
Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. High-Molecular Weight DNA Isolation and Labeling
2.3. Optical Genome Mapping and Rare Variant Pipeline
3. Results
3.1. Patient Characteristics
3.2. Optical Genome Mapping Results
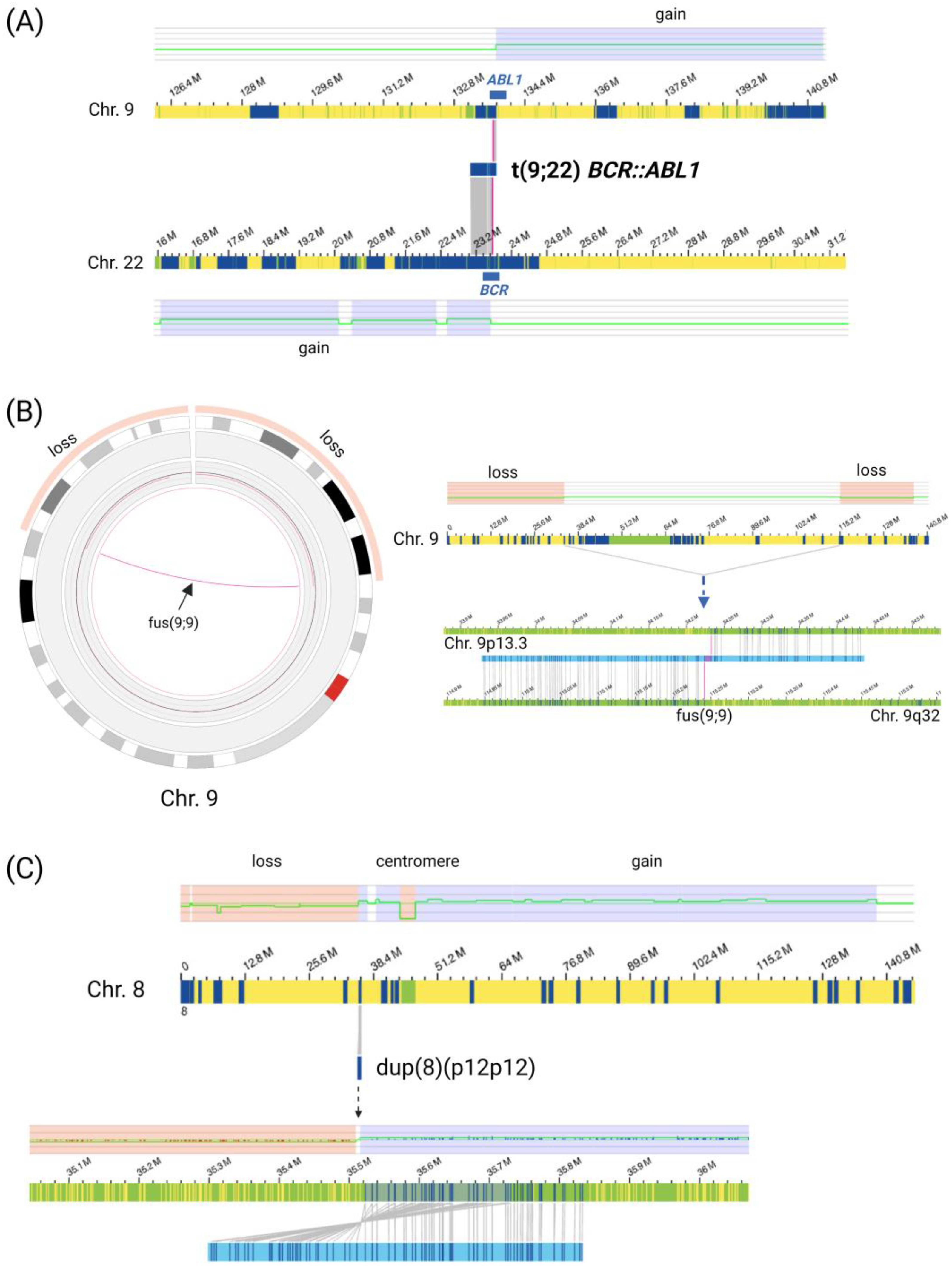
