
Recommendations for Interpreting and Reporting Silent Carrier and Disease-Modifying Variants in SMA Testing Workflows
 , , , and
, , , and
Abstract
:1. Spinal Muscular Atrophy Disease Etiology
2. SMA Diagnostic and Carrier Screening Testing
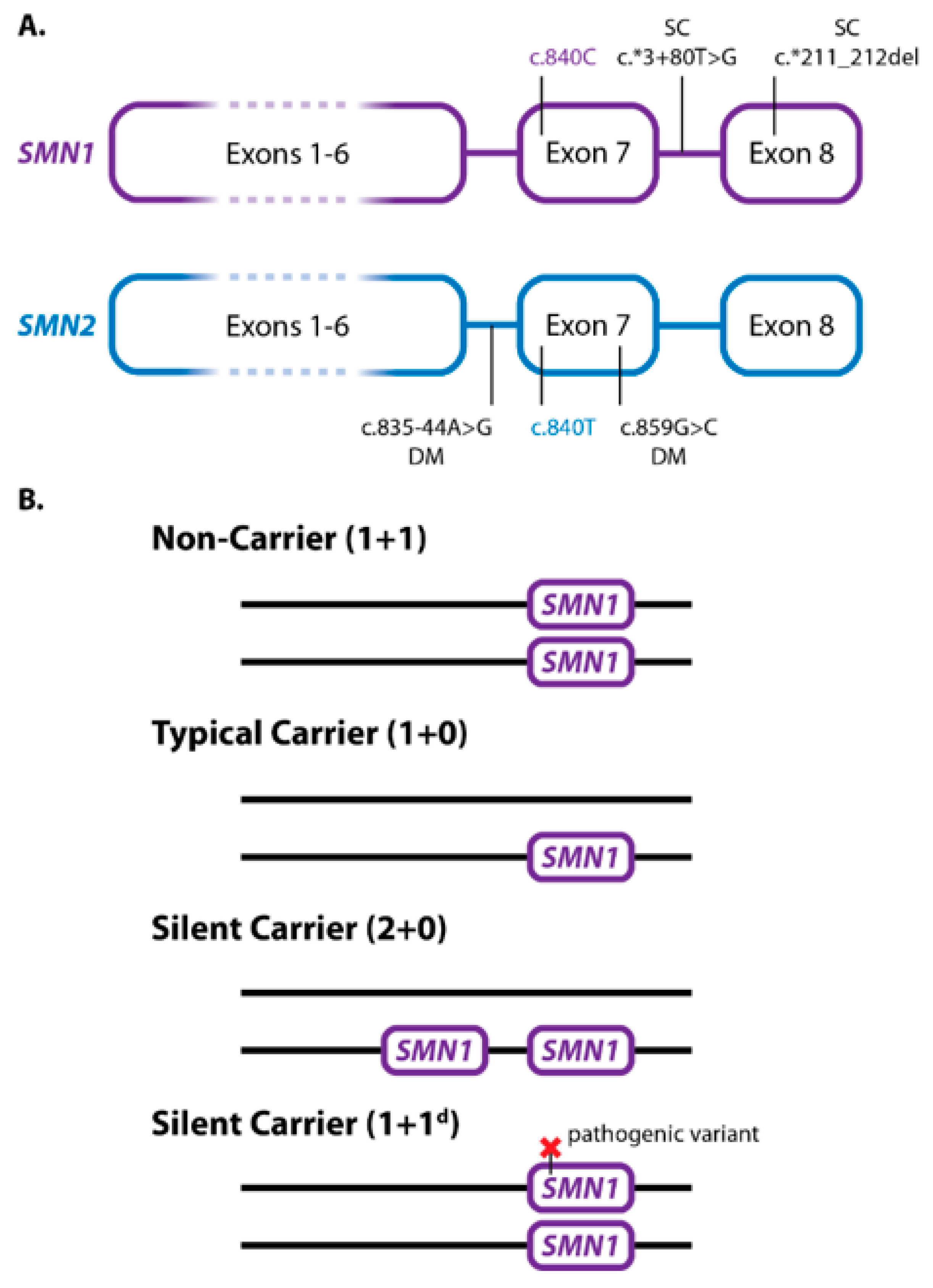
3. SMA Carrier Genotypes, Testing, and Reporting
4. Disease Prognosis Genotypes, Testing, and Reporting
5. Newborn Screening for SMA
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stabley, D.L.; Harris, A.W.; Holbrook, J.; Chubbs, N.J.; Lozo, K.W.; Crawford, T.O.; Swoboda, K.J.; Funanage, V.L.; Wang, W.; Mackenzie, W.; et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol. Genet. Genom. Med. 2015, 3, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Tizzano, E.F. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017, 24, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, E.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar]
- Wadman, R.I.; Stam, M.; Gijzen, M.; Lemmink, H.H.; Snoeck, I.N.; Wijngaarde, C.A.; Braun, K.P.J.; Schoenmakers, M.A.C.G.; van den Berg, L.H.; Dooijes, D.; et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0–4. J. Neurol. Neurosurg. Psychiatry 2017, 88, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Alías, L.; Bernal, S.; Fuentes-Prior, P.; Barceló, M.J.; Also, E.; Hernandez, R.M.; Rodríguez-Alvarez, F.J.; Martín, Y.; Aller, E.; Grau, E.; et al. Mutation update of spinal muscular atrophy in Spain: Molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum. Genet. 2009, 125, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Prior, T.W.; Nagan, N.; Sugarman, E.A.; Batish, S.D.; Braastad, C. Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 2011, 13, 686–694. [Google Scholar] [CrossRef]
- Hendrickson, B.C.; Donohoe, C.; Akmaev, V.R.; Sugarman, E.A.; Labrousse, P.; Boguslavskiy, L.; Flynn, K.; Rohlfs, E.M.; Walker, A.; Allitto, B.; et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 2009, 46, 641–644. [Google Scholar] [CrossRef]
- Luo, M.; Liu, L.; Peter, I.; Zhu, J.; Scott, S.A.; Zhao, G.; Eversley, C.; Kornreich, R.; Desnick, R.J.; Edelmann, L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet. Med. 2014, 16, 149–156. [Google Scholar] [CrossRef]
- Alías, L.; Bernal, S.; Calucho, M.; Martínez, E.; March, F.; Gallano, P.; Fuentes-Prior, P.; Abuli, A.; Serra-Juhe, C.; Tizzano, E.F. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur. J. Hum. Genet. 2018, 26, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Pérez, L.; Costa-Roger, M.; Leno-Colorado, J.; Bernal, S.; Alias, L.; Codina-Solà, M.; Martínez-Cruz, D.; Castiglioni, C.; Bertini, E.; Travaglini, L.; et al. Deep Molecular Characterization of Milder Spinal Muscular Atrophy Patients Carrying the c.859G>C Variant in SMN2. Int. J. Mol. Sci. 2022, 23, 8289. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Vezain, M.; Saugier-Veber, P.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frébourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef] [PubMed]
