
Nonsense-Mediated Decay Targeted RNA (ntRNA): Proposal of a ntRNA–miRNA–lncRNA Triple Regulatory Network Usable as Biomarker of Prognostic Risk in Patients with Kidney Cancer


Abstract
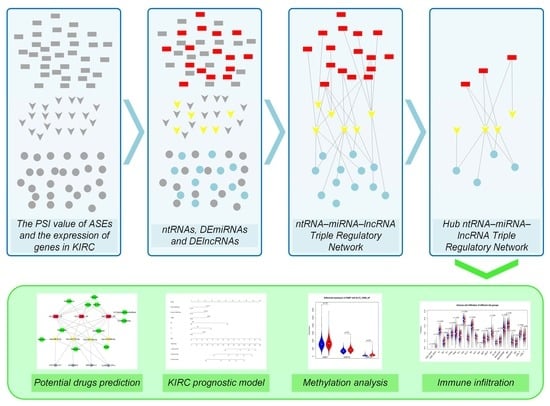
:
1. Introduction
2. Materials and Methods
2.1. Data Preparation and Processing
2.2. Definition and Identification of the Differentially Expressed ntRNAs That Are Independent of Genes and Detection of Differentially Expressed Genes (DEGs)
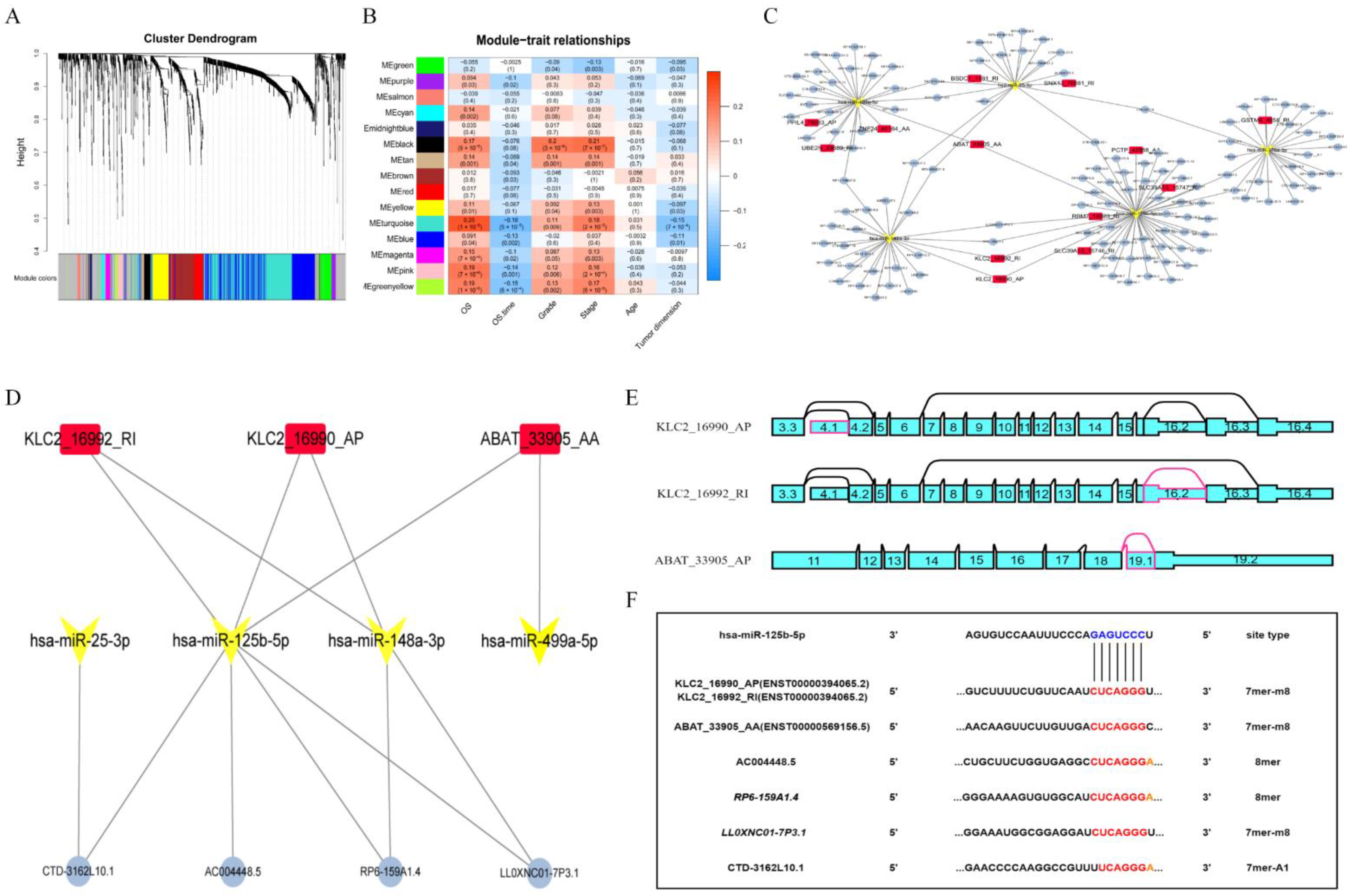
2.3. Establish a Triple Regulatory Network in KIRC
2.4. Establishment of the Risk Prediction Model for the Hub Triple Regulatory Network
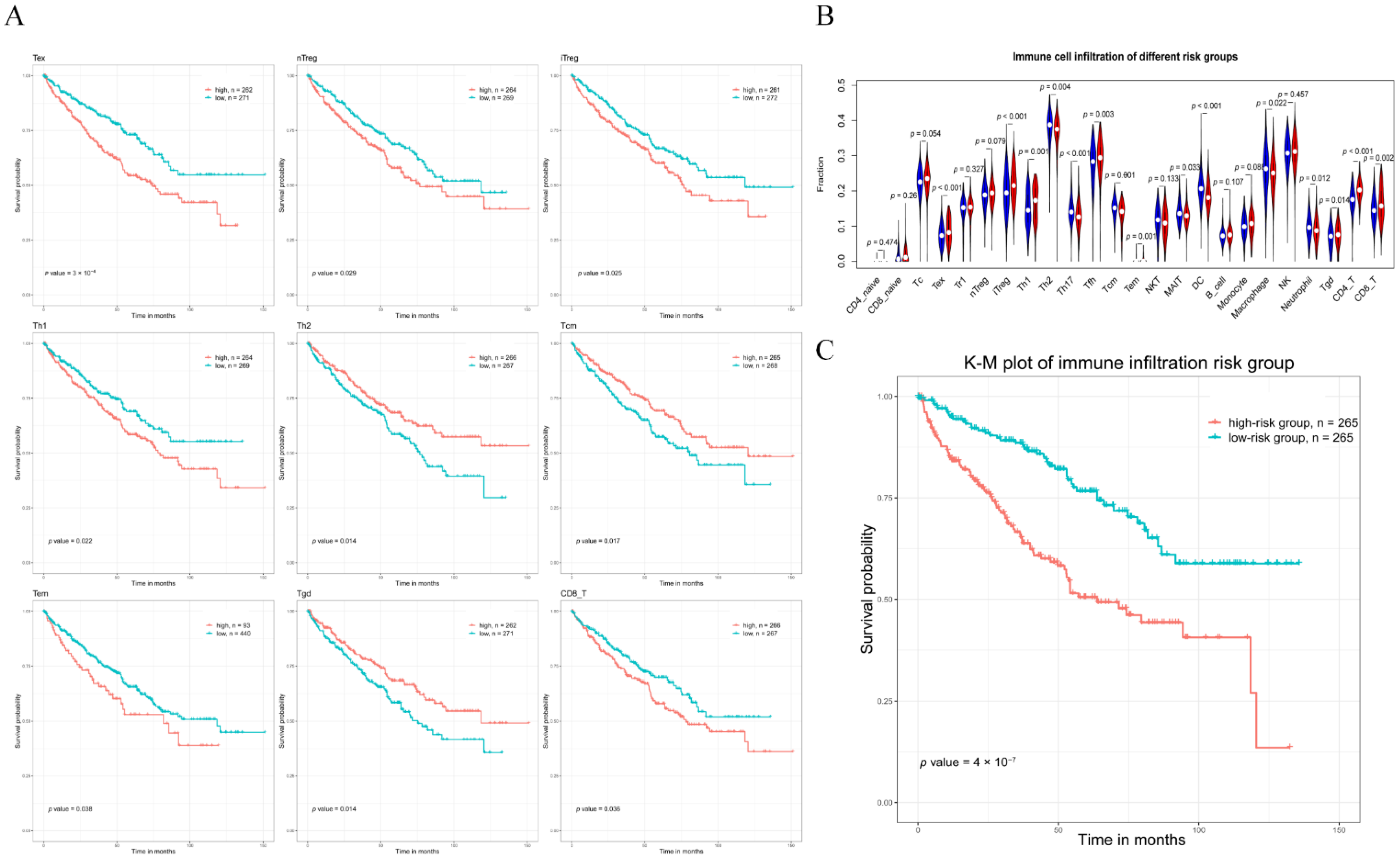
2.5. Establishment of Risk Prediction Model for the Immune Cells Associated with the Network
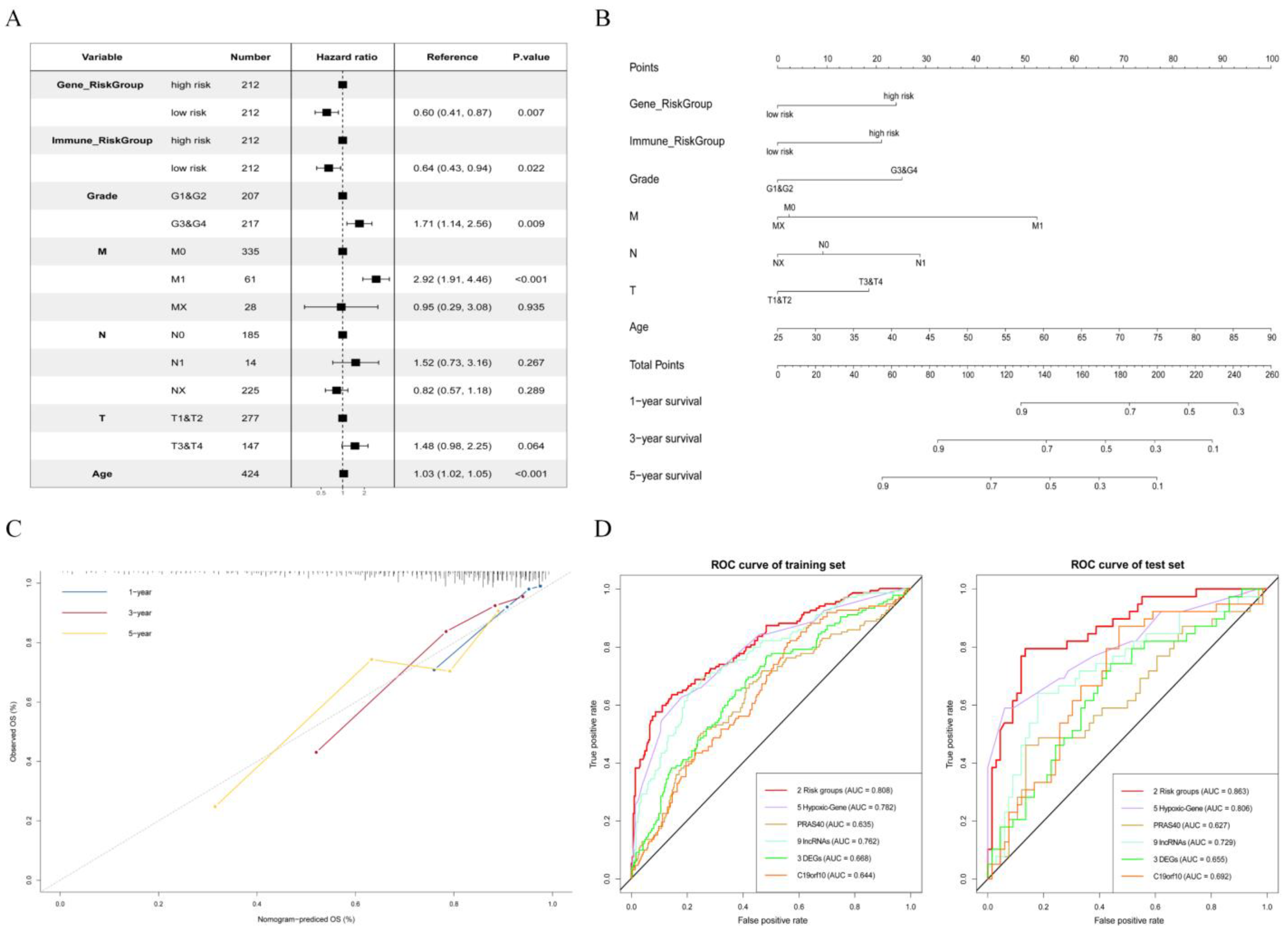
2.6. Using Core Gene and Immune Groups to Construct and Verify Cox Model
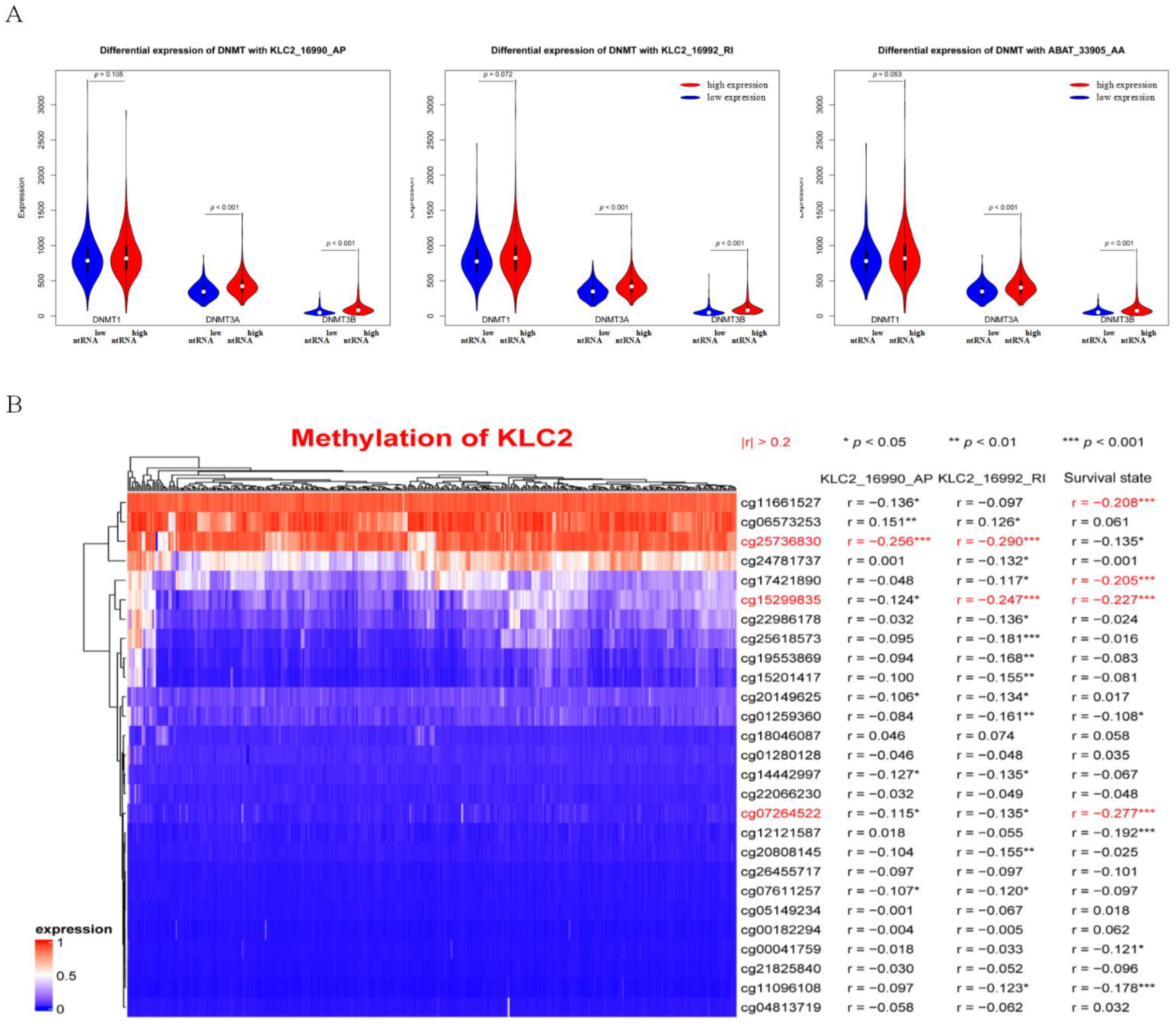
2.7. Methylation Analysis of ntRNAs in Hub Triple Regulatory Network
3. Results
3.1. Summary of Datasets
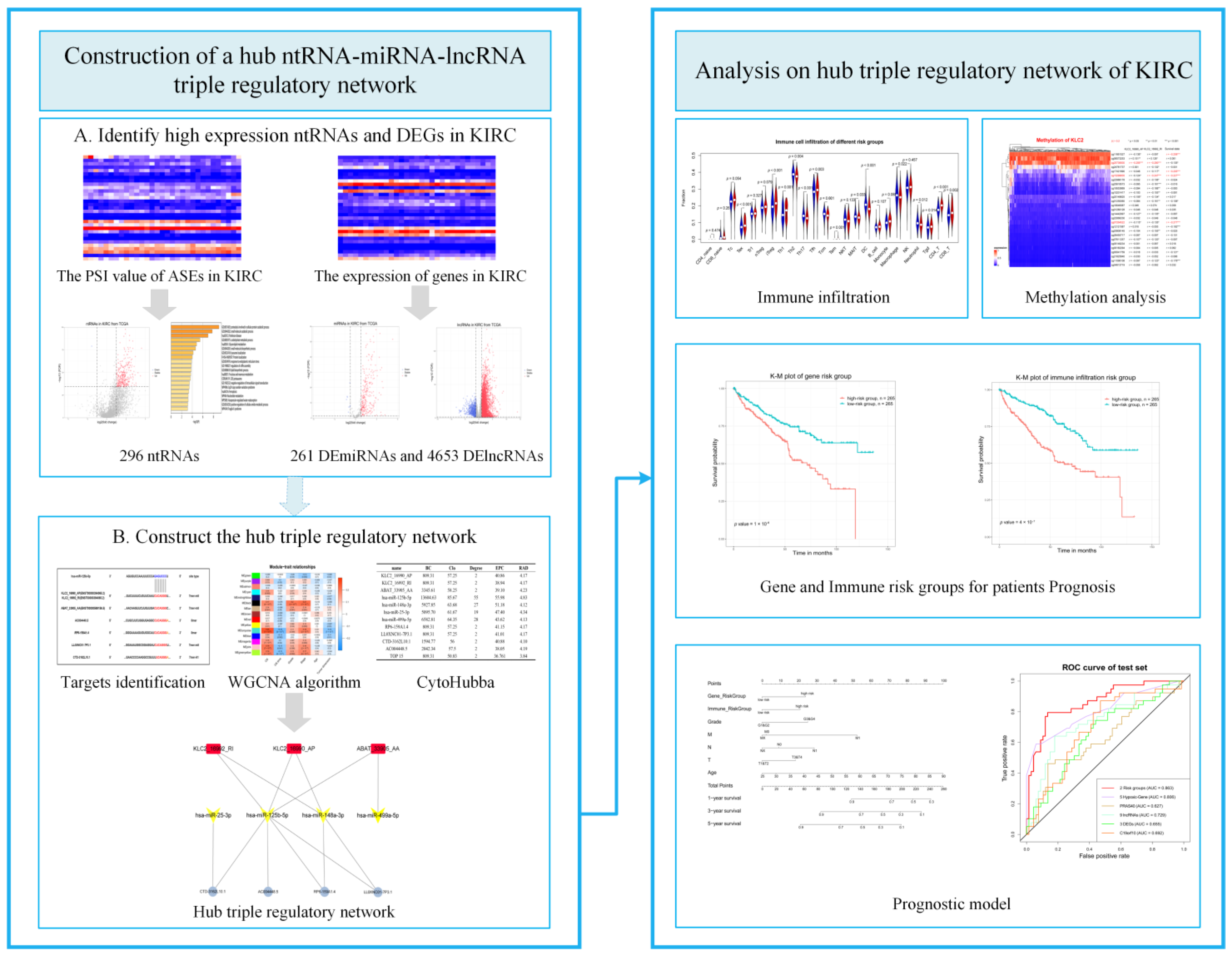
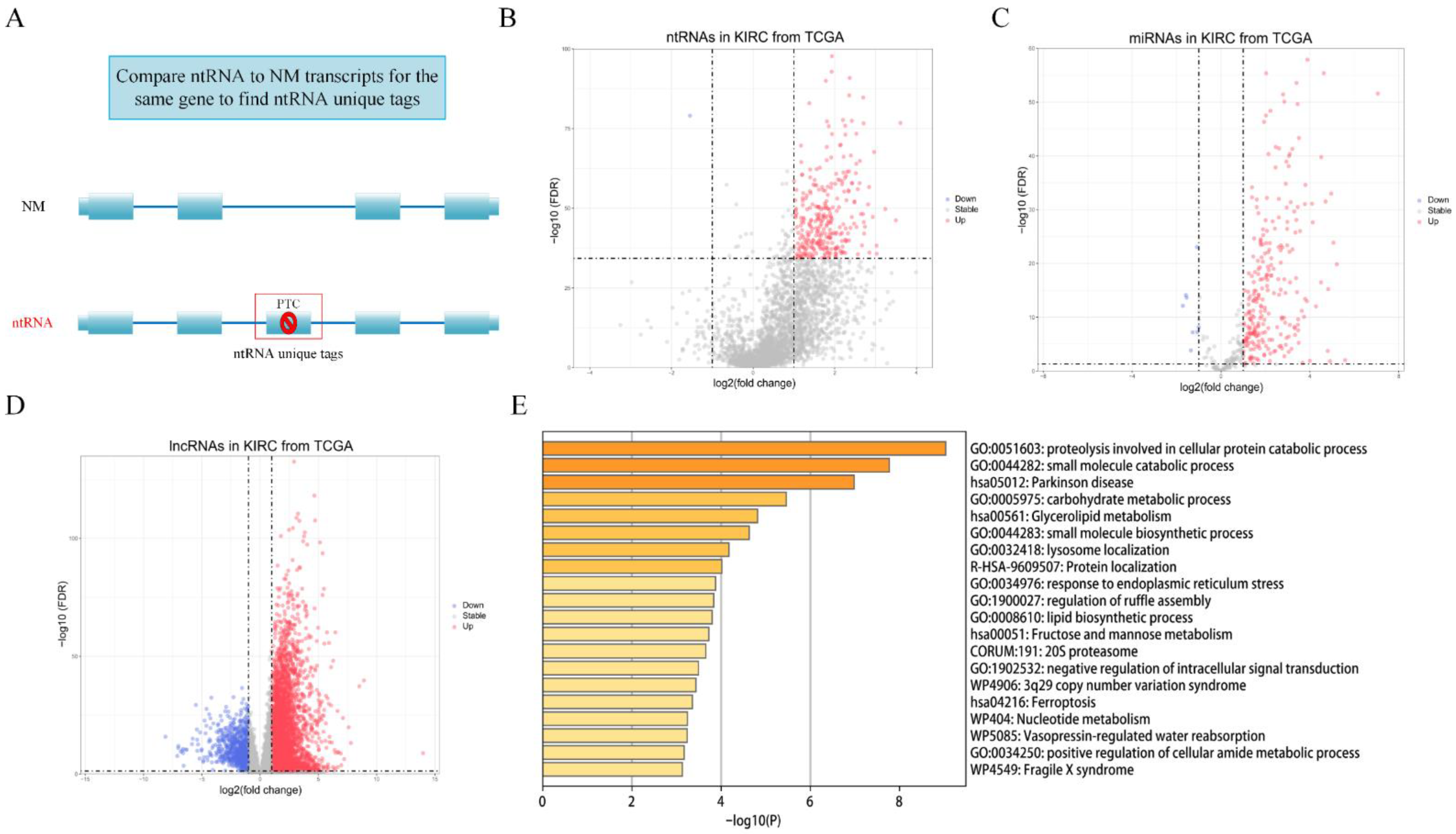
3.2. Identification of Differentially Expressed ntRNAs and DEGs