3.2.1. Gross Structural Rearrangements and Aneuploidies
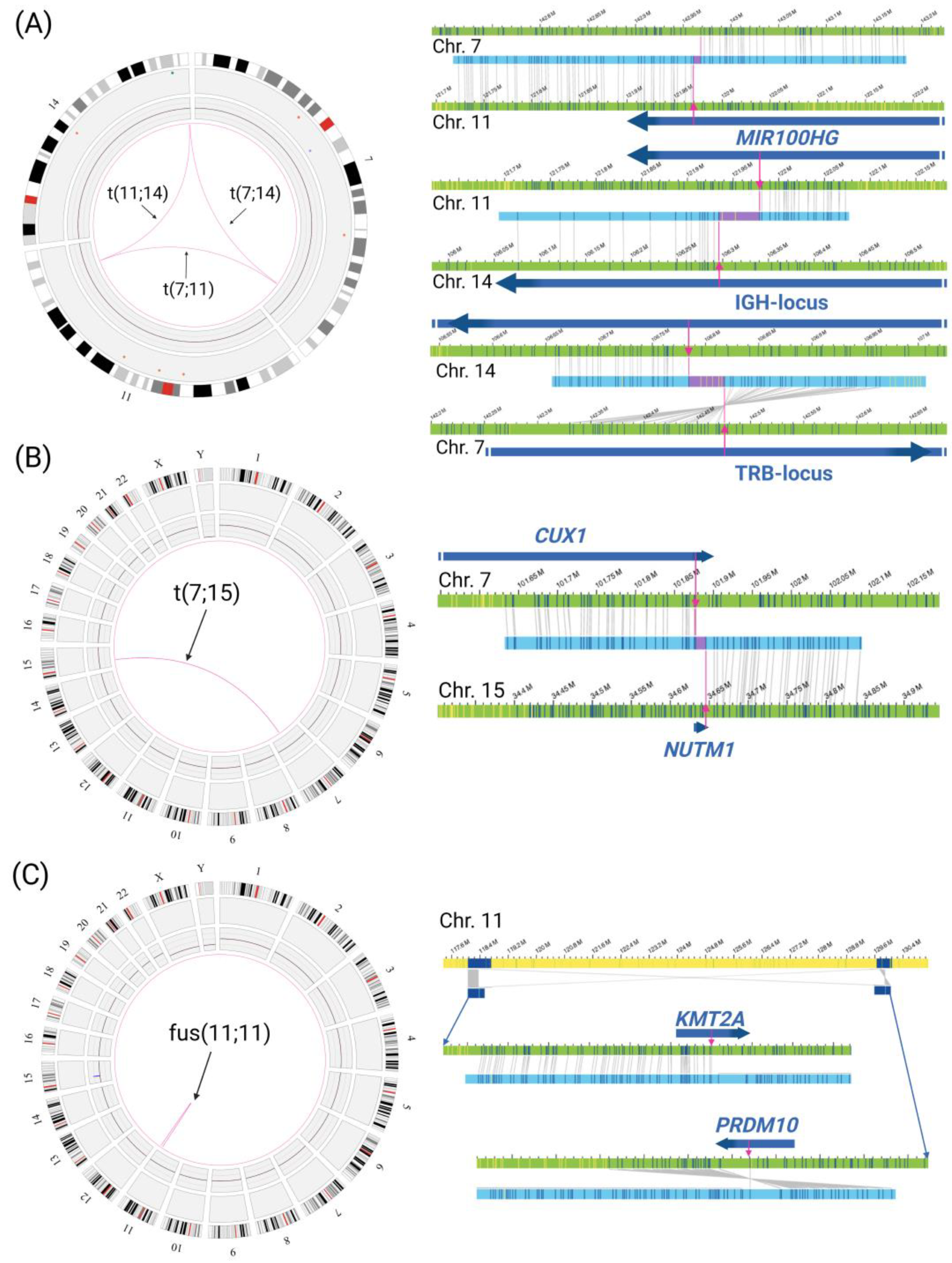
3.2.2. Gene Fusions
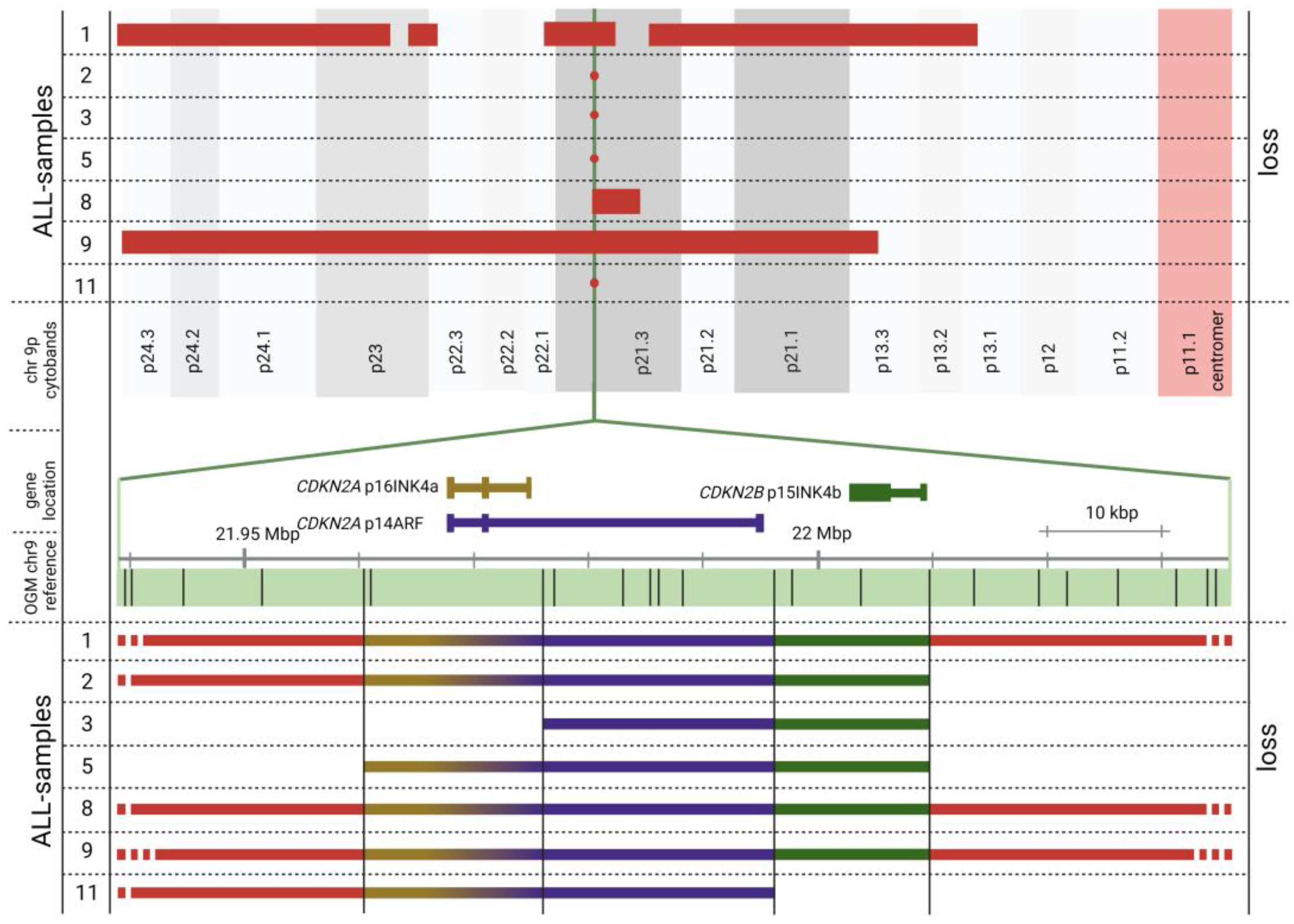
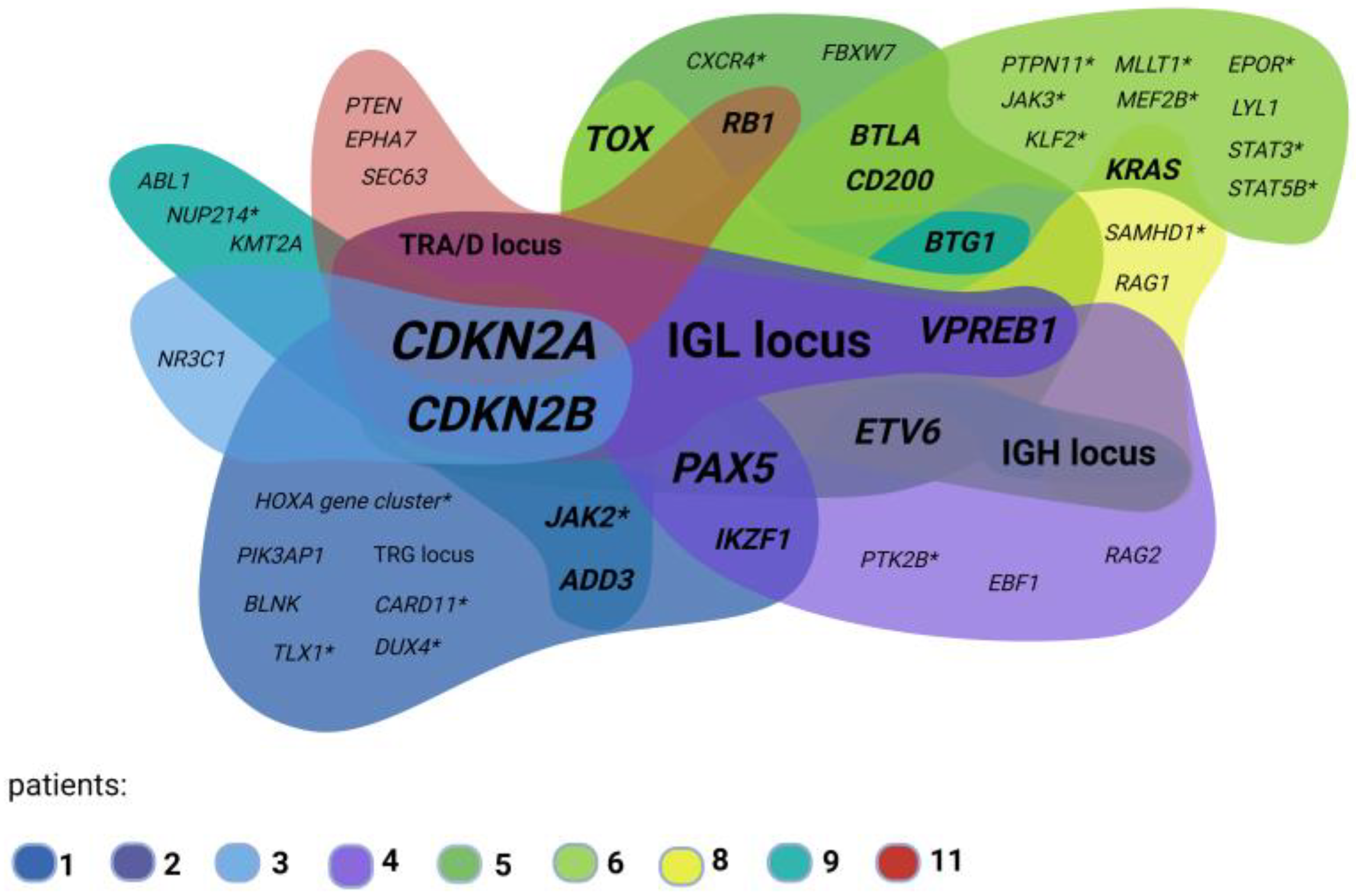
3.2.3. OGM Confirms Recurrent ALL Copy Number Alterations
4. Discussion
4.1. OGM Can Detect Rare Gross Structural Rearrangements
4.2. Rare Gene Fusions
4.3. CDKN2A/2B Loss
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. Adult Acute Lymphoblastic Leukemia Treatment (PDQ®)–Health Professional Version. Available online: https://www.cancer.gov/types/leukemia/hp/adult-all-treatment-pdq (accessed on 20 September 2022).
- Leukemia and Lymphoma Society. ALL Subtypes|Leukemia and Lymphoma Society. Available online: https://www.lls.org/leukemia/acute-lymphoblastic-leukemia/diagnosis/all-subtypes (accessed on 20 September 2022).
