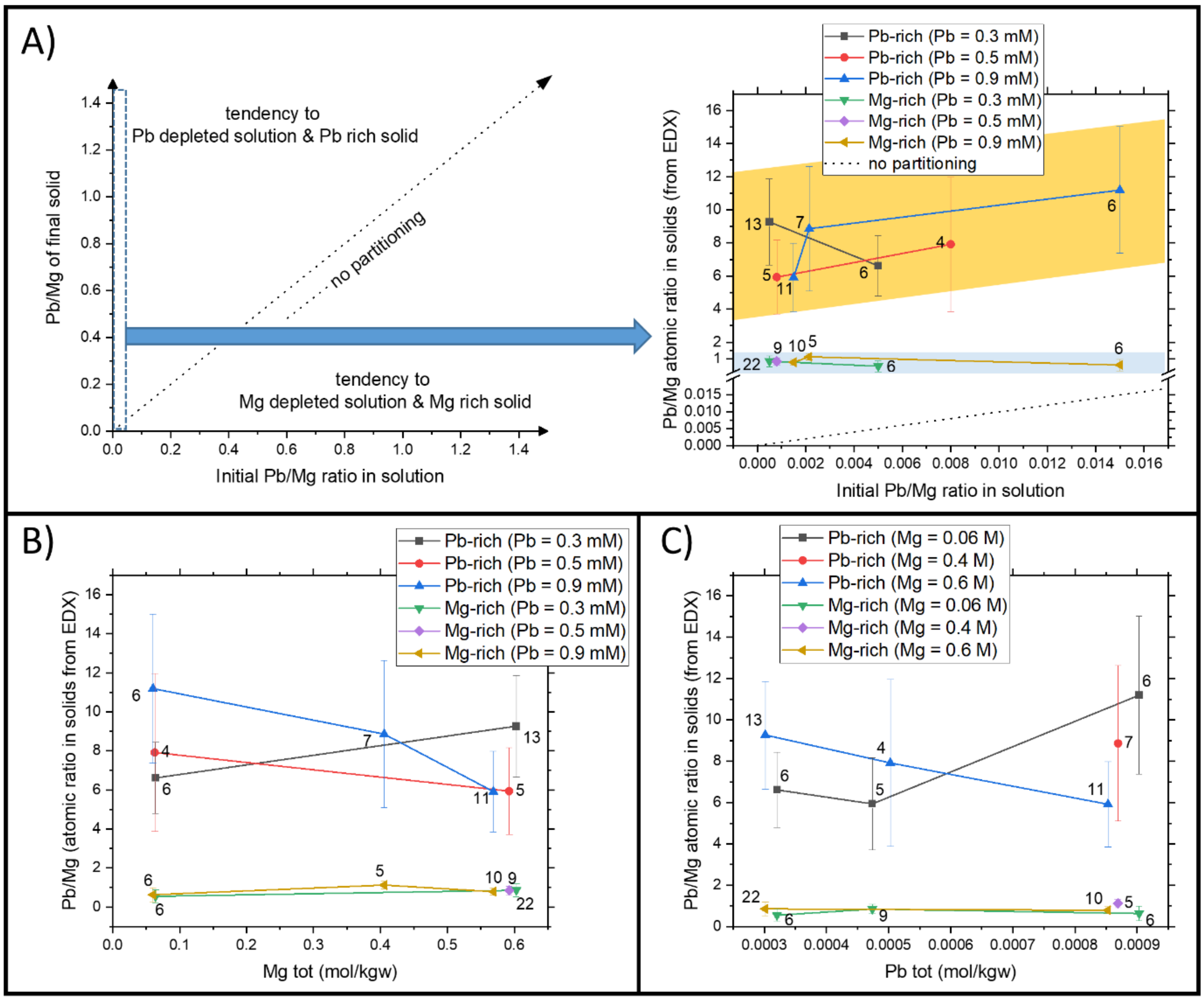
4.2. The Precipitation in Far from Equilibrium Conditions for the System Mg-PbII-CO2-H2O
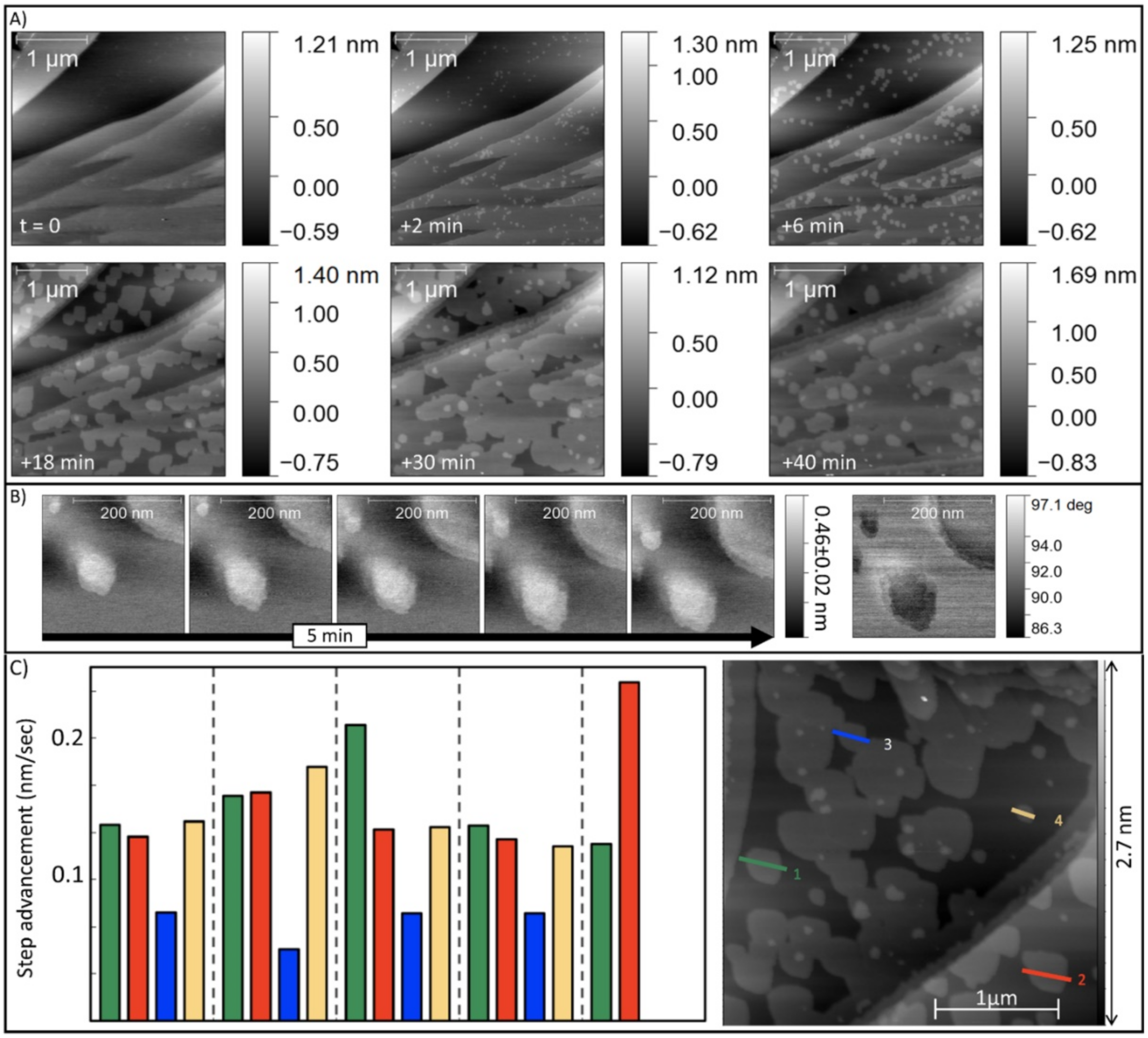
4.3. The Growth of {10.4} Surfaces of Magnesite in Presence and Absence of Pb2+
5. Discussion
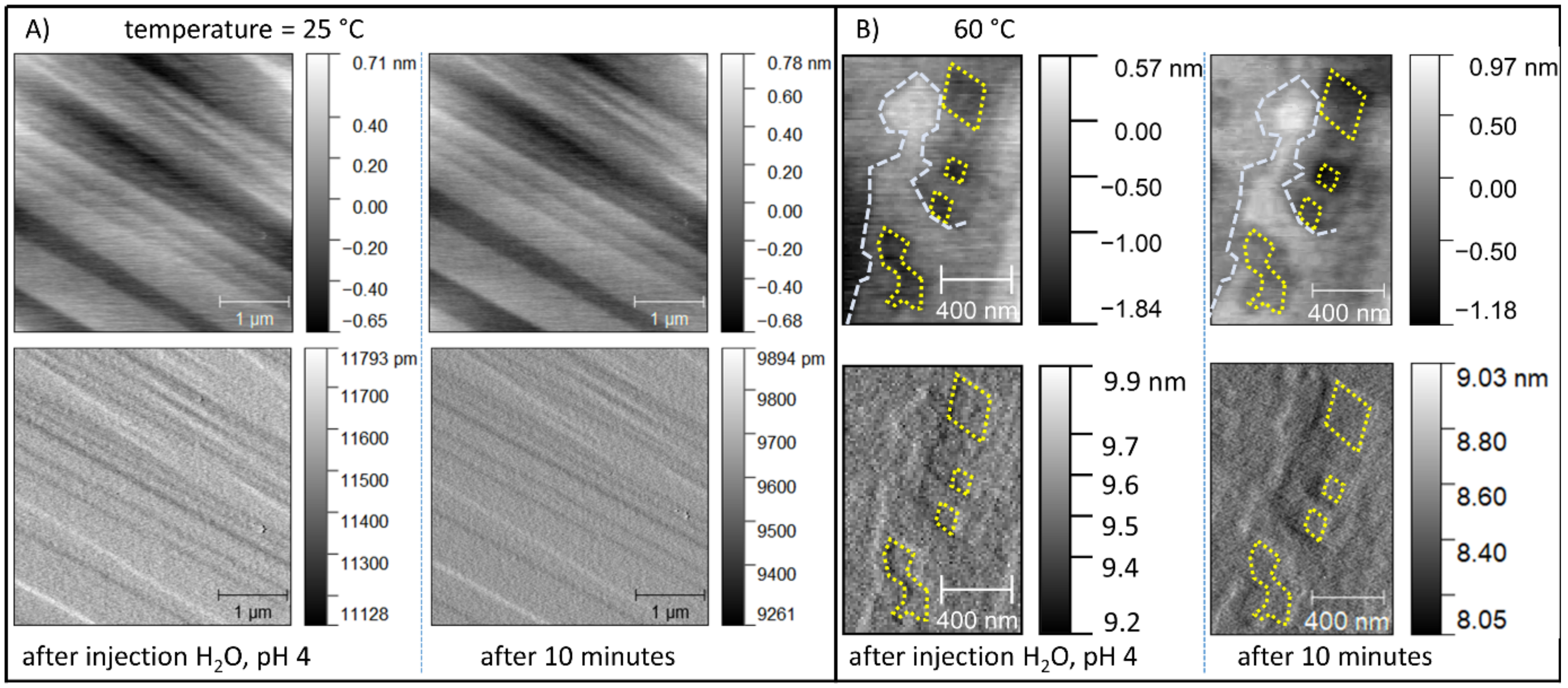
5.1. Thermodynamic vs. Kinetic Control on Magnesite Dissolution
5.2. The Consequences of the Sluggish Kinetics of Magnesite Aiming at Pb2+(aq) Uptake
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godelitsas, A.; Astilleros, J.M.; Hallam, K.; Harissopoulos, S.; Putnis, A. Interaction of Calcium Carbonates with Lead in Aqueous Solutions. Environ. Sci. Technol. 2003, 37, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Godelitsas, A.; Astilleros, J.M. Dissolution, sorption/(re)precipitation, formation of solid solutions and crystal growth phenomena on mineral surfaces: Implications for the removal of toxic metals from the environment. Eur. Mineral. Union Notes Mineral. 2010, 10, 289–324. [Google Scholar] [CrossRef]
- Prieto, M.; Astilleros, J.M.; Fernández-Díaz, L. Environmental Remediation by Crystallization of Solid Solutions. Elements 2013, 9, 195–201. [Google Scholar] [CrossRef]
- González-Núñez, R.; Rigol, A.; Vidal, M. Assessing the efficacy over time of the addition of industrial by-products to remediate contaminated soils at a pilot-plant scale. Environ. Monit. Assess. 2017, 189, 155. [Google Scholar] [CrossRef]
- Davis, A.D.; Webb, C.J.; Sorensen, J.L.; Dixon, D.J.; Hudson, R. Geochemical thermodynamics of cadmium removal from water with limestone. Environ. Earth Sci. 2018, 77, 1–5. [Google Scholar] [CrossRef]
- Füllenbach, L.C.; Perez, J.P.H.; Freeman, H.M.; Thomas, A.N.; Mayanna, S.; Parker, J.E.; Göttlicher, J.; Steininger, R.; Radnik, J.; Benning, L.G.; et al. Nanoanalytical Identification of Siderite Dissolution-Coupled Pb Removal Mechanisms from Oxic and Anoxic Aqueous Solutions. Acs Earth Space Chem. 2020, 4, 1966–1977. [Google Scholar] [CrossRef]