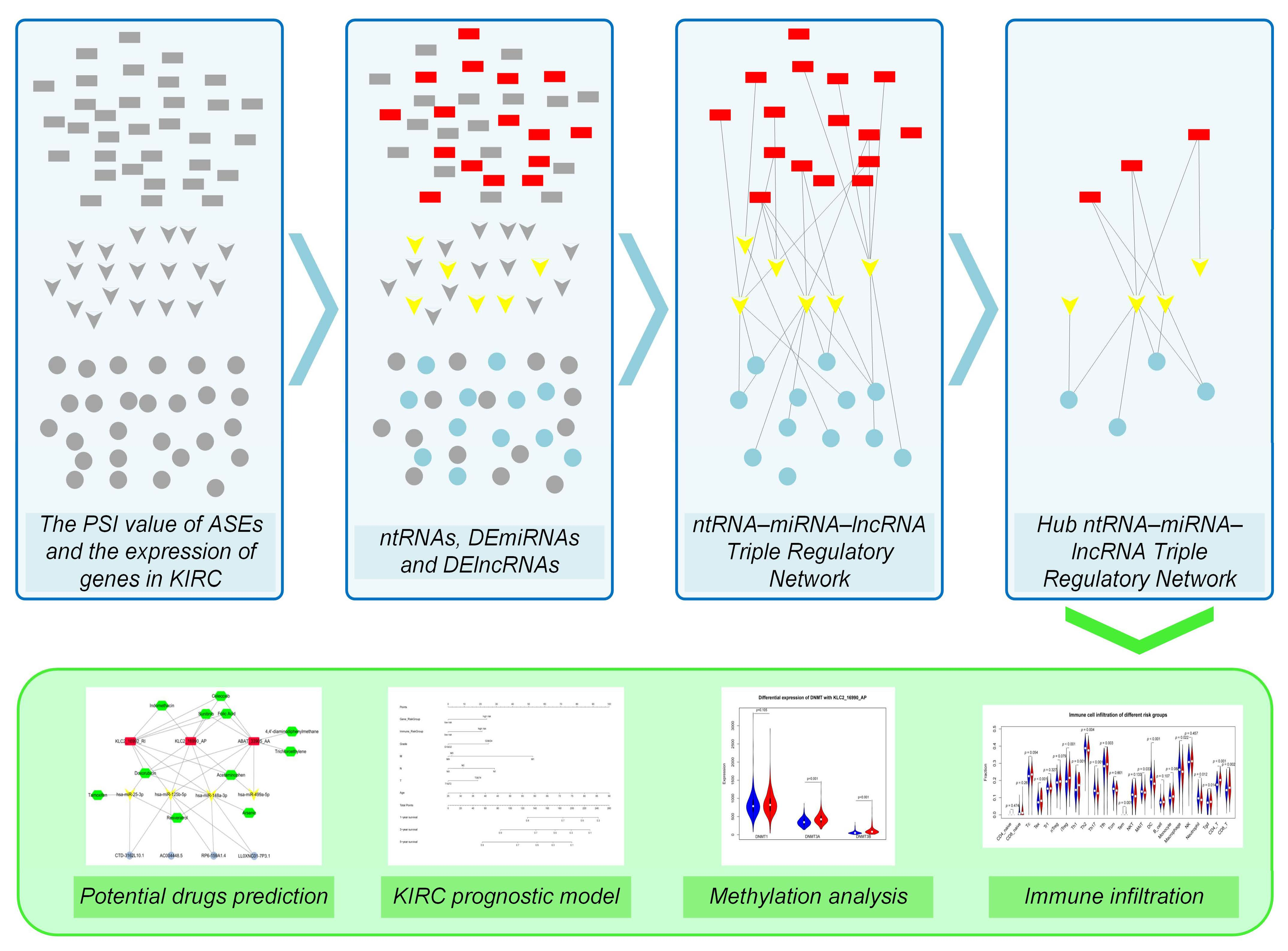
3.3. Construction of the Hub ntRNA–miRNA–lncRNA Triple Regulatory Network in KIRC
3.4. Construction of a Prognostic Risk Score Model Based on the Hub Triple Regulatory Network
3.5. Correlation between Immune Infiltration and the Hub Triple Regulatory Network in KIRC
3.6. Establishment and Validation of a Nomogram for OS Prediction in KIRC
3.7. DNA Methylation Involved in Regulating the Expression of KLC2_16990_AP and KLC2_16992_RI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Amin, A.; Armstrong, A.J.; Eble, J.N.; Ficarra, V.; Lopez-Beltran, A.; Martignoni, G.; Rini, B.I.; Kutikov, A. Understanding pathologic variants of renal cell carcinoma: Distilling therapeutic opportunities from biologic complexity. Eur. Urol. 2015, 67, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Nerich, V.; Hugues, M.; Paillard, M.J.; Borowski, L.; Nai, T.; Stein, U.; Nguyen Tan Hon, T.; Montcuquet, P.; Maurina, T.; Mouillet, G.; et al. Clinical impact of targeted therapies in patients with metastatic clear-cell renal cell carcinoma. Onco Targets Ther. 2014, 7, 365–374. [Google Scholar] [CrossRef]
