

The Mutational Spectrum of Pre- and Post-Neoadjuvant Chemotherapy Triple-Negative Breast Cancers
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
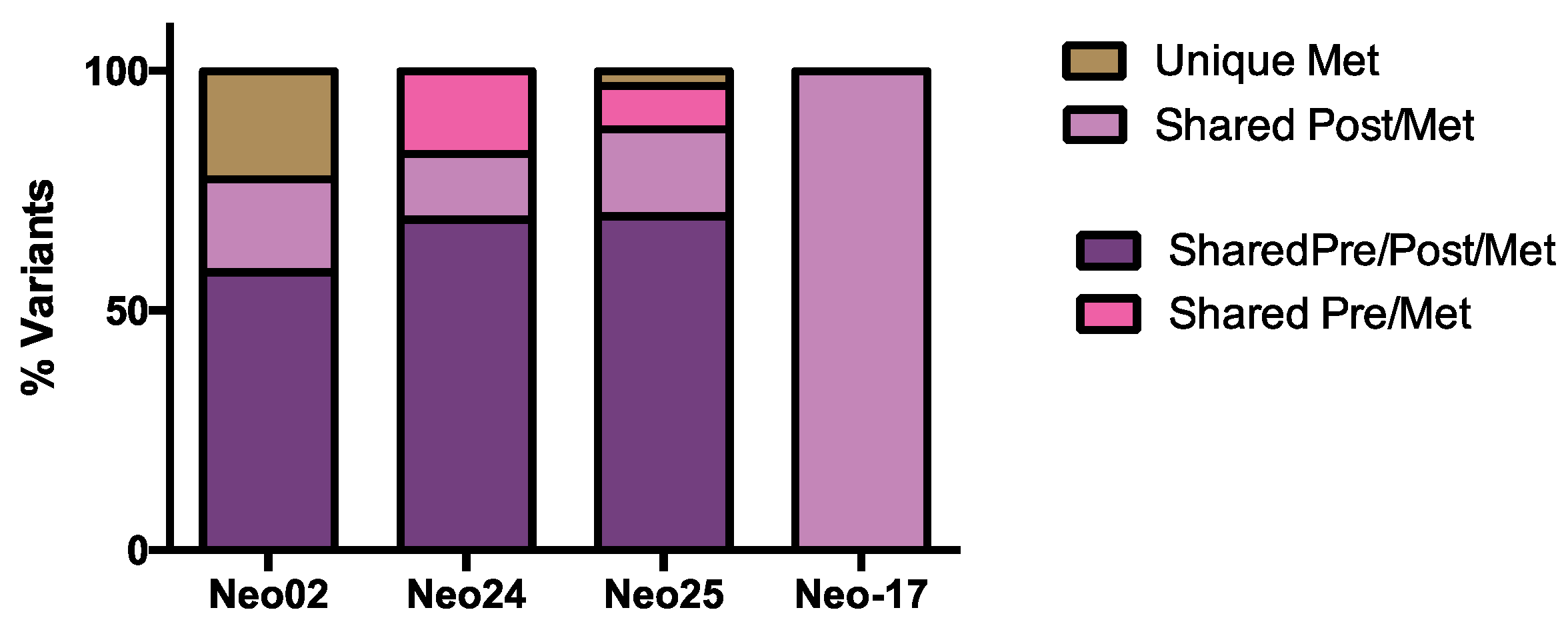{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Accrual
2.2. Sample Processing
2.3. Whole Exome Sequencing and Somatic Variant Annotation
3. Results
3.1. Clinical Results
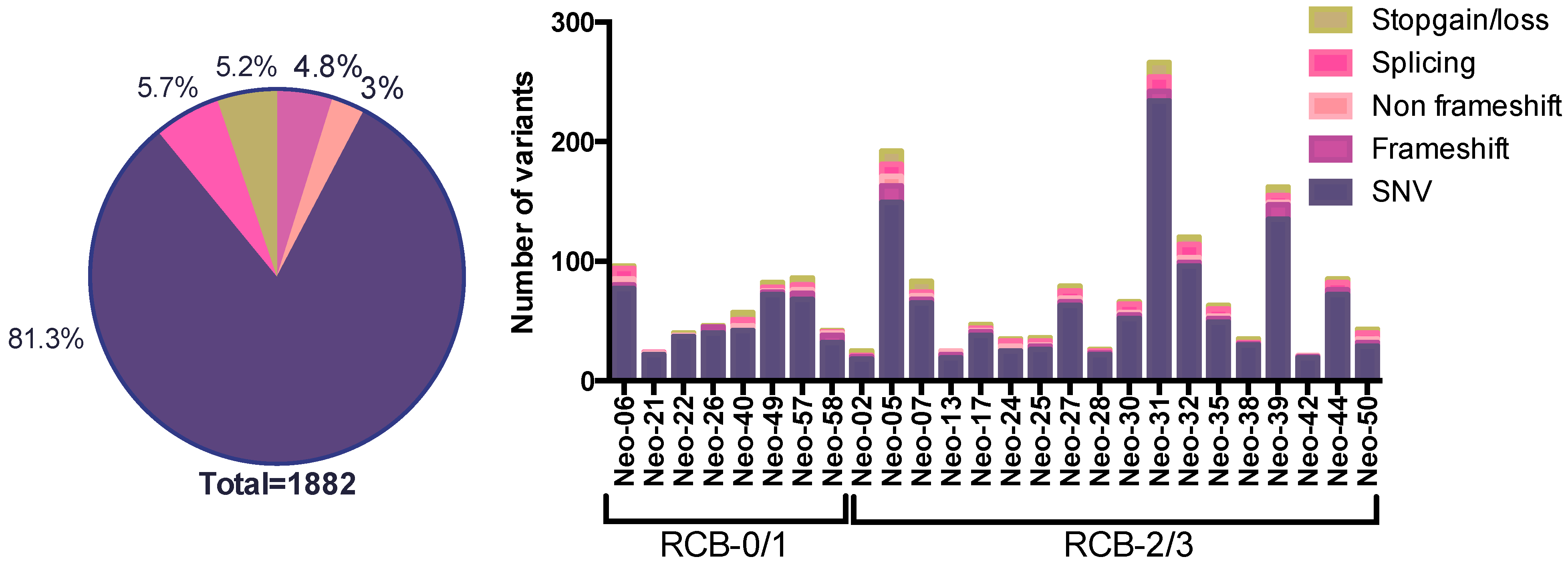
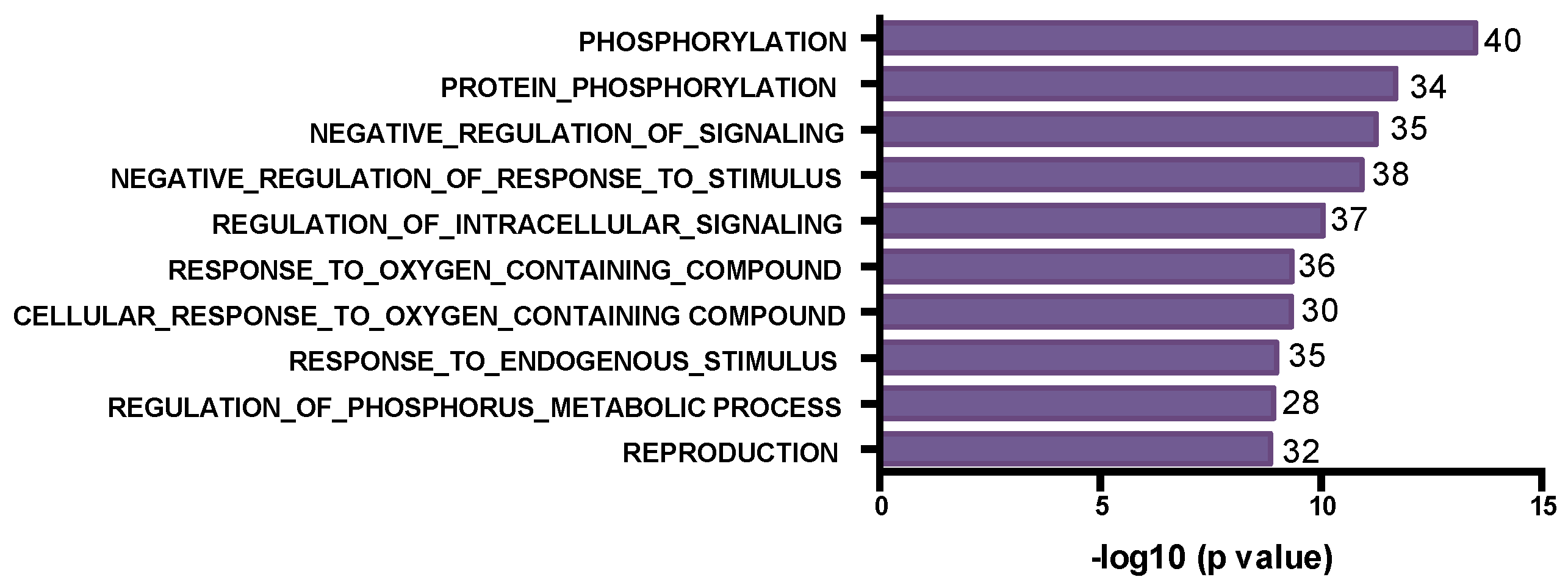
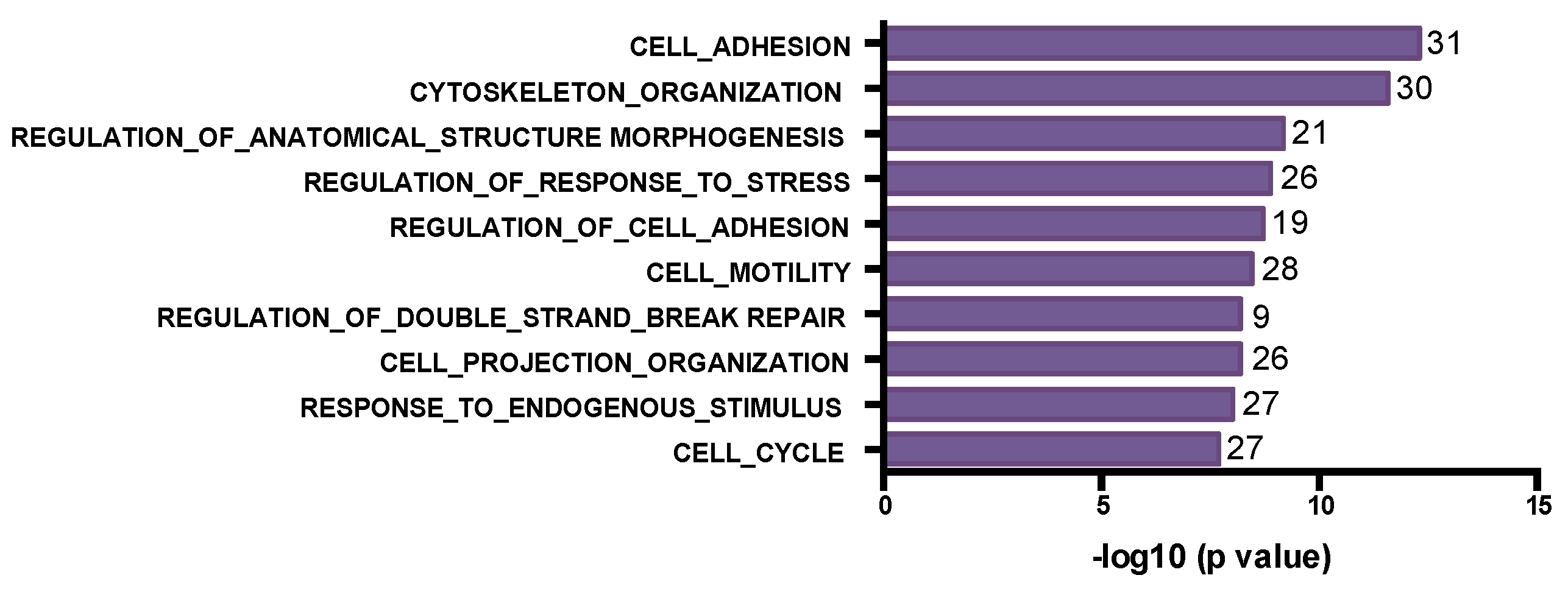
3.2. The SNV Mutational Spectrum of TNBCs
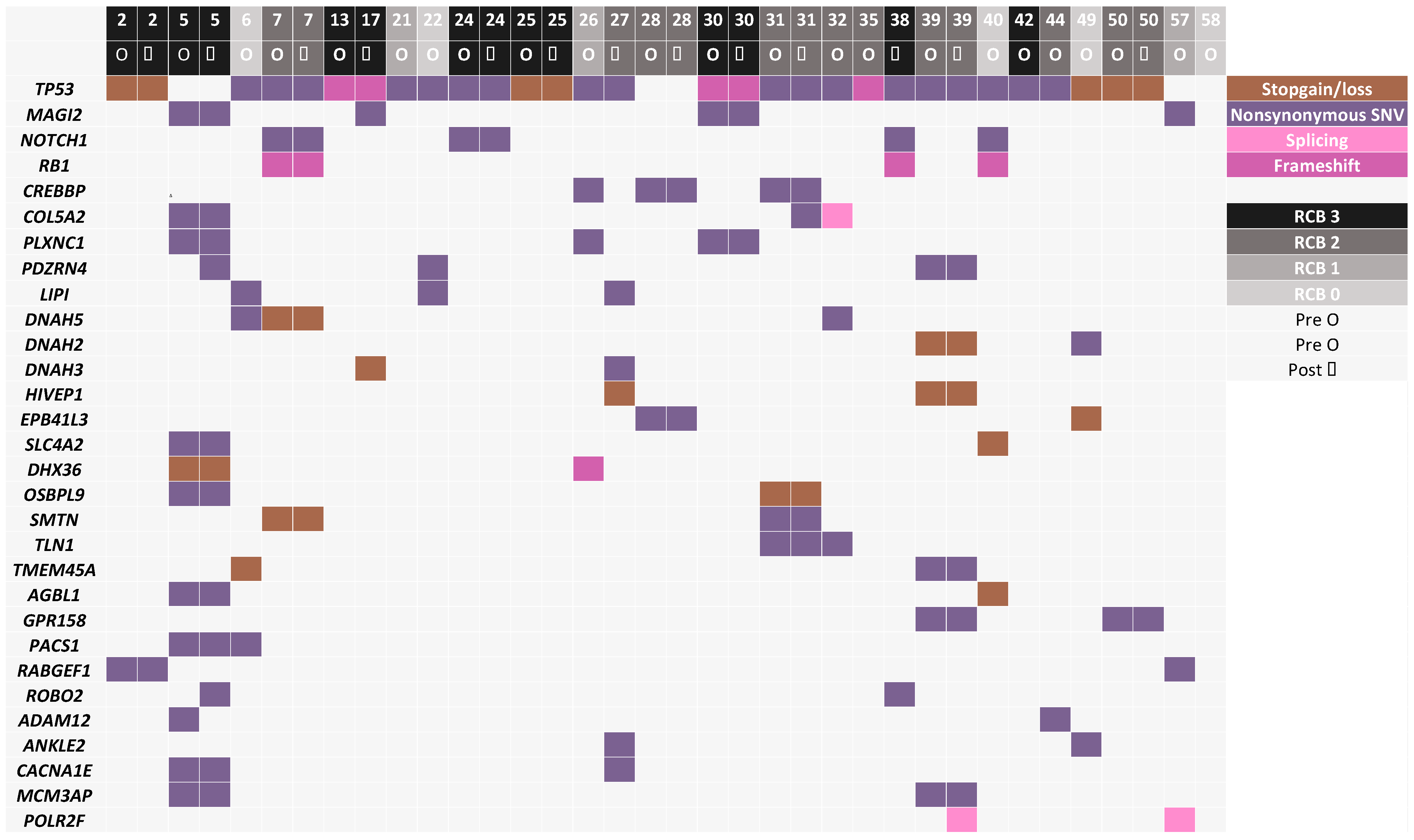
3.3. Mutated Genes Associated with Response to Chemotherapy
3.4. Post-Chemotherapy Genomic Landscape
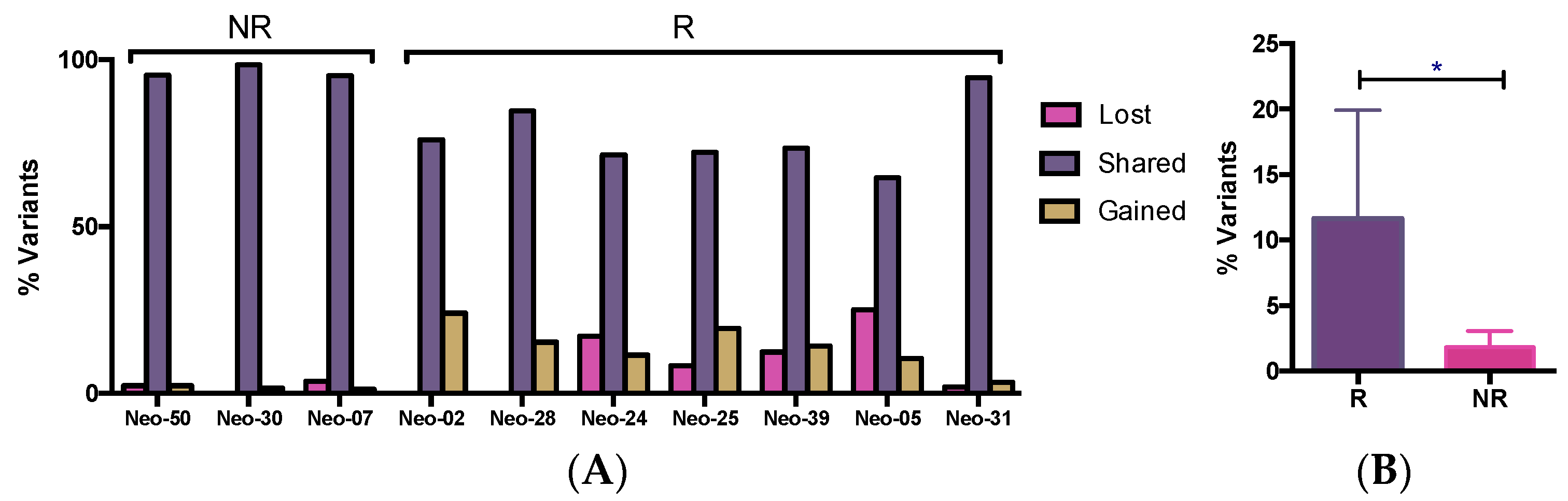
3.5. WES Analysis of Matched Pre/Post-Tumors
3.6. The Dynamics of Tumor Progression
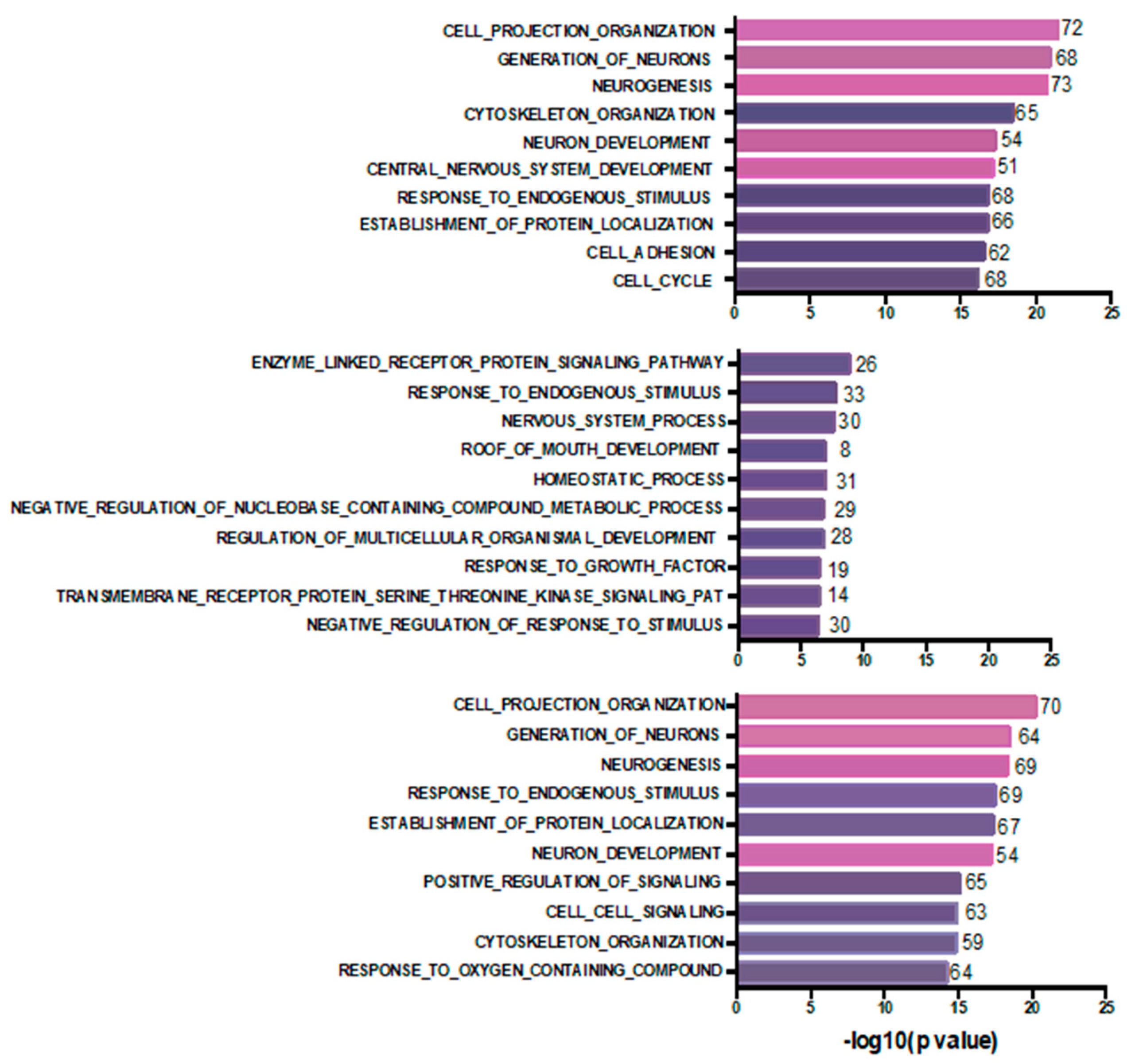
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Mandapati, A.; Lukong, K.E. Triple negative breast cancer: Approved treatment options and their mechanisms of action. J. Cancer Res. Clin. Oncol. 