- Roza Llera, A.; Jimenez, A.; Fernández-Díaz, L. Removal of Pb from Water: The Effectiveness of Gypsum and Calcite Mixtures. Minerals 2021, 11, 66. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; Wiley: Hoboken, NJ, USA, 1996; p. 1040. [Google Scholar]
- Di Lorenzo, F.; Ruiz-Agudo, C.; Churakov, S.V. The key effects of polymorphism during PbII uptake by calcite and aragonite. CrystEngComm 2019, 21, 6145–6155. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Cametti, G.; Vanhecke, D.; Churakov, S.V. The Role of Interfaces in Controlling Pb2+ removal by calcium carbonate minerals. Cryst. Growth Des. 2020, 20, 6157–6169. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Mackenzie, F.T. The dolomite problem: Control of precipitation kinetics by temperature and saturation state. Am. J. Sci. 1999, 2, 257–288. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, F.; Rodríguez-Galán, R.M.; Prieto, M. Kinetics of the solvent-mediated transformation of hydromagnesite into magnesite at different temperatures. Miner. Mag. 2014, 78, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, F.; Prieto, M. Dissolution–Recrystallization of (Mg,Fe)CO3 during Hydrothermal Cycles: FeII/FeIII Conundrums in the Carbonation of Ferromagnesian Minerals. Cryst. Growth Des. 2017, 17, 4170–4182. [Google Scholar] [CrossRef]
- Xu, J.; Yan, C.; Zhang, F.; Konishi, H.; Xu, H.; Teng, H.H. Testing the cation-hydration effect on the crystallization of Ca–Mg–CO3 systems. Proc. Natl. Acad. Sci. USA 2013, 110, 17750–17755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Xu, J.; Teng, H.H. Evolution of calcite growth morphology in the presence of magnesium: Implications for the dolomite problem. Geochim. Cosmochim. Acta 2016, 172, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Di Tommaso, D.; De Leeuw, N.H. Structure and dynamics of the hydrated magnesium ion and of the solvated magnesium carbonates: Insights from first principles simulations. Phys. Chem. Chem. Phys. 2009, 12, 894–901. [Google Scholar] [CrossRef]
- Hamm, L.M.; Wallace, A.F.; Dove, P.M. Molecular Dynamics of Ion Hydration in the Presence of Small Carboxylated Molecules and Implications for Calcification. J. Phys. Chem. B 2010, 114, 10488–10495. [Google Scholar] [CrossRef]