- Mancini, M.; Zazzara, M.; Zattoni, F. Stem cells, biomarkers and genetic profiling: Approaching future challenges in Urology. Urologia 2016, 83, 4–13. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013, 47, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.K.; Lin, H.Y.; Chen, F.C. NMD Classifier: A reliable and systematic classification tool for nonsense-mediated decay events. PLoS ONE 2017, 12, e0174798. [Google Scholar] [CrossRef]
- Isken, O.; Maquat, L.E. The multiple lives of NMD factors: Balancing roles in gene and genome regulation. Nat. Rev. Genet. 2008, 9, 699–712. [Google Scholar] [CrossRef]
- Hu, F. Analysis of Nonsense-Mediated Decay Targeted RNA (nt-RNA) in High-Throughput Sequencing Data; Chinese University of Hong Kong: Hong Kong, China, 2015. [Google Scholar]
- Garcia-Moreno, J.F.; Romao, L. Perspective in Alternative Splicing Coupled to Nonsense-Mediated mRNA Decay. Int. J. Mol. Sci. 2020, 21, 9424. [Google Scholar] [CrossRef]
- He, M.; Hu, F. TF–RBP–AS Triplet Analysis Reveals the Mechanisms of Aberrant Alternative Splicing Events in Kidney Cancer: Implications for Their Possible Clinical Use as Prognostic and Therapeutic Biomarkers. Int. J. Mol. Sci. 2021, 22, 8789. [Google Scholar] [CrossRef]