2022, 149, 3701–3719. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The Triple Negative Paradox: Primary Tumor Chemosensitivity of Breast Cancer Subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Denkert, C.; von Minckwitz, G. Neoadjuvant treatment of breast cancer—Clinical and research perspective. Breast 2015, 24, S73–S77. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. KEYNOTE-522 Investigators. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Bonadio, R.C.; Tarantino, P.; Testa, L.; Punie, K.; Pernas, S.; Barrios, C.; Curigliano, G.; Tolaney, S.M.; Barroso-Sousa, R. Management of patients with early-stage triple-negative breast cancer following pembrolizumab-based neoadjuvant therapy: What are the evidences? Cancer Treat. Rev. 2022, 110, 102459. [Google Scholar] [CrossRef]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles’ heel? Cancer Lett. 2020, 497, 100–111. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Bonnefoi, H.; Piccart, M.; Bogaerts, J.; Mauriac, L.; Fumoleau, P.; Brain, E.; Petit, T.; Rouanet, P.; Jassem, J.; Blot, E.; et al. TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1-00): A randomised phase 3 trial. Lancet Oncol. 2011, 12, 527–539. [Google Scholar] [CrossRef]
- Fernández-Cuesta, L.; Oakman, C.; Falagan-Lotsch, P.; Smoth, K.-S.; Quinaux, E.; Buyse, M.; Dolci, M.S.; De Azambuja, E.; Hainaut, P.; Dell’Orto, P.; et al. Prognostic and predictive value of TP53mutations in node-positive breast cancer patients treated with anthracycline- or anthracycline/taxane-based adjuvant therapy: Results from the BIG 02-98 phase III trial. Breast Cancer Res. 2012, 14, R70. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Puhalla, S.; Domchek, S.M.; Friedlander, M.; Kaufman, B.; Robson, M.; Telli, M.L.; Diéras, V.; Han, H.S.; E Garber, J.; et al. A randomized Phase II study of veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in 1/2 metastatic breast cancer: Design and rationale. Futur. Oncol. 2017, 13, 307–320. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Futur. Oncol. 2017, 13, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Mahecha, A.; Lafleur, J.; Pelmus, M.; Seguin, C.; Lan, C.; Discepola, F.; Kovacina, B.; Christodoulopoulos, R.; Salvucci, O.; Mihalcioiu, C.; et al. The identification of challenges in tissue collection for biomarker studies: The Q-CROC-03 neoadjuvant breast cancer translational trial experience. Mod. Pathol. 2017, 30, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Cavallone, L.; Aguilar-Mahecha, A.; Lafleur, J.; Brousse, S.; Aldamry, M.; Roseshter, T.; Lan, C.; Alirezaie, N.; Bareke, E.; Majewski, J.; et al. Prognostic and predictive value of circulating tumor DNA during neoadjuvant chemotherapy for triple negative breast cancer. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Ng, C.K.; Lim, R.S.; Jiang, T.; Kumar, S.; Li, X.; Wali, V.B.; Piscuoglio, S.; Gerstein, M.B.; Chagpar, A.B.; et al. Reliability of Whole-Exome Sequencing for Assessing Intratumor Genetic Heterogeneity. Cell Rep. 2018, 25, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O‘Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Symmans, W.F.; Peintinger, F.; Hatzis, C.; Rajan, R.; Kuerer, H.; Valero, V.; Assad, L.; Poniecka, A.; Hennessy, B.; Green, M.; et al. Measurement of Residual Breast Cancer Burden to Predict Survival After Neoadjuvant Chemotherapy. J. Clin. Oncol. 2007, 25, 4414–4422. [Google Scholar] [CrossRef]
- Symmans, W.F.; Wei, C.; Gould, R.; Yu, X.; Zhang, Y.; Liu, M.; Walls, A.; Bousamra, A.; Ramineni, M.; Sinn, B.; et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J. Clin. Oncol. 2017, 35, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular Profiling of the Residual Disease of Triple-Negative Breast Cancers after Neoadjuvant Chemotherapy Identifies Actionable Therapeutic Targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef]
