
Competing Sorption of Se(IV) and Se(VI) on Schwertmannite


Abstract
:1. Introduction
2. Materials and Methods
2.1. Schwertmannite Samples
2.2. XRD, FTIR, BET and SEM Analyses
2.3. Selenium Sorption to Different Schwertmannites
2.4. Determination of Aqueous Selenium Species, Sulphate and Total Dissolved Iron
2.5. Selenium Solid-Phase Speciation
3. Results
3.1. Characterisation of Schwertmannite Specimen
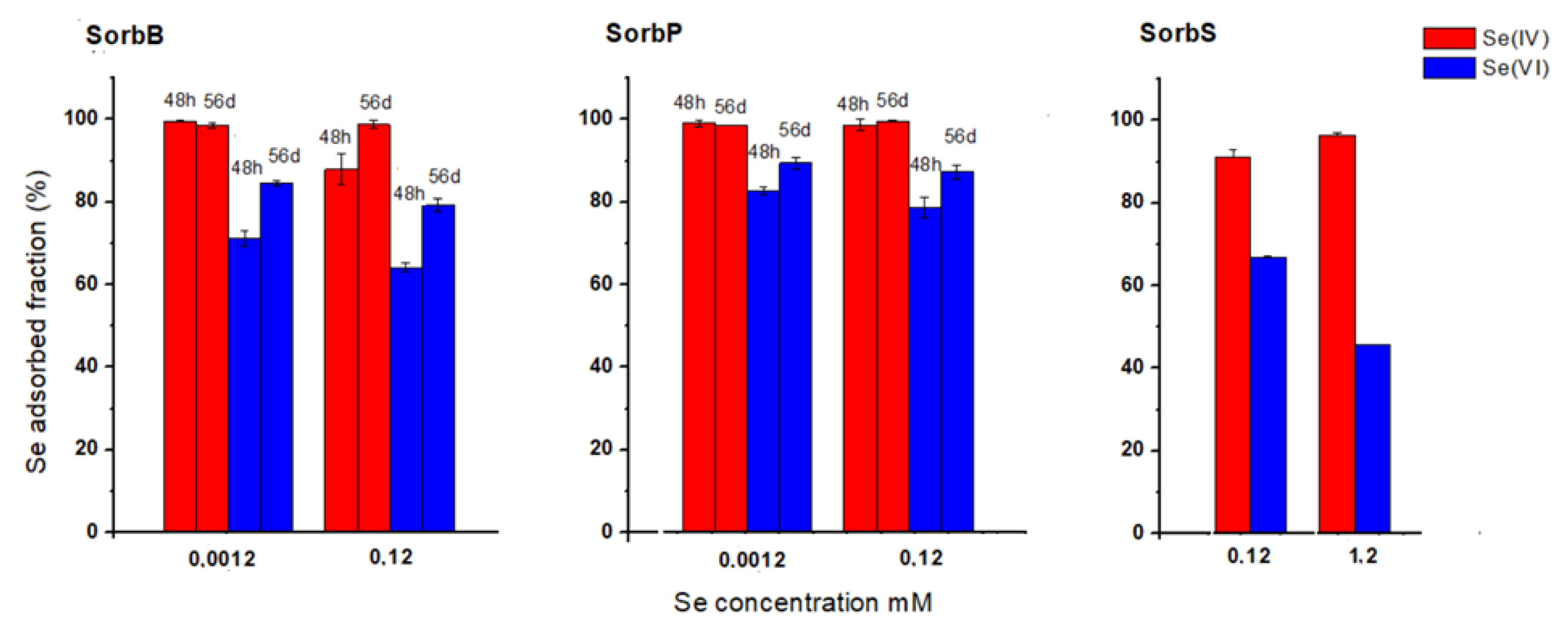
3.2. Interaction between Se Species, Sulphur and Schwertmannites
3.2.1. Aqueous Phase
3.2.2. Solid Phase
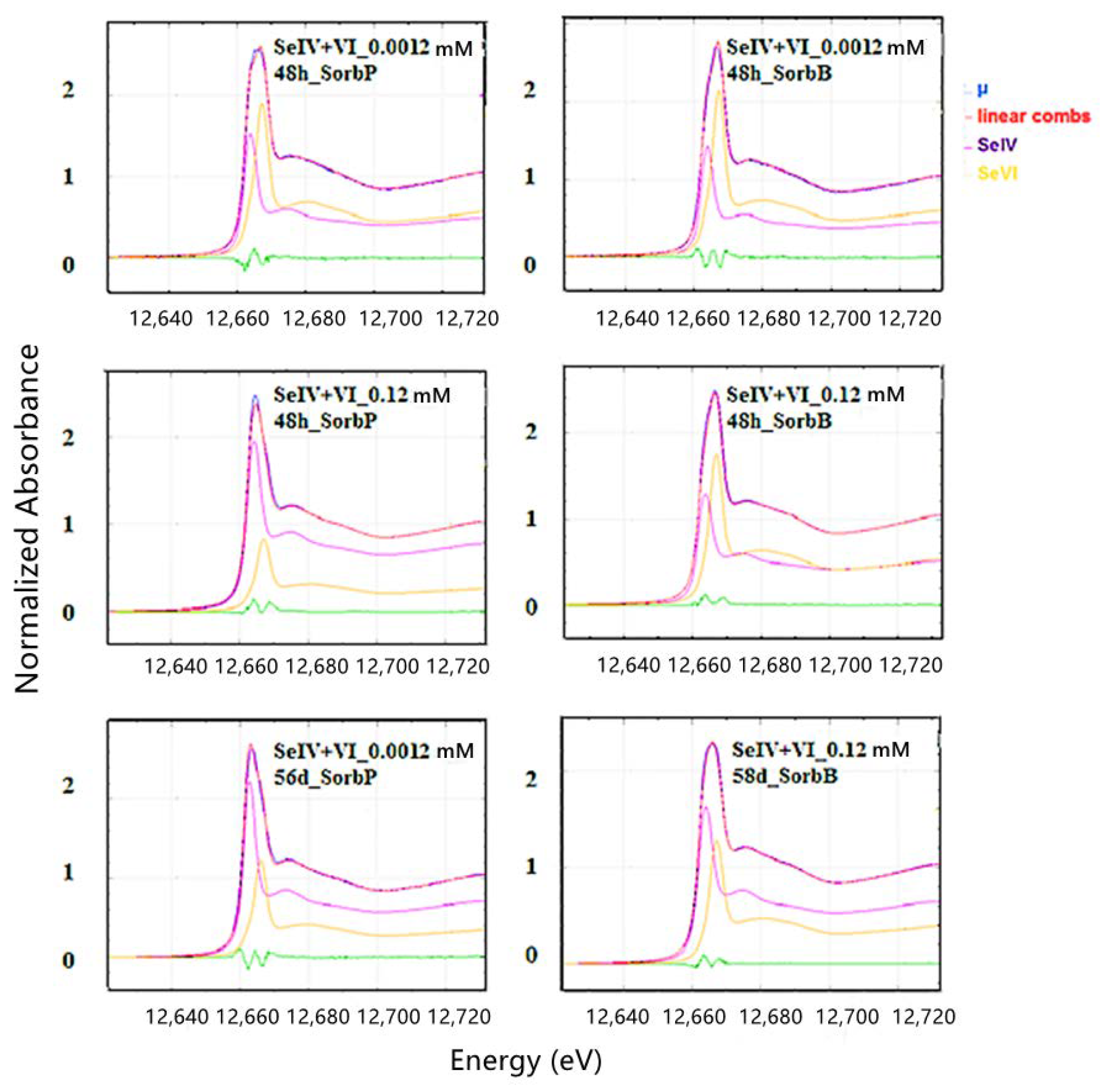
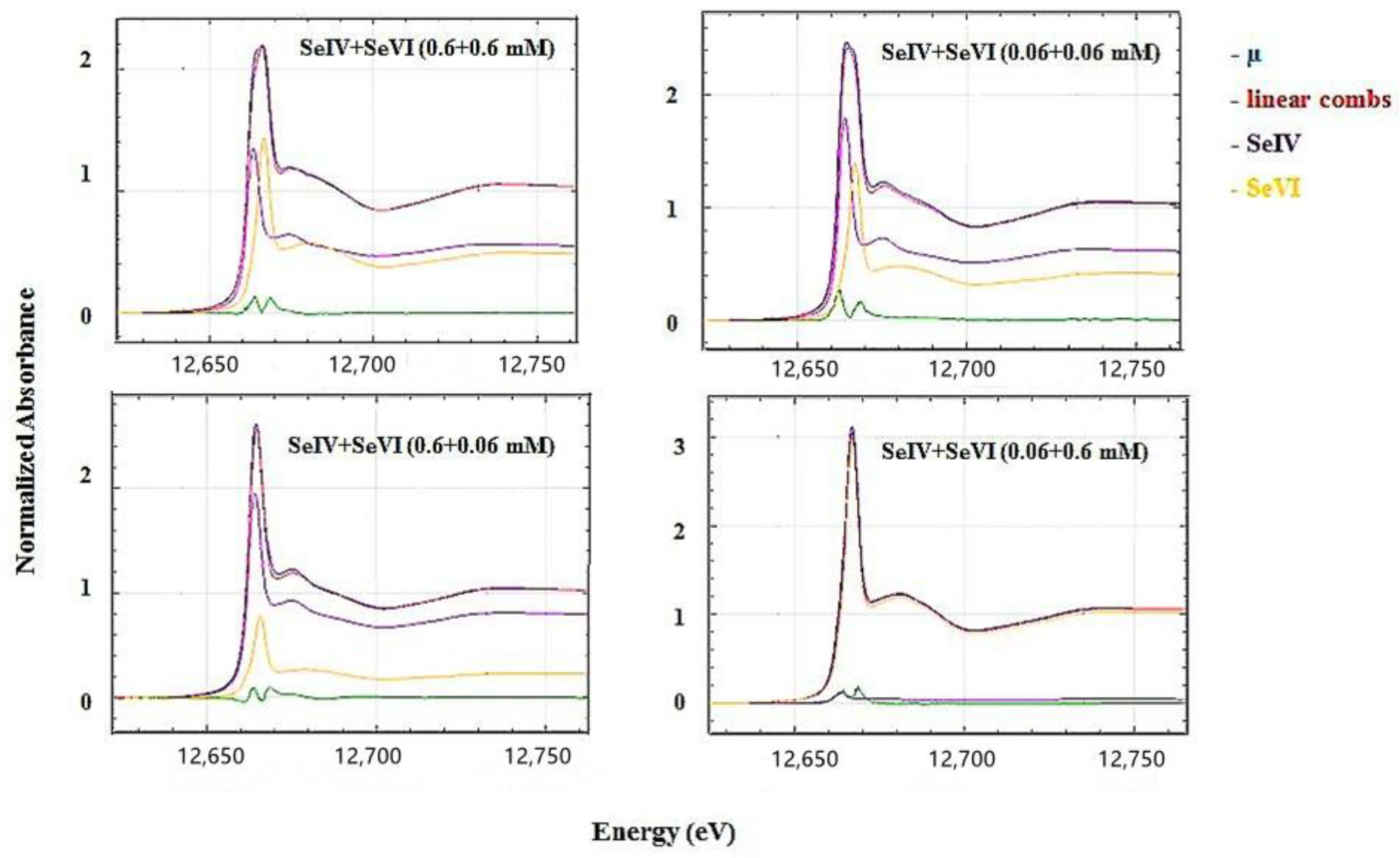
3.3. XANES and EXAFS Analyses
3.3.1. Single Species
3.3.2. Mixed Species
4. Discussion
4.1. Binding Mechanisms of Se(IV) and Se(VI) onto Schwetmannite
4.2. Competitive Effect on Partitioning of Se(IV) and Se(VI) into Schwetmannite
- KF: Freundlich adsorption coefficient (Ln.mol(1−n)/g)
- nF: Freundlich exponent (≤1)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fordyce, F.M. Selenium Deficiency and Toxicity in the Environment. In Essentials of Medical Geology: Revised Edition; Selinus, O., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 375–416. [Google Scholar]
- Ong, C.G.; Tanji, K.K. Evaporative concentration of trace elements in a multicell agricultural evaporation pond. J. Agric. Food Chem. 1993, 41, 1507–1510. [Google Scholar] [CrossRef]
- Twidwell, L.; Mccloskey, J.; Miranda, P.; Gale, M. Technologies and potential technologies for removing arsenic from process and mine wastewater. In Proceedings of the REWAS’99 Global Symposium on Recycling, Waste Treatment and Clean Technology, San Sebastian, Spain, 5–9 September 1999; Volume 2. [Google Scholar]
- Stefaniak, J.; Dutta, A.; Verbinnen, B.; Shakya, M.; Rene, E.R. Selenium removal from mining and process wastewater: A systematic review of available technologies. J. Water Supply Res. Technol. 2018, 67, 903–918. [Google Scholar] [CrossRef]