- Sibley, C.R. Regulation of gene expression through production of unstable mRNA isoforms. Biochem. Soc. Trans. 2014, 42, 1196–1205. [Google Scholar] [CrossRef]
- French, C.E.; Wei, G.; Lloyd, J.P.; Hu, Z.; Brooks, A.N.; Brenner, S.E. Transcriptome analysis of alternative splicing-coupled nonsense-mediated mRNA decay in human cells reveals broad regulatory potential. bioRxiv 2020. [Google Scholar] [CrossRef]
- Munoz, U.; Puche, J.E.; Hannivoort, R.; Lang, U.E.; Cohen-Naftaly, M.; Friedman, S.L. Hepatocyte growth factor enhances alternative splicing of the Kruppel-like factor 6 (KLF6) tumor suppressor to promote growth through SRSF1. Mol. Cancer Res. 2012, 10, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Burnett, M.; Rodolico, V.; Shen, F.; Leng, R.; Zhang, M.; Eisenstat, D.D.; Sergi, C. PathVisio Analysis: An Application Targeting the miRNA Network Associated with the p53 Signaling Pathway in Osteosarcoma. Biocell 2021, 45, 17. [Google Scholar] [CrossRef]
- Li, M.; Marin-Muller, C.; Bharadwaj, U.; Chow, K.H.; Yao, Q.; Chen, C. MicroRNAs: Control and loss of control in human physiology and disease. World J. Surg. 2009, 33, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef]