- Costa-Roger, M.; Blasco-Pérez, L.; Cuscó, I.; Tizzano, E.F. The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy. Int. J. Mol. Sci. 2021, 22, 9029. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Deng, S.; Chiriboga, C.A.; Kay, D.M.; Irumudomon, O.; Laureta, E.; Delfiner, L.; Treidler, S.O.; Anziska, Y.; Sakonju, A.; et al. Newborn Screening for Spinal Muscular Atrophy in New York State: Clinical Outcomes from the First 3 Years. Neurology 2022. [Google Scholar] [CrossRef]
- Cuscó, I.; Bernal, S.; Blasco-Pérez, L.; Calucho, M.; Alias, L.; Fuentes-Prior, P.; Tizzano, E.F. Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurol. Genet. 2020, 6, e530. [Google Scholar] [CrossRef]
- Chen, X.; Sanchis-Juan, A.; French, C.E.; Connell, A.J.; Delon, I.; Kingsbury, Z.; Chawla, A.; Halpern, A.L.; Taft, R.J.; Bentley, D.R.; et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 2020, 22, 945–953. [Google Scholar] [CrossRef]
- Blasco-Pérez, L.; Paramonov, I.; Leno, J.; Bernal, S.; Alías, L.; Fuentes-Prior, P.; Cuscó, I.; Tizziano, E.F. Beyond copy number: A new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Hum. Mutat. 2021, 42, 787–795. [Google Scholar] [CrossRef]
- Gambardella, A.; Mazzei, R.; Toscano, A.; Annesi, G.; Pasqua, A.; Annesi, F.; Quattrone, F.; Oliveri, R.I.; Valentino, P.; Bono, F.; et al. Spinal muscular atrophy due to an isolated deletion of exon 8 of the telomeric survival motor neuron gene. Ann. Neurol. 1998, 44, 836–839. [Google Scholar] [CrossRef]
- Scheffer, H.; Cobben, J.M.; Matthijs, G.; Wirth, B. Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur. J. Hum. Genet. 2001, 9, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niba, E.T.E.; Nishio, H.; Wijaya, Y.O.S.; Lai, P.S.; Tozawa, T.; Chiyonobu, T.; Yamadera, M.; Okamoto, K.; Awano, H.; Takeshima, Y.; et al. Clinical phenotypes of spinal muscular atrophy patients with hybrid SMN gene. Brain Dev. 2021, 43, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Cusco, I.; Barceló, M.; Del Rio, E.; Martín, Y.; Hernández-Chico, C.; Bussaglia, E.; Baiget, M.; Tizzano, E. Characterisation of SMN hybrid genes in Spanish SMA patients: De novo, homozygous and compound heterozygous cases. Hum. Genet. 2001, 108, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.N.; Larson, J.L.; Filipovic-Sadic, S.; Laosinchai-Wolf, W.; Huang, Y.-W.; Ko, T.-M.; Abbott, K.M.; Lemmink, H.H.; Toivonen, M.; Schleutker, J.; et al. Multisite Evaluation and Validation of a Sensitive Diagnostic and Screening System for Spinal Muscular Atrophy that Reports SMN1 and SMN2 Copy Number, along with Disease Modifier and Gene Duplication Variants. J. Mol. Diagn. 2021, 23, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, W.K.; Hamilton, D.; Kuhle, S. SMA carrier testing: A meta-analysis of differences in test performance by ethnic group. Prenat. Diagn. 2014, 34, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Yun, S.A.; Roh, E.Y.; Yoon, J.H.; Shin, S.; Ki, C.S. Carrier Frequency of Spinal Muscular Atrophy in a Large-scale Korean Population. Ann. Lab. Med. 2020, 40, 326–330. [Google Scholar] [CrossRef]