- Marouane, B.; Klug, M.; As, S.K.; Engel, J.; Reichel, S.; Janneck, E.; Peiffer, S. The potential of granulated schwertmannite adsorbents to remove oxyanions (SeO32−, SeO42−, MoO42−, PO43−, Sb(OH)6−) from contaminated water. J. Geochem. Explor. 2020, 223, 106708. [Google Scholar] [CrossRef]
- Regenspurg, S.; Peiffer, S. Arsenate and chromate incorporation in schwertmannite. Appl. Geochem. 2005, 20, 1226–1239. [Google Scholar] [CrossRef]
- Paikaray, S.; Göttlicher, J.; Peiffer, S. Removal of As(III) from acidic waters using schwertmannite: Surface speciation and effect of synthesis pathway. Chem. Geol. 2011, 283, 134–142. [Google Scholar] [CrossRef]
- Antelo, J.; Fiol, S.; Gondar, D.; López, R.; Arce, F. Comparison of arsenate, chromate and molybdate binding on schwertmannite: Surface adsorption vs anion-exchange. J. Colloid Interface Sci. 2012, 386, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Mohan, D.; Pittman, C.U. Arsenate adsorption on three types of granular schwertmannite. Water Res. 2013, 47, 2938–2948. [Google Scholar] [CrossRef]
- Janneck, E.; Burghardt, D.; Simon, E.; Peiffer, S.; Paul, M.; Koch, T. Development of an Adsorbent Comprising Schwertmannite and its Utilization in Mine Water Treatment. In Proceedings of the International Mine Water Association, Santiago de Chile, Chile, 21–24 April 2015. [Google Scholar]
- Hayes, K.F.; Roe, A.L.; Brown, G.E.; Hodgson, K.O.; Leckie, J.O.; Parks, G.A. In Situ X-ray Absorption Study of Surface Complexes: Selenium Oxyanions on α-FeOOH. Science 1987, 238, 783–786. [Google Scholar] [CrossRef]
- Rietra, R.P.; Hiemstra, T.; van Riemsdijk, W. Comparison of Selenate and Sulfate Adsorption on Goethite. J. Colloid Interface Sci. 2001, 240, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.; Marmier, N.; Lomenech, C.; Giffaut, E.; Ehrhardt, J. Competition between selenium (IV) and silicic acid on the hematite surface. Chemosphere 2009, 75, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Min, J.H.; Lee, J.K.; Baik, M.H.; Choi, J.W.; Shin, H.S. Effects of pH and anions on the sorption of selenium ions onto magnetite. J. Environ. Radioact. 2012, 104, 1–6. [Google Scholar] [CrossRef]