- Gökbuget, N.; Baldus, C.; Brüggemann, M.; Hauswirth, A.W.; Schanz, U.; Machherndl-Spandl, S.; Stelljes, M.; Topp, M. Akute lymphatische Leukämie (ALL): Onkopedia Leitlinien. Available online: https://www.onkopedia.com/de/onkopedia/guidelines/akute-lymphatische-leukaemie-all/@@guideline/html/index.html (accessed on 20 September 2022).
- Sallan, S.E. Myths and lessons from the adult/pediatric interface in acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2006, 2006, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Mroczek, A.; Zawitkowska, J.; Kowalczyk, J.; Lejman, M. Comprehensive Overview of Gene Rearrangements in Childhood T-Cell Acute Lymphoblastic Leukaemia. Int. J. Mol. Sci. 2021, 22, 808. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Pei, D.; Sandlund, J.T.; Jeha, S.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Shurtleff, S.; Onciu, M.; Cheng, C.; et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: Improved outcome with contemporary therapy. Leukemia 2012, 26, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G. Genetics and prognosis of ALL in children vs adults. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, B.; Baughn, L.B.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, A.; Akkari, Y.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. A National Multicenter Evaluation of the Clinical Utility of Optical Genome Mapping for Assessment of Genomic Aberrations in Acute Myeloid Leukemia. medRxiv 2020. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Gerding, W.M. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Baughn, L.B.; Akkari, Y.M.N.; Chartrand, S.; LaBarge, B.; Claxton, D.F.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical Genome Mapping in Acute Myeloid Leukemia: A Multicenter Evaluation. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef] [PubMed]
- Rack, K.; De Bie, J.; Ameye, G.; Gielen, O.; Demeyer, S.; Cools, J.; De Keersmaecker, K.; Vermeesch, J.R.; Maertens, J.; Segers, H.; et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am. J. Hematol. 2022, 97, 548–561. [Google Scholar] [CrossRef]
- Bionano Genomics. 30110-Bionano-Solve-Theory-of-Operation-Structural-Variant-Calling. Available online: https://bionanogenomics.com/wp-content/uploads/2018/04/30110-Bionano-Solve-Theory-of-Operation-Structural-Variant-Calling.pdf (accessed on 28 November 2022).
- Stancuioaica, M.C.; Popescu, B.; Popa, C.; Jardan, C.; Dragomir, M.; Coriu, D. Coexpression of p190 and p210 Fusion Transcripts in Ph+ Acute Lymphoblastic Leukemia. A Case Report. Clin. Lymphoma Myeloma Leuk. 2019, 19, S194–S195. [Google Scholar] [CrossRef]
- Chapiro, E.; Russell, L.J.; Struski, S.; Cavé, H.; Radford-Weiss, I.; Valle, V.D.; Lachenaud, J.; Brousset, P.; Bernard, O.A.; Harrison, C.J.; et al. A new recurrent translocation t(11;14)(q24;q32) involving IGH@ and miR-125b-1 in B-cell progenitor acute lymphoblastic leukemia. Leukemia 2010, 24, 1362–1364. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Shago, M.; Abla, O.; Hitzler, J.; Weitzman, S.; Abdelhaleem, M. Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr. Blood Cancer 2016, 63, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 2016, 48, 569–574. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ohki, K.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yaguchi, A.; Terada, K.; Saito, Y.; Yoshimi, A.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017, 102, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graux, C.; Cools, J.; Michaux, L.; Vandenberghe, P.; Hagemeijer, A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: From thymocyte to lymphoblast. Leukemia 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Türkmen, S.; Timmermann, B.; Bartels, G.; Gröger, D.; Meyer, C.; Schwartz, S.; Haferlach, C.; Rieder, H.; Gökbuget, N.; Hoelzer, D.; et al. Involvement of the MLL gene in adult T-lymphoblastic leukemia. Genes Chromosomes Cancer 2012, 51, 1114–1124. [Google Scholar] [CrossRef]