- Koskamp, J.A.; Ruiz-Hernandez, S.E.; Di Tommaso, D.; Elena, A.M.; De Leeuw, N.H.; Wolthers, M. Reconsidering Calcium Dehydration as the Rate-Determining Step in Calcium Mineral Growth. J. Phys. Chem. C 2019, 123, 26895–26903. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Alvarez-Lloret, P.; Chass, G.A.; Di Tommaso, D. Interatomic potentials of Mg ions in aqueous solutions: Structure and dehydration kinetics. Eur. J. Miner. 2019, 31, 275–287. [Google Scholar] [CrossRef]
- Lindner, M.; Saldi, G.D.; Carrocci, S.; Bénézeth, P.; Schott, J.; Jordan, G. On the growth of anhydrous Mg-bearing carbonates—Implications from norsethite growth kinetics. Geochim. Cosmochim. Acta 2018, 238, 424–437. [Google Scholar] [CrossRef]
- Lindner, M.; Saldi, G.D.; Jordan, G.; Schott, J. On the effect of aqueous barium on magnesite growth—A new route for the precipitation of the ordered anhydrous Mg-bearing double carbonate norsethite. Chem. Geol. 2017, 460, 93–105. [Google Scholar] [CrossRef]
- Lindner, M.; Jordan, G. On the growth of witherite and its replacement by the Mg-bearing double carbonate norsethite: Implications for the dolomite problem. Am. Miner. 2018, 103, 252–259. [Google Scholar] [CrossRef]
- Pimentel, C.; Pina, C.M.; Martinez, C.M.P. The formation of the dolomite-analogue norsethite: Reaction pathway and cation ordering. Geochim. Cosmochim. Acta 2014, 142, 217–223. [Google Scholar] [CrossRef]
- Pimentel, C.; Pina, C.M. Reaction pathways towards the formation of dolomite-analogues at ambient conditions. Geochim. Cosmochim. Acta 2016, 178, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, W. Transformation of amorphous precursor to crystalline carbonate: Insights from Mg isotopes in the dolomite-analogue mineral norsethite [BaMg(CO3)2]. Geochim. Cosmochim. Acta 2020, 272, 1–20. [Google Scholar] [CrossRef]
- Bersani, D.; Campani, E.; Casoli, A.; Lottici, P.; Marino, I.-G. Spectroscopic study of the degradation products in the holy water fonts in Santa Maria della Steccata Church in Parma (Italy). Anal. Chim. Acta 2008, 610, 74–79. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3−A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Book 6, Section A; US Geological Survey: Reston, VA, USA, 2013; Chapter 43.