- Di Cosimo, S.; Appierto, V.; Silvestri, M.; Pruneri, G.; Vingiani, A.; Perrone, F.; Busico, A.; Folli, S.; Scaperrotta, G.; de Braud, F.G.; et al. Targeted-Gene Sequencing to Catch Triple Negative Breast Cancer Heterogeneity before and after Neoadjuvant Chemotherapy. Cancers 2019, 11, 1753. [Google Scholar] [CrossRef]
- Hancock, B.A.; Chen, Y.-H.; Solzak, J.P.; Ahmad, M.N.; Wedge, D.C.; Brinza, D.; Scafe, C.; Veitch, J.; Gottimukkala, R.; Short, W.; et al. Profiling molecular regulators of recurrence in chemorefractory triple-negative breast cancers. 2019, 21, 1–15. Breast Cancer Res. 2019, 21, 87. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Powles, R.L.; Wali, V.B.; Li, X.; Barlow, W.E.; Nahleh, Z.; Thompson, A.M.; Godwin, A.K.; Hatzis, C.; Pusztai, L. Analysis of Pre- and Posttreatment Tissues from the SWOG S0800 Trial Reveals an Effect of Neoadjuvant Chemotherapy on the Breast Cancer Genome. Clin. Cancer Res. 2020, 26, 1977–1984. [Google Scholar] [CrossRef]