- Das, S.; Hendry, J.M.; Essilfie-Dughan, J. Adsorption of selenate onto ferrihydrite, goethite, and lepidocrocite under neutral pH conditions. Appl. Geochem. 2013, 28, 185–193. [Google Scholar] [CrossRef]
- Jordan, N.; Ritter, A.; Scheinost, A.C.; Weiss, S.; Schild, D.; Hübner, R. Selenium(IV) Uptake by Maghemite (γ-Fe2O3). Environ. Sci. Technol. 2014, 48, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Börsig, N.; Scheinost, A.C.; Shaw, S.; Schild, D.; Neumann, T. Uptake mechanisms of selenium oxyanions during the ferrihydrite-hematite recrystallization. Geochim. Cosmochim. Acta 2017, 206, 236–253. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Finck, N.; Heberling, F.; Pruessmann, T.; Liu, C.; Lützenkirchen, J. Adsorption of Selenium and Strontium on Goethite: EXAFS Study and Surface Complexation Modeling of the Ternary Systems. Environ. Sci. Technol. 2017, 51, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Waychunas, G.A.; Xu, N.; Fuller, C.C.; Davis, J.A.; Bigham, J.M. XAS study of AsO43− and SeO42− substituted schwertmannites. Phys. B Condens. Matter 1995, 208–209, 481–483. [Google Scholar] [CrossRef]
- Khamphila, K.; Kodama, R.; Sato, T.; Otake, T. Adsorption and Post Adsorption Behavior of Schwertmannite with Various Oxyanions. J. Miner. Mater. Charact. Eng. 2017, 5, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.; Ungureanu, G.; Boaventura, R.; Botelho, C. Science of the Total Environment Selenium contaminated waters : An overview of analytical methods, treatment options and recent advances in sorption methods. Sci. Total Environ. 2015, 521–522, 246–260. [Google Scholar] [CrossRef]
