

Whole-Exome Sequencing Reveals High Mutational Concordance between Primary and Matched Recurrent Triple-Negative Breast Cancers
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Patient Samples
2.2. WES and Variant Calling
3. Results
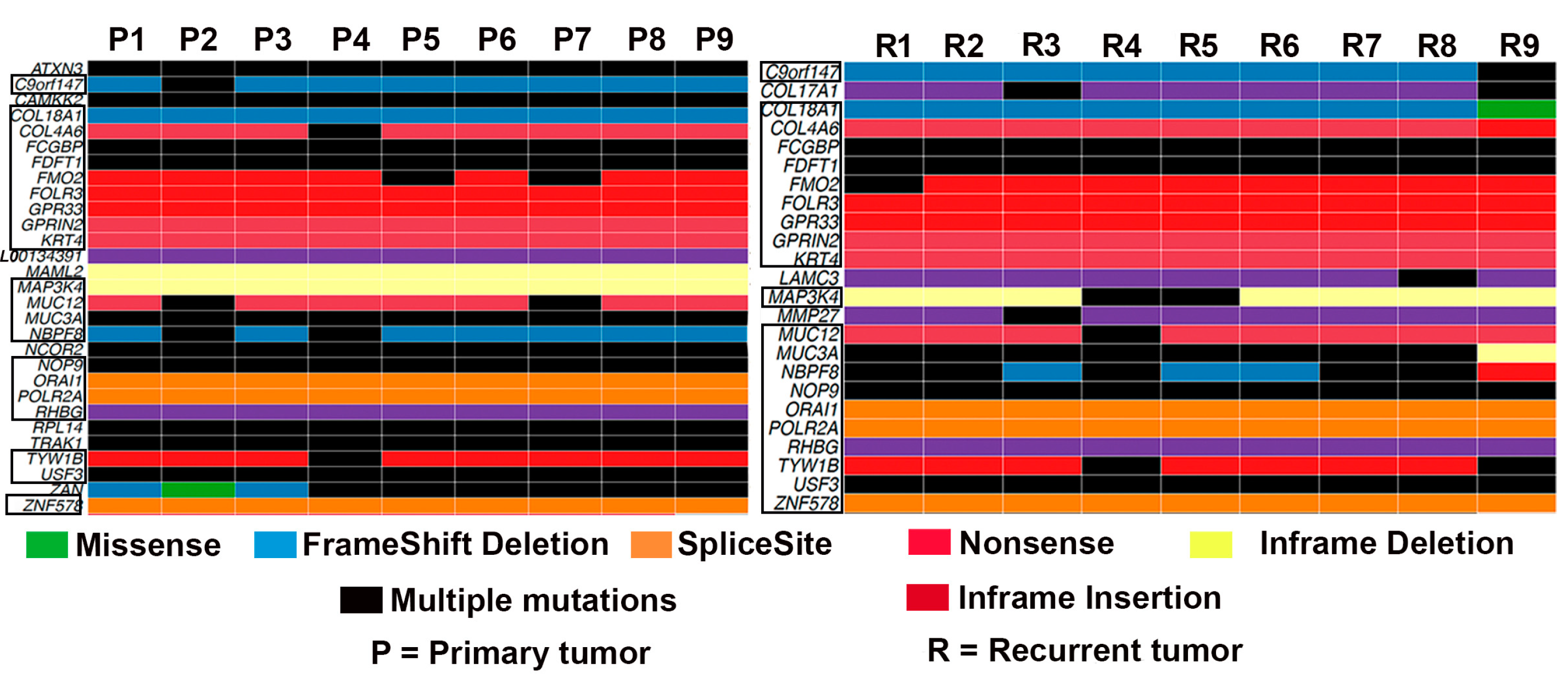
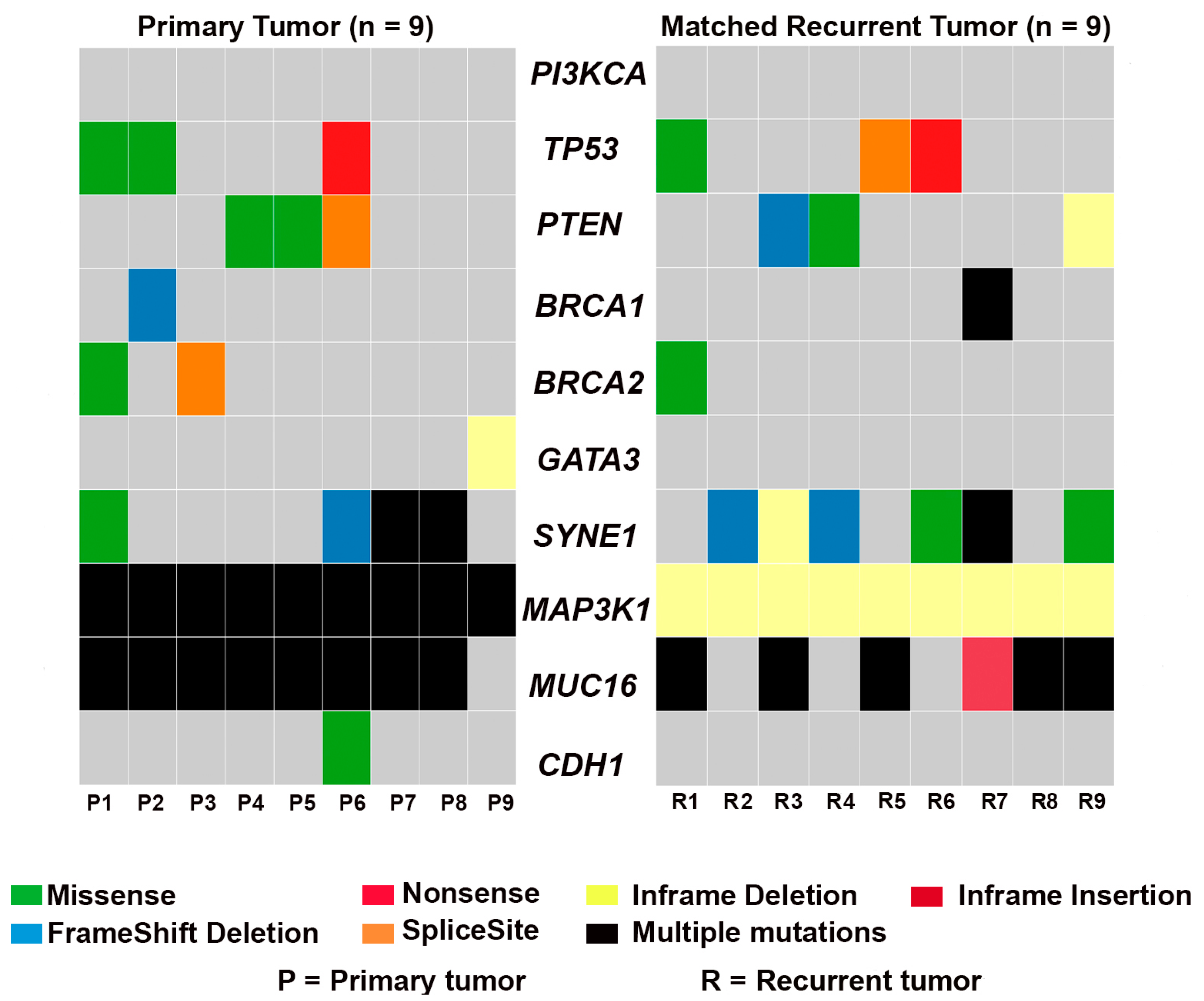
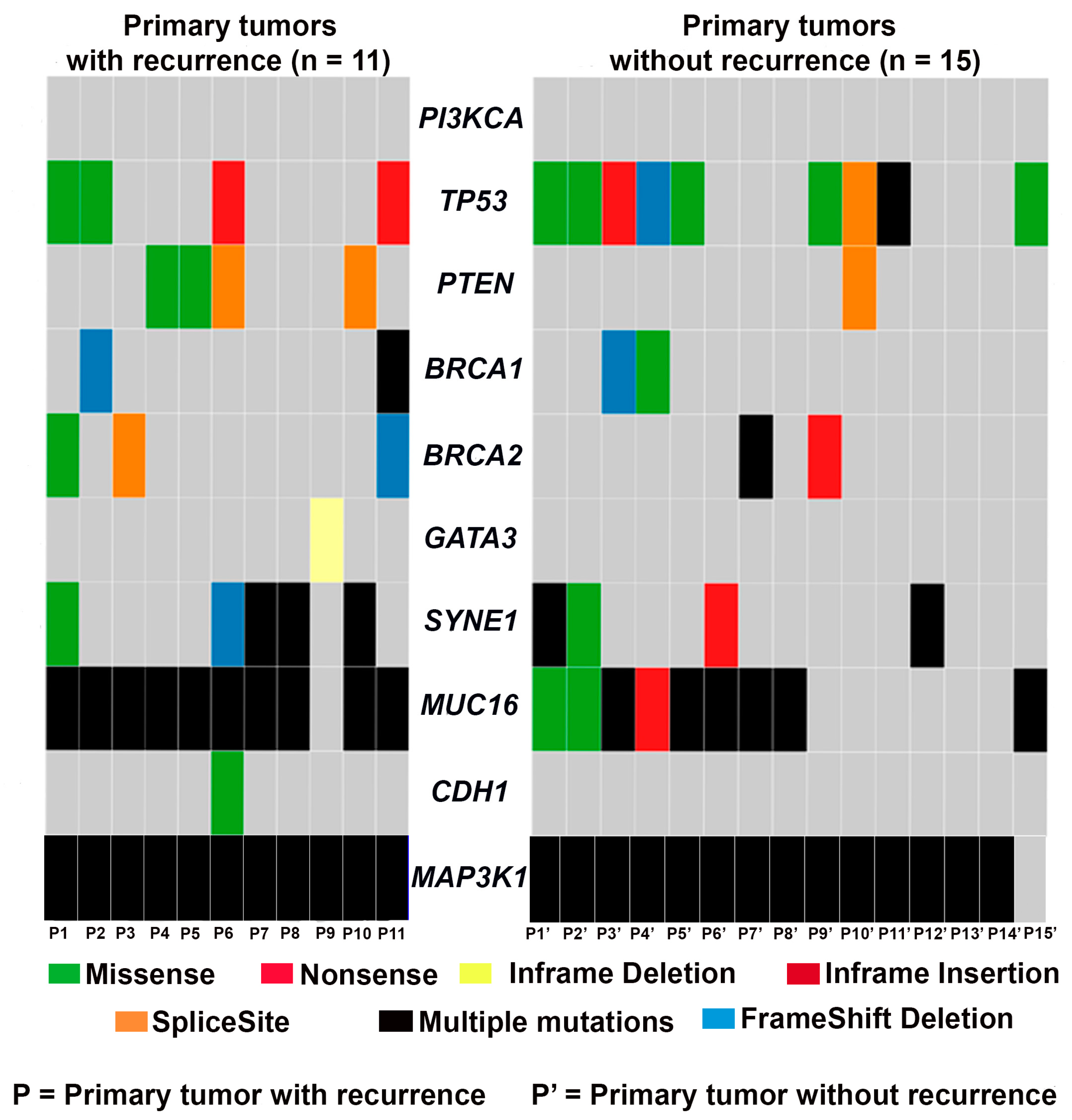
3.1. Mutational Landscape of TNBC
3.2. Frequency of Recurrent Gene Mutations in TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9 (Suppl. 2), S73–S81. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef]
- O’Reilly, D.; Sendi, M.A.; Kelly, C.M. Overview of recent advances in metastatic triple negative breast cancer. World J. Clin. Oncol. 2021, 12, 164–182. [Google Scholar] [CrossRef]
- Yao, Y.; Chu, Y.; Xu, B.; Hu, Q.; Song, Q. Risk factors for distant metastasis of patients with primary triple-negative breast cancer. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Jin, J.; Gao, Y.; Zhang, J.; Wang, L.; Wang, B.; Cao, J.; Shao, Z.; Wang, Z. Incidence, pattern and prognosis of brain metastases in patients with metastatic triple negative breast cancer. BMC Cancer 2018, 18, 446. [Google Scholar] [CrossRef]
- Beltran, H.; Eng, K.; Mosquera, J.M.; Sigaras, A.; Romanel, A.; Rennert, H.; Kossai, M.; Pauli, C.; Faltas, B.; Fontugne, J.; et al. Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response. JAMA Oncol. 2015, 1, 466–474. [Google Scholar] [CrossRef]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef]
- Kroigard, A.B.; Larsen, M.J.; Thomassen, M.; Kruse, T.A. Molecular Concordance Between Primary Breast Cancer and Matched Metastases. Breast J. 2016, 22, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Niikura, N.; Ogiya, R.; Yasojima, H.; Watanabe, K.I.; Kanbayashi, C.; Tsuneizumi, M.; Matsui, A.; Fujisawa, T.; Iwasa, T.; et al. Distinct gene expression profiles between primary breast cancers and brain metastases from pair-matched samples. Sci. Rep. 2019, 9, 13343. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.E.; Kim, N.I.; Lee, J.S.; Park, M.H.; Kang, K. Differentially Expressed Genes in Matched Normal, Cancer, and Lymph Node Metastases Predict Clinical Outcomes in Patients With Breast Cancer. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Vareslija, D.; Priedigkeit, N.; Fagan, A.; Purcell, S.; Cosgrove, N.; O’Halloran, P.J.; Ward, E.; Cocchiglia, S.; Hartmaier, R.; Castro, C.A.; et al. Transcriptome Characterization of Matched Primary Breast and Brain Metastatic Tumors to Detect Novel Actionable Targets. J. Natl. Cancer Inst. 2019, 111, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Song, M.; Chouchane, L.; Ma, X. Functional Genomic Analysis of Breast Cancer Metastasis: Implications for Diagnosis and Therapy. Cancers 2021, 13, 3276. [Google Scholar] [CrossRef]
- Kim, K.P.; Kim, J.E.; Hong, Y.S.; Ahn, S.M.; Chun, S.M.; Hong, S.M.; Jang, S.J.; Yu, C.S.; Kim, J.C.; Kim, T.W. Paired Primary and Metastatic Tumor Analysis of Somatic Mutations in Synchronous and Metachronous Colorectal Cancer. Cancer Res. Treat. 2017, 49, 161–167. [Google Scholar] [CrossRef]
- Aftimos, P.; Oliveira, M.; Irrthum, A.; Fumagalli, D.; Sotiriou, C.; Gal-Yam, E.N.; Robson, M.E.; Ndozeng, J.; Di Leo, A.; Ciruelos, E.M.; et al. Genomic and Transcriptomic Analyses of Breast Cancer Primaries and Matched Metastases in AURORA, the Breast International Group (BIG) Molecular Screening Initiative. Cancer Discov. 2021, 11, 2796–2811. [Google Scholar] [CrossRef]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Lukyanova, E.N.; Kalinin, D.V.; Pokrovsky, A.V.; Dmitriev, A.A.; Koroban, N.V.; Pudova, E.A.; Fedorova, M.S.; Volchenko, N.N.; Stepanov, O.A.; et al. Exome analysis of carotid body tumor. BMC Med. Genom. 2018, 11, 17. [Google Scholar] [CrossRef]
- Bonin, S.; Hlubek, F.; Benhattar, J.; Denkert, C.; Dietel, M.; Fernandez, P.L.; Hofler, G.; Kothmaier, H.; Kruslin, B.; Mazzanti, C.M.; et al. Multicentre validation study of nucleic acids extraction from FFPE tissues. Virchows Arch. 2010, 457, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wood, H.M.; Belvedere, O.; Conway, C.; Daly, C.; Chalkley, R.; Bickerdike, M.; McKinley, C.; Egan, P.; Ross, L.; Hayward, B.; et al. Using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-fixed paraffin-embedded specimens. Nucleic Acids Res. 2010, 38, e151. [Google Scholar] [CrossRef]