- Lopez-Lopez, D.; Loucera, C.; Carmona, R.; Aquino, V.; Salgado, J.; Pasalodos, S.; Miranda, M.; Alonso, Á.; Dopazo, J. SMN1 copy-number and sequence variant analysis from next-generation sequencing data. Hum. Mutat. 2020, 41, 2073–2077. [Google Scholar] [CrossRef]
- Prior, T.W.; Nagan, N.; Sugarman, E.A.; Batish, S.D.; Braastad, C. ADDENDUM: Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 2016, 18, 752, Addendum to Genet. Med. 2011, 13, 686–694. [Google Scholar] [CrossRef]
- Feng, Y.; Ge, X.; Meng, L.; Scull, J.; Li, J.; Tian, X.; Zhang, T.; Jin, W.; Cheng, H.; Wang, X.; et al. The next generation of population-based spinal muscular atrophy carrier screening: Comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet. Med. 2017, 19, 936–944. [Google Scholar] [CrossRef]
- Kaseniit, K.E.; Haque, I.S.; Goldberg, J.D.; Shulman, L.P.; Muzzey, D. Genetic ancestry analysis on >93,000 individuals undergoing expanded carrier screening reveals limitations of ethnicity-based medical guidelines. Genet. Med. 2020, 22, 1694–1702. [Google Scholar] [CrossRef]
- Davidson, D.; Kaseniit, K.; Haque, I. Duplication Tag SNP g.27134T>G should not be considered diagnostic of SMA carrier status. In Proceedings of the ACMG Annual Meeting, Phoenix, AZ, USA, 22–24 March 2017. [Google Scholar]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum. Genet. 2019, 138, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, H.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1, Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.H.; Sun, J.; Krainer, A.R.; Hua, Y.; Prior, T.W. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int. J. Mol. Sci. 2021, 22, 7896. [Google Scholar] [CrossRef]
- Keinath, M.C.; Prior, D.E.; Prior, T.W. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl. Clin. Genet. 2021, 14, 11–25. [Google Scholar] [CrossRef]
- Maretina, M.A.; Zheleznyakova, G.Y.; Lanko, K.M.; Egorova, A.A.; Baranov, V.S.; Kiselev, A.V. Molecular Factors Involved in Spinal Muscular Atrophy Pathways as Possible Disease-modifying Candidates. Curr. Genom. 2018, 19, 339–355. [Google Scholar] [CrossRef]
- Taylor, J.L.; Lee, F.K.; Yazdanpanah, G.K.; Staropoli, J.F.; Liu, M.; Carulli, J.P.; Sun, C.; Dobrowolski, S.F.; Hannon, W.H.; Vogt, R.F. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin. Chem. 2015, 61, 412–419. [Google Scholar] [CrossRef]
- Pane, M.; Donati, M.A.; Cutrona, C.; De Sanctis, R.; Pirinu, M.; Coratti, G.; Ricci, M.; Palermo, C.; Berti, B.; Leone, D.; et al. Neurological assessment of newborns with spinal muscular atrophy identified through neonatal screening. Eur. J. Pediatr. 2022, 181, 2821–2829. [Google Scholar] [CrossRef]