- Farazi, T.A.; Spitzer, J.I.; Morozov, P.; Tuschl, T. miRNAs in human cancer. J. Pathol. 2011, 223, 102–115. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Kong, C.; Liu, X.; Bi, J.; Li, Z.; Li, Z.; Zhu, Y.; Zhang, Z. GAS5 functions as a ceRNA to regulate hZIP1 expression by sponging miR-223 in clear cell renal cell carcinoma. Am. J. Cancer Res. 2018, 8, 1414. [Google Scholar]
- Zhou, L.; Ye, J.; Wen, F.; Yu, H. Identification of Novel Prognostic Signatures for Clear Cell Renal Cell Carcinoma Based on ceRNA Network Construction and Immune Infiltration Analysis. Dis. Markers 2022, 2022, 4033583. [Google Scholar] [CrossRef] [PubMed]
- Machnik, M.; Cylwa, R.; Kiełczewski, K.; Biecek, P.; Liloglou, T.; Mackiewicz, A.; Oleksiewicz, U. The expression signature of cancer-associated KRAB-ZNF factors identified in TCGA pan-cancer transcriptomic data. Mol. Oncol. 2019, 13, 701–724. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Liu, L.; Li, J.; Hu, Q.; Sun, R. Expression and prognostic significance of m6A-related genes in lung adenocarcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e919644. [Google Scholar] [CrossRef]