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Cent. Eur. J. Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Morse, J.W.; Arvidson, R.S.; Lüttge, A. Calcium Carbonate Formation and Dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef]
- Naviaux, J.D.; Subhas, A.V.; Rollins, N.E.; Dong, S.; Berelson, W.M.; Adkins, J.F. Temperature dependence of calcite dissolution kinetics in seawater. Geochim. Cosmochim. Acta 2019, 246, 363–384. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Berelson, W.M.; Rollins, N.E.; Subhas, A.V.; Naviaux, J.D.; Celestian, A.J.; Liu, X.; Turaga, N.; Kemnitz, N.J.; Byrne, R.H.; et al. Aragonite dissolution kinetics and calcite/aragonite ratios in sinking and suspended particles in the North Pacific. Earth Planet. Sci. Lett. 2019, 515, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pokrovsky, O.S.; Golubev, S.V.; Schott, J.; Castillo, A. Calcite, dolomite and magnesite dissolution kinetics in aqueous solutions at acid to circumneutral pH, 25 to 150 °C and 1 to 55 atm pCO2: New constraints on CO2 sequestration in sedimentary basins. Chem. Geol. 2009, 265, 20–32. [Google Scholar] [CrossRef]
- Gautier, J.-M.; Oelkers, E.H.; Schott, J. Are quartz dissolution rates proportional to B.E.T. surface areas? Geochim. Cosmochim. Acta 2001, 65, 1059–1070. [Google Scholar] [CrossRef]
- Tester, J.W.; Worley, W.G.; Robinson, B.A.; Grigsby, C.O.; Feerer, J.L. Correlating quartz dissolution kinetics in pure water from 25 to 625 °C. Geochim. Cosmochim. Acta 1994, 58, 2407–2420. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Suess, H.E.; Urey, H.C. Abundances of the Elements. Rev. Mod. Phys. 1956, 28, 53–74. [Google Scholar] [CrossRef]
- Putnis, A. Effects of kinetics and mechanisms of crystal growth on ion-partitioning in solid solution–aqueous solution (SS–AS) systems. Eur. Mineral. Union Notes Mineral. 2010, 10, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Renard, F.; Røyne, A.; Putnis, C.V. Timescales of interface-coupled dissolution-precipitation reactions on carbonates. Geosci. Front. 2019, 10, 17–27. [Google Scholar] [CrossRef]