- Souza, P.V.S.; Pinto, W.B.V.D.R.; Ricarte, A.; Badia, B.D.M.L.; Seneor, D.D.; Teixeira, D.T.; Caetano, L.; Gonçalves, E.A.; Chieia, M.A.T.; Farias, I.B.; et al. Clinical and radiological profile of patients with spinal muscular atrophy type 4. Eur. J. Neurol. 2021, 28, 609–619. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Ethnicity | Carrier Frequency | 2 Copies SMN1 Exon 7 | 3 Copies SMN1 Exon 7 | 2 Copies SMN1, Variant Status “Negative” | 2 Copies SMN1, Variant Status “Positive” |
|---|---|---|---|---|---|
| Ashkenazi Jewish | 1:56 a | 1:514 a | 1:5899 a | 1:580 b | ~1 b |
| Asian | 1:50 a | 1:719 a | 1:5185 a | 1:779 c | 1:57 c |
| African American/Black | 1:71 a | 1:132 a | 1:6997 a | 1:375 d | 1:39 d |
| Caucasian/European | 1:45 a | 1:604 a | 1:4719 a | 1:814 c | 1:12 c |
| Hispanic | 1:83 a | 1:641 a | 1:7574 a | 1:906 d | 1:99 d |
| Spanish | 1:40 e | 1:781 e | Not Reported | 1:888 e | ~1 e |
| Israeli Jewish | 1:38 a | 1:450 a | 1:4004 a | Not Reported | Not Reported |
| Asian Indian | 1:50 a | 1:428 a | 1:5252 a | Not Reported | Not Reported |
| Iranian | 1:16 a | 1:96 a | 1:1604 a | Not Reported | Not Reported |
| Example Results | SMN1 Copies | c.*3+80T>G | c.*211_ *212del | Interpretation |
|---|---|---|---|---|
| Case 1 | 1 | Not indicated | Not indicated | Carrier The SMN1 copy number indicates a carrier of SMA. Genetic counseling is recommended and carrier testing should be made available to other at-risk family members. |
| Case 2 | 2 | Positive | Negative | Increased Carrier Risk The SMN1 copy number is two, ruling out a typical carrier genotype (1 + 0). However, the presence of one or more variants indicates an increased risk of being a silent carrier. The residual risk of SMA carrier status based on genotype alone is between 1:99 to ~1 depending on ethnicity. Ethnic-specific risk values based on these results are provided (see Table 1, last column). Parental testing should be considered to elucidate the presence of a silent carrier (2 + 0). Genetic counseling is recommended and carrier testing should be made available to other at-risk family members. |
| Case 3 | 2 | Positive | Positive | Increased Carrier Risk Refer to Case 2 for example language. |
| Case 4 | 2 | Negative | Negative | Reduced Carrier Risk The SMN1 copy number and variant status indicate reduced, but not eliminated, carrier risk. The residual risk of SMA carrier status based on genotype alone is between 1:375 and 1:906 depending on ethnicity. Ethnic-specific risk values based on these results are provided (see Table 1, 2nd to last column). Genetic counseling is recommended. |