- Pincus, L.N.; Rudel, H.E.; Petrović, P.V.; Gupta, S.; Westerhoff, P.; Muhich, C.L.; Zimmerman, J.B. Exploring the Mechanisms of Selectivity for Environmentally Significant Oxo-Anion Removal during Water Treatment: A Review of Common Competing Oxo-Anions and Tools for Quantifying Selective Adsorption. Environ. Sci. Technol. 2020, 54, 9769–9790. [Google Scholar] [CrossRef] [PubMed]
- Janneck, E.; Arnold, I.; Koch, T.; Meyer, J.; Burghardt, D.; Ehinger, S. Microbial synthesis of schwertmannite from lignite mine water and its utilization for removal of arsenic from mine waters and for production of iron pigments. In Proceedings of the International Mine Water Association, Sydney, NS, Canada, 5–9 September 2010. [Google Scholar]
- Tabatabai, M.A. A Rapid Method for Determination of Sulfate in Water Samples. Environ. Lett. 1974, 7, 237–243. [Google Scholar] [CrossRef]
- Rehr, J.J.; Albers, R.C. Theoretical approaches to x-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621–654. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ressler, T. WinXAS: A New Software Package not only for the Analysis of Energy-Dispersive XAS Data. J. Phys. IV Fr. 1997, 7, C2-269–C2-270. [Google Scholar] [CrossRef]
- Paikaray, S.; Peiffer, S. Abiotic schwertmannite transformation kinetics and the role of sorbed As(III). Appl. Geochem. 2012, 27, 590–597. [Google Scholar] [CrossRef]
- Fukushi, K.; Sato, T.; Yanase, N. Solid-Solution Reactions in As(V) Sorption by Schwertmannite. Environ. Sci. Technol. 2003, 37, 3581–3586. [Google Scholar] [CrossRef]
- Burton, E.D.; Bush, R.T.; Johnston, S.G.; Watling, K.M.; Hocking, R.K.; Sullivan, L.A.; Parker, G.K. Sorption of Arsenic(V) and Arsenic(III) to Schwertmannite. Environ. Sci. Technol. 2009, 43, 9202–9207. [Google Scholar] [CrossRef]
- Bigham, J.M.; Schwertmann, U.; Carlson, L.; Murad, E. A poorly crystallized oxyhydroxysulfate of iron formed by bacterial oxidation of Fe(II) in acid mine waters. Geochim. Cosmochim. Acta 1990, 54, 2743–2758. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides; WILEY-VCH Verlag GmbH & Co.KGaA: Weinheim, Germany, 2003; Volume 39, ISBN 3527302743. [Google Scholar]