- Miao, Y.R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.Y.; Wang, H.; Guo, A.Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Vlachos, I.S.; Karagkouni, D.; Georgakilas, G.; Kanellos, I.; Vergoulis, T.; Zagganas, K.; Tsanakas, P.; Floros, E.; Dalamagas, T. DIANA-LncBase v2: Indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016, 44, D231–D238. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Ye, F.; Han, F.; Wang, X.; Jiang, H.; Zhang, J. A Novel Radiogenomics Biomarker Based on Hypoxic-Gene Subset: Accurate Survival and Prognostic Prediction of Renal Clear Cell Carcinoma. Front. Oncol. 2021, 11, 739815. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhu, Y.; Liu, Y.; Niu, P.; Chen, H.; Deng, J.; Shi, D. High PRAS40 mRNA expression and its role in prognosis of clear cell renal cell carcinoma. Transl. Urol. 2020, 9, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Qi-Dong, X.; Yang, X.; Lu, J.L.; Liu, C.Q.; Sun, J.X.; Li, C.; Wang, S.G. Development and Validation of a Nine-Redox-Related Long Noncoding RNA Signature in Renal Clear Cell Carcinoma. Oxid. Med. Cell. Longev. 2020, 2020, 6634247. [Google Scholar] [CrossRef]
- Huang, H.; Zhu, L.; Huang, C.; Dong, Y.; Fan, L.; Tao, L.; Peng, Z.; Xiang, R. Identification of Hub Genes Associated with Clear Cell Renal Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Oncol. 2021, 11, 726655. [Google Scholar] [CrossRef]
- Lu, Y.; Liao, X.; Wang, T.; Hong, X.; Li, Z. The Clinical Relevance and Tumor Promoting Function of C19orf10 in Kidney Renal Clear Cell Carcinoma. Front. Oncol. 2021, 11, 725959. [Google Scholar] [CrossRef]
- Wolf, J.; Auw-Haedrich, C.; Schlecht, A.; Boneva, S.; Mittelviefhaus, H.; Lapp, T.; Agostini, H.; Reinhard, T.; Schlunck, G.; Lange, C.A.K. Transcriptional characterization of conjunctival melanoma identifies the cellular tumor microenvironment and prognostic gene signatures. Sci. Rep. 2020, 10, 17022. [Google Scholar] [CrossRef]
- Wang, D.; Chen, J.; Ding, Y.; Kong, H.; You, H.; Zhao, Y.; Wei, H.; Liu, Y. miR-188-5p Promotes Tumor Growth by Targeting CD2AP Through PI3K/AKT/mTOR Signaling in Children with Acute Promyelocytic Leukemia. Onco Targets Ther. 2020, 13, 6681–6697. [Google Scholar] [CrossRef]
- Chen, J.; Qian, Z.; Li, F.; Li, J.; Lu, Y. Integrative Analysis of Microarray Data to Reveal Regulation Patterns in the Pathogenesis of Hepatocellular Carcinoma. Gut Liver 2017, 11, 112–120. [Google Scholar] [CrossRef]