| Case 5 | 3 | At genetic counselor’s discretion | At genetic counselor’s discretion | Reduced Carrier Risk The SMN1 copy number indicates a significantly reduced, but not eliminated, carrier risk. The residual risk of SMA carrier status based on genotype is low. Ethnic-specific risk values based on these results are provided (see Table 1, Column 4). Genetic counseling is recommended. |
| SMN2 Copy Number | c.859G>C Variant Status | Interpretation and Reporting Example |
|---|---|---|
| 1 | Negative | SMA (Type 0 probable) a Most individuals with SMA and one SMN2 copy present with Type 0 congenital disease. While the relationship between SMN2 copy number and disease outcomes is strongly correlated, it is not absolute, and individual exceptions do occur. Genetic counseling is recommended. |
| 2 | Negative | SMA (Type 1 probable) a Most individuals with SMA and two SMN2 copies present with Type 1 SMA. Refer to other examples with Negative c.859G>C Variant Status for example language. |
| 2 | Detected in one copy | SMA (Type 2/3 probable) b,c Whereas most individuals with SMA and two SMN2 copies present with Type 1 SMA, the presence of the c.859G>C variant in one SMN2 copy is associated with reduced severity consistent with SMA Type 2/3. Genetic counseling is recommended. |
| 2 | Detected in two copies | SMA (Type 3/4 probable) c,d Whereas most individuals with SMA and two SMN2 copies present with Type 1 SMA, the presence of the c.859G>C variant in two SMN2 copies is associated with reduced severity consistent with SMA Type 3/4. Genetic counseling is recommended. |
| 3 | Negative | SMA (Type 2/3 probable) a Refer to other examples with negative c.859G>C variant status for an example language. |
| 3 | Detected in one copy | SMA (Type 3 probable) c,e Whereas most individuals with SMA and three SMN2 copies present with Type 2/3 SMA, the presence of the c.859G>C variant in one SMN2 copy is associated with reduced severity consistent with SMA Type 3. Genetic counseling is recommended. |
| ≥4 | Negative | SMA (Type 3/4 probable) a Refer to other examples with negative c.859G>C variant status for example language. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milligan, J.N.; Blasco-Pérez, L.; Costa-Roger, M.; Codina-Solà, M.; Tizzano, E.F. Recommendations for Interpreting and Reporting Silent Carrier and Disease-Modifying Variants in SMA Testing Workflows. Genes 2022, 13, 1657. https://doi.org/10.3390/genes13091657
Milligan JN, Blasco-Pérez L, Costa-Roger M, Codina-Solà M, Tizzano EF. Recommendations for Interpreting and Reporting Silent Carrier and Disease-Modifying Variants in SMA Testing Workflows. Genes. 2022; 13(9):1657. https://doi.org/10.3390/genes13091657
Chicago/Turabian StyleMilligan, John N., Laura Blasco-Pérez, Mar Costa-Roger, Marta Codina-Solà, and Eduardo F. Tizzano. 2022. "Recommendations for Interpreting and Reporting Silent Carrier and Disease-Modifying Variants in SMA Testing Workflows" Genes 13, no. 9: 1657. https://doi.org/10.3390/genes13091657