- Su, C.; Suarez, D.L. Selenate and Selenite Sorption on Iron Oxides. Soil Sci. Soc. Am. J. 2000, 64, 101–111. [Google Scholar] [CrossRef]
- Wang, X.; Ying, H.; Zhao, W.; Feng, X.; Tan, W.; Beyer, K.A.; Huang, Q.; Liu, F.; Zhu, M. Molecular-Scale Understanding of Sulfate Exchange from Schwertmannite by Chromate Versus Arsenate. Environ. Sci. Technol. 2021, 55, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, C.; Feng, X.; Zhu, M. Sulfate Local Coordination Environment in Schwertmannite. Environ. Sci. Technol. 2015, 49, 10440–10448. [Google Scholar] [CrossRef]
- Jönsson, J.; Persson, P.; Sjöberg, S.; Lövgren, L. Schwertmannite precipitated from acid mine drainage: Phase transformation, sulphate release and surface properties. Appl. Geochem. 2005, 20, 179–191. [Google Scholar] [CrossRef]
- Fernandez-Martinez, A.; Timón, V.; Román-Ross, G.; Cuello, G.; Daniels, J.; Ayora, C. The structure of schwertmannite, a nanocrystalline iron oxyhydroxysulfate. Am. Mineral. AMER Miner. 2010, 95, 1312–1322. [Google Scholar] [CrossRef]
- Chubar, N.; Gerda, V.; Szlachta, M. Mechanism of Selenite Removal by a Mixed Adsorbent Based on Fe–Mn Hydrous Oxides Studied Using X-ray Absorption Spectroscopy. Environ. Sci. Technol. 2014, 48, 13376–13383. [Google Scholar] [CrossRef] [PubMed]
- Manceau, A.; Charlet, L. The Mechanism of Selenate Adsorption on Goethite and Hydrous Ferric Oxide. J. Colloid Interface Sci. 1994, 168, 87–93. [Google Scholar] [CrossRef]
- Peak, D. Adsorption mechanisms of selenium oxyanions at the aluminum oxide/water interface. J. Colloid Interface Sci. 2006, 303, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Peak, D.; Sparks, D.L. Mechanisms of selenate adsorption on iron oxides and hydroxides. Environ. Sci. Technol. 2002, 36, 1460–1466. [Google Scholar] [CrossRef]