- García, R.; Pérez, R. Dynamic atomic force microscopy methods. Surf. Sci. Rep. 2002, 47, 197–301. [Google Scholar] [CrossRef]
- Callagon, E.B.R.; Lee, S.S.; Eng, P.J.; Laanait, N.; Sturchio, N.C.; Nagy, K.L.; Fenter, P. Heteroepitaxial growth of cadmium carbonate at dolomite and calcite surfaces: Mechanisms and rates. Geochim. Cosmochim. Acta 2017, 205, 360–380. [Google Scholar] [CrossRef] [Green Version]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Dove, P.M.; Han, N.; De Yoreo, J.J. Mechanisms of classical crystal growth theory explain quartz and silicate dissolution behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 15357–15362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastero, L.; Bruno, M.; Aquilano, D. Habit Change of Monoclinic Hydroxyapatite Crystals Growing from Aqueous Solution in the Presence of Citrate Ions: The Role of 2D Epitaxy. Crystals 2018, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Bénézeth, P.; Saldi, G.D.; Dandurand, J.-L.; Schott, J. Experimental determination of the solubility product of magnesite at 50 to 200 °C. Chem. Geol. 2011, 286, 21–31. [Google Scholar] [CrossRef]
- Saldi, G.D.; Jordan, G.; Schott, J.; Oelkers, E.H. Magnesite growth rates as a function of temperature and saturation state. Geochim. Cosmochim. Acta 2009, 73, 5646–5657. [Google Scholar] [CrossRef]
- De Yoreo, J.J.; Vekilov, P.G. Principles of Crystal Nucleation and Growth. Rev. Miner. Geochem. 2003, 54, 57–93. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable Prenucleation Calcium Carbonate Clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radha, A.; Fernandez-Martinez, A.; Hu, Y.; Jun, Y.-S.; Waychunas, G.A.; Navrotsky, A. Energetic and structural studies of amorphous Ca1−xMgxCO3 × nH2O (0 ≤ x ≤ 1). Geochim. Cosmochim. Acta 2012, 90, 83–95. [Google Scholar] [CrossRef]