- Goetz, M.P.; Kalari, K.R.; Suman, V.J.; Moyer, A.M.; Yu, J.; Visscher, D.W.; Dockter, T.J.; Vedell, P.T.; Sinnwell, J.P.; Tang, X.; et al. Tumor Sequencing and Patient-Derived Xenografts in the Neoadjuvant Treatment of Breast Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djw306. [Google Scholar] [CrossRef] [PubMed]
- Sirois, I.; Aguilar-Mahecha, A.; Lafleur, J.; Fowler, E.; Vu, V.; Scriver, M.; Buchanan, M.; Chabot, C.; Ramanathan, A.; Balachandran, B.; et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol. Cancer Res. 2019, 17, 2492–2507. [Google Scholar] [CrossRef] [PubMed]
- Verret, B.; Sourisseau, T.; Stefanovska, B.; Mosele, F.; Tran-Dien, A.; André, F. The Influence of Cancer Molecular Subtypes and Treatment on the Mutation Spectrum in Metastatic Breast Cancers. Cancer Res. 2020, 80, 3062–3069. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Chandrashekar, D.S.; Varga, Z.; Sobottka, B.; Janssen, E.; Gandhi, K.; Kowalski, J.; Kiraz, U.; Varambally, S.; Aneja, R. Whole-Exome Sequencing Reveals High Mutational Concordance between Primary and Matched Recurrent Triple-Negative Breast Cancers. Genes 2023, 14, 1690. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Mahecha, A.; Alirezaie, N.; Lafleur, J.; Bareke, E.; Przybytkowski, E.; Lan, C.; Cavallone, L.; Salem, M.; Pelmus, M.; Aleynikova, O.; et al. The Mutational Spectrum of Pre- and Post-Neoadjuvant Chemotherapy Triple-Negative Breast Cancers. Genes 2024, 15, 27. https://doi.org/10.3390/genes15010027
Aguilar-Mahecha A, Alirezaie N, Lafleur J, Bareke E, Przybytkowski E, Lan C, Cavallone L, Salem M, Pelmus M, Aleynikova O, et al. The Mutational Spectrum of Pre- and Post-Neoadjuvant Chemotherapy Triple-Negative Breast Cancers. Genes. 2024; 15(1):27. https://doi.org/10.3390/genes15010027
Chicago/Turabian StyleAguilar-Mahecha, Adriana, Najmeh Alirezaie, Josiane Lafleur, Eric Bareke, Ewa Przybytkowski, Cathy Lan, Luca Cavallone, Myriam Salem, Manuela Pelmus, Olga Aleynikova, and et al. 2024. "The Mutational Spectrum of Pre- and Post-Neoadjuvant Chemotherapy Triple-Negative Breast Cancers" Genes 15, no. 1: 27. https://doi.org/10.3390/genes15010027