- Song, J.; Jia, S.Y.; Ren, H.T.; Wu, S.H.; Han, X. Application of a high-surface-area schwertmannite in the removal of arsenate and arsenite. Int. J. Environ. Sci. Technol. 2015, 12, 1559–1568. [Google Scholar] [CrossRef]
- Bailey, R.T. Review: Selenium contamination, fate, and reactive transport in groundwater in relation to human health. Hydrogeol. J. 2016, 25, 1191–1217. [Google Scholar] [CrossRef]
- Ullah, H.; Liu, G.; Yousaf, B.; Ali, M.U.; Irshad, S.; Abbas, Q.; Ahmad, R. A comprehensive review on environmental transformation of selenium: Recent advances and research perspectives. Environ. Geochem. Health 2019, 41, 1003–1035. [Google Scholar] [CrossRef]
- Wasewar, K.L.; Prasad, B.; Gulipalli, S. Adsorption of Selenium Using Bagasse Fly Ash. Clean Soil Air Water 2009, 37, 534–543. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef] [Green Version]
- USEPA. Drinking Water Advisory: Consumer Acceptability Advice and Health Effects Analysis on Sulfate. EPA-822-R-03-007; USEPA: Washington, DC, USA, 2003.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Se(IV) mM | Se(VI) mM | Se(IV) + Se(VI) mM | Reaction Time | |
|---|---|---|---|---|
| SorbS | 0.12 1.2 | 0.12 1.2 | 0.06 + 0.06 0.6 + 0.6 0.6 + 0.06 0.06 + 0.6 | 48 h |
| SorbP | 0.0012 | 0.0012 | 0.0006 + 0.0006 | 48 h 56 d |
| 0.12 | 0.12 | 0.06 + 0.06 | 48 h | |
| SorbB | 0.0012 | 0.0012 | 0.0006 + 0.0006 | 48 h |
| 0.12 | 0.12 | 0.06 + 0.06 | 48 h 56 d |
| CN | R (Å) | σ2 (Å) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C of Se Species (mM) | 0.0012 | 0.12 | 1.2 | 0.0012 | 0.12 | 1.2 | 0.0012 | 0.12 | 1.2 | ||
| 48 h | SorbB | Se(IV)—O | 2.8 | 2.8 | - | 1.70 | 1.70 | - | 0.0010 | 0.0014 | - |
| SorbP | 3.0 | 2.8 | - | 1.70 | 1.69 | - | 0.0018 | 0.0017 | - | ||
| SorbS | - | 2.9 | 2.5 | - | 1.68 | 1.70 | - | 0.0016 | 0.0014 | ||
| SorbB | Se(IV)—Fe | 2.0 | 1.9 | - | 3.35 | 3.35 | - | 0.0077 | 0.0071 | - | |