- de Sena Brandine, G.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000Research 2019, 8, 1874. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Niyomnaitham, S.; Parinyanitikul, N.; Roothumnong, E.; Jinda, W.; Samarnthai, N.; Atikankul, T.; Suktitipat, B.; Thongnoppakhun, W.; Limwongse, C.; Pithukpakorn, M. Tumor mutational profile of triple negative breast cancer patients in Thailand revealed distinctive genetic alteration in chromatin remodeling gene. PeerJ 2019, 7, e6501. [Google Scholar] [CrossRef]
- Yodsurang, V.; Tanikawa, C.; Miyamoto, T.; Lo, P.H.Y.; Hirata, M.; Matsuda, K. Identification of a novel p53 target, COL17A1, that inhibits breast cancer cell migration and invasion. Oncotarget 2017, 8, 55790–55803. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Mukohara, T. PI3K mutations in breast cancer: Prognostic and therapeutic implications. Breast Cancer 2015, 7, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.T.; Angus, S.P.; Johnson, G.L. MAP3K1, Genomic Alterations in Cancer and Function in Promoting Cell Survival or Apoptosis. Genes Cancer 2013, 4, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Glas, A.M.; Wessels, L.F.; Witteveen, A.T.; Peterse, J.L.; van’t Veer, L.J. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 15901–15905. [Google Scholar] [CrossRef] [PubMed]
- Moreno, F.; Gayarre, J.; Lopez-Tarruella, S.; Del Monte-Millan, M.; Picornell, A.C.; Alvarez, E.; Garcia-Saenz, J.A.; Jerez, Y.; Marquez-Rodas, I.; Echavarria, I.; et al. Concordance of Genomic Variants in Matched Primary Breast Cancer, Metastatic Tumor, and Circulating Tumor DNA: The MIRROR Study. JCO Precis Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Roy-Chowdhuri, S.; de Melo Gagliato, D.; Routbort, M.J.; Patel, K.P.; Singh, R.R.; Broaddus, R.; Lazar, A.J.; Sahin, A.; Alvarez, R.H.; Moulder, S.; et al. Multigene clinical mutational profiling of breast carcinoma using next-generation sequencing. Am. J. Clin. Pathol. 2015, 144, 713–721. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Hauselmann, I.; Borsig, L. Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 2014, 4, 28. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Chakraborty, S.; Ponnusamy, M.P.; Lakshmanan, I.; Jain, M.; Batra, S.K. Mucins in the pathogenesis of breast cancer: Implications in diagnosis, prognosis and therapy. Biochim. Biophys. Acta 2011, 1815, 224–240. [Google Scholar] [CrossRef]
- Rakha, E.A.; Boyce, R.W.; Abd El-Rehim, D.; Kurien, T.; Green, A.R.; Paish, E.C.; Robertson, J.F.; Ellis, I.O. Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer. Mod. Pathol. 2005, 18, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Aithal, A.; Rauth, S.; Kshirsagar, P.; Shah, A.; Lakshmanan, I.; Junker, W.M.; Jain, M.; Ponnusamy, M.P.; Batra, S.K. MUC16 as a novel target for cancer therapy. Expert Opin. Ther. Targets 2018, 22, 675–686. [Google Scholar] [CrossRef]
- Haridas, D.; Chakraborty, S.; Ponnusamy, M.P.; Lakshmanan, I.; Rachagani, S.; Cruz, E.; Kumar, S.; Das, S.; Lele, S.M.; Anderson, J.M.; et al. Pathobiological implications of MUC16 expression in pancreatic cancer. PLoS ONE 2011, 6, e26839. [Google Scholar] [CrossRef]
- Klug, T.L.; Bast, R.C., Jr.; Niloff, J.M.; Knapp, R.C.; Zurawski, V.R., Jr. Monoclonal antibody immunoradiometric assay for an antigenic determinant (CA 125) associated with human epithelial ovarian carcinomas. Cancer Res. 1984, 44, 1048–1053. [Google Scholar]
- Lakshmanan, I.; Ponnusamy, M.P.; Das, S.; Chakraborty, S.; Haridas, D.; Mukhopadhyay, P.; Lele, S.M.; Batra, S.K. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene 2012, 31, 805–817. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Salfity, S.; Seshacharyulu, P.; Rachagani, S.; Thomas, A.; Das, S.; Majhi, P.D.; Nimmakayala, R.K.; Vengoji, R.; Lele, S.M.; et al. MUC16 Regulates TSPYL5 for Lung Cancer Cell Growth and Chemoresistance by Suppressing p53. Clin. Cancer Res. 2017, 23, 3906–3917. [Google Scholar] [CrossRef]