- Stefánsson, A.; Bénézeth, P.; Schott, J. Potentiometric and spectrophotometric study of the stability of magnesium carbonate and bicarbonate ion pairs to 150 °C and aqueous inorganic carbon speciation and magnesite solubility. Geochim. Cosmochim. Acta 2014, 138, 21–31. [Google Scholar] [CrossRef]
- Wen, H.; Li, L. An upscaled rate law for magnesite dissolution in heterogeneous porous media. Geochim. Cosmochim. Acta 2017, 210, 289–305. [Google Scholar] [CrossRef]
- Hänchen, M.; Prigiobbe, V.; Baciocchi, R.; Mazzotti, M. Precipitation in the Mg-carbonate system—Effects of temperature and CO2 pressure. Chem. Eng. Sci. 2008, 63, 1012–1028. [Google Scholar] [CrossRef]
- Ballirano, P.; De Vito, C.; Mignardi, S.; Ferrini, V. Phase transitions in the MgCO2H2O system and the thermal decomposition of dypingite, Mg5(CO3)4(OH)2 × 5H2O: Implications for geosequestration of carbon dioxide. Chem. Geol. 2013, 340, 59–67. [Google Scholar] [CrossRef]
- Fricker, K.J.; Park, A.-H.A. Effect of H2O on Mg (OH)2 carbonation pathways for combined CO2 capture and storage. Chem. Eng. Sci. 2013, 100, 332–341. [Google Scholar] [CrossRef]
- Harrison, A.L.; Mavromatis, V.; Oelkers, E.H.; Bénézeth, P. Solubility of the hydrated Mg-carbonates nesquehonite and dypingite from 5 to 35 °C: Implications for CO2 storage and the relative stability of Mg-carbonates. Chem. Geol. 2019, 504, 123–135. [Google Scholar] [CrossRef]
- Lu, L.; Guest, J.S.; Peters, C.A.; Zhu, X.; Rau, G.H.; Ren, Z.J. Wastewater treatment for carbon capture and utilization. Nat. Sustain. 2018, 1, 750–758. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Lorenzo, F.; Arnold, T.; Churakov, S.V. Pb2+ Uptake by Magnesite: The Competition between Thermodynamic Driving Force and Reaction Kinetics. Minerals 2021, 11, 415. https://doi.org/10.3390/min11040415
Di Lorenzo F, Arnold T, Churakov SV. Pb2+ Uptake by Magnesite: The Competition between Thermodynamic Driving Force and Reaction Kinetics. Minerals. 2021; 11(4):415. https://doi.org/10.3390/min11040415
Chicago/Turabian StyleDi Lorenzo, Fulvio, Tobias Arnold, and Sergey V. Churakov. 2021. "Pb2+ Uptake by Magnesite: The Competition between Thermodynamic Driving Force and Reaction Kinetics" Minerals 11, no. 4: 415. https://doi.org/10.3390/min11040415