| SorbP | 2.2 | 1.9 | - | 3.35 | 3.34 | - | 0.0079 | 0.0083 | - | ||
| SorbS | - | 2.2 | 1.6 | - | 3.36 | 3.35 | - | 0.0083 | 0.0083 | ||
| SorbB | Se(VI)—O | 3.6 | 3.5 | - | 1.65 | 1.66 | - | 0.0015 | 0.0014 | - | |
| SorbP | 3.7 | 3.5 | - | 1.65 | 1.66 | - | 0.0015 | 0.0016 | - | ||
| SorbS | - | 3.7 | 3.6 | - | 1.65 | 1.66 | - | 0.0014 | 0.0014 | ||
| SorbB | Se(VI)—Fe | 2.2 | 1.8 | - | 3.34 | 3.34 | - | 0.0060 | 0.0060 | - | |
| SorbP | 2.1 | 1.8 | - | 3.35 | 3.34 | - | 0.0060 | 0.0062 | - | ||
| SorbS | - | 1.9 | 1.6 | - | 3.32 | 3.43 | - | 0.0061 | 0.0062 | ||
| 56 d | SorbB | Se(IV)—O | 2.9 | 2.8 | - | 1.69 | 1.70 | - | 0.0010 | 0.0014 | - |
| SorbP | 2.6 | 2.8 | - | 1.70 | 1.70 | - | 0.0010 | 0.0014 | - | ||
| SorbB | Se(IV)—Fe | 2.2 | 1.8 | - | 3.34 | 3.35 | - | 0.0074 | 0.0065 | - | |
| SorbP | 2.2 | 1.8 | - | 3.35 | 3.34 | - | 0.0075 | 0.0065 | - | ||
| SorbB | Se(VI)—O | 3.5 | 3.7 | - | 1.67 | 1.65 | - | 0.0015 | 0.0014 | - | |
| SorbP | 3.5 | 3.6 | - | 1.67 | 1.65 | - | 0.0018 | 0.0014 | - | ||
| SorbB | Se(VI)—Fe | 2.0 | 1.9 | - | 3.35 | 3.33 | - | 0.0060 | 0.0060 | - | |
| SorbP | 2.0 | 1.9 | - | 3.35 | 3.33 | - | 0.0067 | 0.0070 | - | ||
| C of Se (IV) + (VI) (mM) | SeIV—O | SeVI—O | SeIV/VI—Fe | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SorbB | SorbP | SorbS | SorbB | SorbP | SorbS | SorbB | SorbP | SorbS | |||
| 48 h | CN | 0.0012 (1:1) | 1.8 | 1.7 | - | 1.3 | 1.6 | 1.9 | 1.9 | - | |
| 0.12 (1:1) | 1.5 | 2.2 | 1.9 | 1.7 | 0.8 | 1.3 | 1.8 | 1.8 | 2.3 | ||
| 0.66 (10:1) | - | - | 2.6 | - | - | 0.4 | - | - | 2.0 | ||
| 0.66 (1:10) | - | - | 0.4 | - | - | 3.4 | - | - | 2.0 | ||
| 1.2 (1:1) | - | - | 1.5 | - | - | 1.3 | - | - | 1.5 | ||
| R(Å) | 0.0012 (1:1) | 1.70 | 1.70 | - | 1.65 | 1.65 | - | 3.35 | 3.35 | - | |
| 0.12 (1:1) | 1.70 | 1.69 | 1.69 | 1.66 | 1.66 | 1.66 | 3.35 | 3.34 | 3.32 | ||
| 0.66 (10:1) | - | - | 1.69 | - | - | 1.65 | - | - | 3.34 | ||
| 0.66 (1:10) | - | - | 1.69 | - | - | 1.65 | - | - | 3.34 | ||