- Forgione, M.O.; McClure, B.J.; Eadie, L.N.; Yeung, D.T.; White, D.L. KMT2A rearranged acute lymphoblastic leukaemia: Unravelling the genomic complexity and heterogeneity of this high-risk disease. Cancer Lett. 2020, 469, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, B.; Rutherford, K.D.; Gundem, G.; McGovern, E.M.; Millard, N.E.; Arango Ossa, J.E.; Cheung, I.Y.; Gao, T.; Levine, M.F.; Zhang, Y.; et al. Bone Marrow Surveillance of Pediatric Cancer Survivors Identifies Clones that Predict Therapy-Related Leukemia. Clin. Cancer Res. 2022, 28, 1614–1627. [Google Scholar] [CrossRef]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. Hematology 2012, 2012, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Z.; Chase Crawford, J.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Schwab, C.J.; Chilton, L.; Morrison, H.; Jones, L.; Al-Shehhi, H.; Erhorn, A.; Russell, L.J.; Moorman, A.V.; Harrison, C.J. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: Association with cytogenetics and clinical features. Haematologica 2013, 98, 1081–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Gil, C.; Ribera, J.; Ribera, J.M.; Genescà, E. The Yin and Yang-Like Clinical Implications of the CDKN2A/ARF/CDKN2B Gene Cluster in Acute Lymphoblastic Leukemia. Genes 2021, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Roberts, K.G.; Gu, Z.; Shi, L.; Pounds, S.; Pei, D.; Cheng, C.; Dai, Y.; Devidas, M.; Qu, C.; et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 2022, 54, 1376–1389. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Downing, J.R. Genome-wide profiling of genetic alterations in acute lymphoblastic leukemia: Recent insights and future directions. Leukemia 2009, 23, 1209–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangum, D.S.; Downie, J.; Mason, C.C.; Jahromi, M.S.; Joshi, D.; Rodic, V.; Müschen, M.; Meeker, N.; Trede, N.; Frazer, J.K.; et al. VPREB1 deletions occur independent of lambda light chain rearrangement in childhood acute lymphoblastic leukemia. Leukemia 2014, 28, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Mangum, D.S.; Meyer, J.A.; Mason, C.C.; Shams, S.; Maese, L.D.; Gardiner, J.D.; Downie, J.M.; Pei, D.; Cheng, C.; Gleason, A.; et al. Association of Combined Focal 22q11.22 Deletion and IKZF1 Alterations with Outcomes in Childhood Acute Lymphoblastic Leukemia. JAMA Oncol. 2021, 7, 1521–1528. [Google Scholar] [CrossRef]
- Gocho, Y.; Kiyokawa, N.; Ichikawa, H.; Nakabayashi, K.; Osumi, T.; Ishibashi, T.; Ueno, H.; Terada, K.; Oboki, K.; Sakamoto, H.; et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia 2015, 29, 2445–2448. [Google Scholar] [CrossRef] [Green Version]
- Polityko, A.D.; Goncharova, E.; Shamgina, L.; Drozdovskaja, N.; Podleschuk, L.; Abramchik, E.; Jaroshevich, E.; Khurs, O.; Pisarik, I.; Pribushenya, O.; et al. Pallister-Killian syndrome: Rapid decrease of isochromosome 12p frequency during amniocyte subculturing. Conclusion for strategy of prenatal cytogenetic diagnostics. J. Histochem. Cytochem. 2005, 53, 361–364. [Google Scholar] [CrossRef]
- Boer, J.M.; Valsecchi, M.G.; Hormann, F.M.; Antić, Ž.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cavé, H.; Attarbaschi, A.; et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 2021, 35, 2978–2982. [Google Scholar] [CrossRef]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [Green Version]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; Den Boer, M.L.; et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 2019, 104, e455–e459. [Google Scholar] [CrossRef] [Green Version]