- Wang, X.; Guda, C. Integrative exploration of genomic profiles for triple negative breast cancer identifies potential drug targets. Medicine 2016, 95, e4321. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Qi, Y.; Stemke-Hale, K.; Wang, B.; Young, E.J.; Booser, D.J.; Holmes, F.A.; O’Shaughnessy, J.; Hellerstedt, B.; Pippen, J.; et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res. Treat. 2012, 134, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jin, J.; Ji, W.; Guan, X. Therapeutic landscape in mutational triple negative breast cancer. Mol. Cancer 2018, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gomez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Ding, X.; Xu, S.; Zhu, B.; Jia, Q. Gene expression profiling identified TP53(Mut)PIK3CA(Wild) as a potential biomarker for patients with triple-negative breast cancer treated with immune checkpoint inhibitors. Oncol. Lett. 2020, 19, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Atchley, D.P.; Albarracin, C.T.; Lopez, A.; Valero, V.; Amos, C.I.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Arun, B.K. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J. Clin. Oncol. 2008, 26, 4282–4288. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Hartman, A.R.; Kaldate, R.R.; Sailer, L.M.; Painter, L.; Grier, C.E.; Endsley, R.R.; Griffin, M.; Hamilton, S.A.; Frye, C.A.; Silberman, M.A.; et al. Prevalence of BRCA mutations in an unselected population of triple-negative breast cancer. Cancer 2012, 118, 2787–2795. [Google Scholar] [CrossRef]
- von Wahlde, M.K.; Timms, K.M.; Chagpar, A.; Wali, V.B.; Jiang, T.; Bossuyt, V.; Saglam, O.; Reid, J.; Gutin, A.; Neff, C.; et al. Intratumor Heterogeneity of Homologous Recombination Deficiency in Primary Breast Cancer. Clin. Cancer Res. 2017, 23, 1193–1199. [Google Scholar] [CrossRef]
- Papadimitriou, M.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.; Wesseling, J.; Vd Vijver, M.J.; de Vries, E.G.; van Tinteren, H.; Jonkers, J.; Hauptmann, M.; et al. Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014, 16, R47. [Google Scholar] [CrossRef]