| 1.2 (1:1) | - | - | 1.69 | - | - | 1.65 | - | - | 3.36 | ||
| σ2 (Å) | 0.0012 (1:1) | 0.0010 | 0.0010 | - | 0.0015 | 0.0015 | - | 0.0060 | 0.0060 | - | |
| 0.12 (1:1) | 0.0014 | 0.0017 | 0.0014 | 0.0014 | 0.0016 | 0.0014 | 0.0064 | 0.0071 | 0.0071 | ||
| 0.66 (10:1) | - | - | 0.0014 | - | - | 0.0014 | - | - | 0.0071 | ||
| 0.66 (1:10) | - | - | 0.0014 | - | - | 0.0014 | - | - | 0.0071 | ||
| 1.2 (1:1) | - | - | 0.0014 | - | - | 0.0014 | - | - | 0.0071 | ||
| 56 d | CN | 0.0012 (1:1) | - | 2.2 | - | - | 0.6 | - | - | 2.0 | - |
| 0.12 (1:1) | 2.0 | - | - | 1.1 | - | - | 1.9 | - | - | ||
| R(Å) | 0.0012 (1:1) | - | 1.70 | - | 1.67 | - | - | 3.38 | - | ||
| 0.12 (1:1) | 1.70 | - | - | 1.65 | - | - | 3.35 | - | - | ||
| σ2 (Å) | 0.0012 (1:1) | - | 0.0010 | - | - | 0.0018 | - | - | 0.0071 | - | |
| 0.12 (1:1) | 0.0014 | - | - | 0.0014 | - | - | 0.0065 | - | - | ||
| C of Se (IV) + (VI) (mM) | SorbB | SorbP | SorbS | ||||
|---|---|---|---|---|---|---|---|
| Se(IV) | Se(VI) | Se(IV) | Se(VI) | Se(IV) | Se(VI) | ||
| 48 h | 0.0012 (1:1) | 42 | 57 | 47 | 52 | - | - |
| 0.12 (1:1) | 51 | 49 | 77 | 23 | 60 | 40 | |
| 0.66 (10:1) | - | - | - | - | 85 | 15 | |
| 0.66 (1:10) | - | - | - | - | 06 | 94 | |
| 1.2 (1:1) | - | - | - | - | 56 | 44 | |
| 56 d | 0.0012 (1:1) | n.a | n.a | 66 | 33 | - | - |
| 0.12 (1:1) | 62 | 37 | n.a | n.a | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marouane, B.; Chen, N.; Obst, M.; Peiffer, S. Competing Sorption of Se(IV) and Se(VI) on Schwertmannite. Minerals 2021, 11, 764. https://doi.org/10.3390/min11070764
Marouane B, Chen N, Obst M, Peiffer S. Competing Sorption of Se(IV) and Se(VI) on Schwertmannite. Minerals. 2021; 11(7):764. https://doi.org/10.3390/min11070764
Chicago/Turabian StyleMarouane, Bouchra, Ning Chen, Martin Obst, and Stefan Peiffer. 2021. "Competing Sorption of Se(IV) and Se(VI) on Schwertmannite" Minerals 11, no. 7: 764. https://doi.org/10.3390/min11070764