- Thorsteinsdottir, U.; Sauvageau, G.; Hough, M.R.; Dragowska, W.; Lansdorp, P.M.; Lawrence, H.J.; Largman, C.; Humphries, R.K. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol. Cell. Biol. 1997, 17, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Yin, C.; Jiang, L.; Xu, D.; Zheng, Z.; Wang, Z.; Wang, C.; Zhou, H.; Jiang, X.; Liu, Q.; et al. Amyloid precursor protein has clinical and prognostic significance in AML1-ETO-positive acute myeloid leukemia. Oncol. Lett. 2018, 15, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.V. Potential molecular link between the β-amyloid precursor protein (APP) and hypoxanthine-guanine phosphoribosyltransferase (HGprt) enzyme in Lesch-Nyhan disease and cancer. AIMS Neurosci. 2021, 8, 548–557. [Google Scholar] [CrossRef]
- Wang, R.; Xu, P.; Chang, L.-L.; Zhang, S.-Z.; Zhu, H.-H. Targeted therapy in NPM1-mutated AML: Knowns and unknowns. Front. Oncol. 2022, 12, 972606. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Song, Y.; Gong, X.; Wang, J.; Li, Q.; Liu, K.; Feng, Y.; Hao, Q.; Li, Y.; Wei, H.; et al. Gene Deletions and Prognostic Values in B-Linage Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11, 677034. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, H.; Raum, K.; Markovic, S.; Nowak, V.; Fey, S.; Obländer, J.; Pressler, J.; Böhm, V.; Brüggemann, M.; Wunderle, L.; et al. Genomic CDKN2A/2B deletions in adult Ph+ ALL are adverse despite allogeneic stem cell transplantation. Blood 2018, 131, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, Y.; Li, X.; Zhou, X.; Cao, R.; Li, H.; Li, L.; Lu, Z.; Huang, J.; Fan, Z.; et al. Correlation between deletion of the CDKN2 gene and tyrosine kinase inhibitor resistance in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bride, K.L.; Hu, H.; Tikhonova, A.; Fuller, T.J.; Vincent, T.L.; Shraim, R.; Li, M.M.; Carroll, W.L.; Raetz, E.A.; Aifantis, I.; et al. Rational drug combinations with CDK4/6 inhibitors in acute lymphoblastic leukemia. Haematologica 2022, 107, 1746–1757. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age | Sex | Sample Type | Diagnosis | Initial Diagnosis | GMALL Risk | WHO 2022 Classification | Disease Stage | Blasts 3 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 18 | m | BM 1 | Common B-ALL | 07/21 | High | B-ALL with BCR::ABL1 | Initial | >90% |
| 2 | 60 | f | BM 1 | Common B-ALL | 02/22 | High | B-ALL with BCR::ABL1 | Initial | >90% |
| 3 | 43 | f | BM 1 | Common B-ALL | 08/21 | High | B-ALL with BCR::ABL1 | Progress | >90% |
| 4 | 62 | f | BM 1 | Common B-ALL | 05/22 | High | B-ALL with BCR::ABL1 | Initial | 80% |
| 5 | 34 | m | BM 1 | Common B-ALL | 04/21 | High | B-ALL, NOS 4 | Initial | 85% |
| 6 | 18 | m | BM 1 | Pro B-ALL | 05/21 | High | B-ALL, NOS 4 | Initial | 40% |
| 7 | 35 | f | PB 2 | Pre B-ALL | 04/21 | High | B-ALL, NOS 4 | Initial | >90% |
| 8 | 58 | m | BM 1 | Pro B-ALL | 03/21 | High | B-ALL, NOS 4 | Refractory | 80% |
| 9 | 26 | m | BM 1 | Common B-ALL | 01/22 | Standard | B-ALL, NOS 4 | Initial | >90% |
| 10 | 22 | m | BM 1 | Thymic T-ALL | 06/19 | Standard | T-ALL, NOS 4 | Progress | <5% |
| 11 | 33 | f | BM 1 | Thymic T-ALL | 08/22 | Standard | T-ALL, NOS 4 | Initial | >90% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieler, L.-M.; Nilius-Eliliwi, V.; Schroers, R.; Vangala, D.B.; Nguyen, H.P.; Gerding, W.M. Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL. Genes 2023, 14, 686. https://doi.org/10.3390/genes14030686
Vieler L-M, Nilius-Eliliwi V, Schroers R, Vangala DB, Nguyen HP, Gerding WM. Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL. Genes. 2023; 14(3):686. https://doi.org/10.3390/genes14030686
Chicago/Turabian StyleVieler, Lisa-Marie, Verena Nilius-Eliliwi, Roland Schroers, Deepak Ben Vangala, Huu Phuc Nguyen, and Wanda Maria Gerding. 2023. "Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL" Genes 14, no. 3: 686. https://doi.org/10.3390/genes14030686