- Grigoriadis, A.; Mackay, A.; Reis-Filho, J.S.; Steele, D.; Iseli, C.; Stevenson, B.J.; Jongeneel, C.V.; Valgeirsson, H.; Fenwick, K.; Iravani, M.; et al. Establishment of the epithelial-specific transcriptome of normal and malignant human breast cells based on MPSS and array expression data. Breast Cancer Res. 2006, 8, R56. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tarin, D. Gene expression profiling of human lymph node metastases and matched primary breast carcinomas: Clinical implications. Mol. Oncol. 2007, 1, 172–180. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | Recurrence (Lymph Node or Other) | Recurrence-Free | p-Value |
|---|---|---|---|
| Patient Age, n (%) | |||
| 20–29 | 0 (0.00) | 1 (6.70) | 0.877 |
| 30–39 | 2 (18.18) | 1 (6.70) | |
| 40–49 | 2 (18.18) | 3 (20.00) | |
| 50–59 | 4 (36.36) | 6 (40.00) | |
| 60–69 | 2 (18.18) | 1 (6.70) | |
| 70+ | 1 (9.09) | 3 (20.00) | |
| Tumor Grade, n (%) | |||
| I | 0 (0.00) | 0 (0.00) | 0.492 |
| II | 0 (0.00) | 2 (13.33) | |
| III | 11 (100.00) | 13 (86.67) | |
| Histological Type, n (%) | |||
| NST (Ductal) | 8 (72.72) | 13 (86.67) | 0.521 |
| (With Secretory Differentiation) | 1 (9.09) | 0 (0.00) | |
| Apocrine | 1 (9.09) | 2 (13.33) | |
| Metaplastic | 1 (9.09) | 0 (0.00) | |
| Survival Status, n(%) | |||
| Alive | 6 (54.54) | 11 (73.33) | 0.418 |
| Dead | 5 (45.45) | 4 (26.67) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, J.; Chandrashekar, D.S.; Varga, Z.; Sobottka, B.; Janssen, E.; Gandhi, K.; Kowalski, J.; Kiraz, U.; Varambally, S.; Aneja, R. Whole-Exome Sequencing Reveals High Mutational Concordance between Primary and Matched Recurrent Triple-Negative Breast Cancers. Genes 2023, 14, 1690. https://doi.org/10.3390/genes14091690
Kaur J, Chandrashekar DS, Varga Z, Sobottka B, Janssen E, Gandhi K, Kowalski J, Kiraz U, Varambally S, Aneja R. Whole-Exome Sequencing Reveals High Mutational Concordance between Primary and Matched Recurrent Triple-Negative Breast Cancers. Genes. 2023; 14(9):1690. https://doi.org/10.3390/genes14091690
Chicago/Turabian StyleKaur, Jaspreet, Darshan S. Chandrashekar, Zsuzsanna Varga, Bettina Sobottka, Emiel Janssen, Khanjan Gandhi, Jeanne Kowalski, Umay Kiraz, Sooryanarayana Varambally, and Ritu Aneja. 2023. "Whole-Exome Sequencing Reveals High Mutational Concordance between Primary and Matched Recurrent Triple-Negative Breast Cancers" Genes 14, no. 9: 1690. https://doi.org/10.3390/genes14091690