- Chowdhury, B.; Porter, E.G.; Stewart, J.C.; Ferreira, C.R.; Schipma, M.J.; Dykhuizen, E.C. PBRM1 Regulates the Expression of Genes Involved in Metabolism and Cell Adhesion in Renal Clear Cell Carcinoma. PLoS ONE 2016, 11, e0153718. [Google Scholar] [CrossRef]
- Bonnet, E.; Michoel, T.; Van de Peer, Y. Prediction of a gene regulatory network linked to prostate cancer from gene expression, microRNA and clinical data. Bioinformatics 2010, 26, i638–i644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Cheng, X.; Pei, Y.; Fu, J.; Lyu, Z.; Peng, H.; Yao, Q.; Jiang, Y.; Luo, L.; Zhuo, H. Data from a comparative proteomic analysis of tumor-derived lung-cancer CD105(+) endothelial cells. Data Brief 2016, 7, 927–939. [Google Scholar] [CrossRef]
- Sakai, Y.; Honda, M.; Matsui, S.; Komori, O.; Murayama, T.; Fujiwara, T.; Mizuno, M.; Imai, Y.; Yoshimura, K.; Nasti, A.; et al. Development of novel diagnostic system for pancreatic cancer, including early stages, measuring mRNA of whole blood cells. Cancer Sci. 2019, 110, 1364–1388. [Google Scholar] [CrossRef] [PubMed]
- Catchpole, G.; Platzer, A.; Weikert, C.; Kempkensteffen, C.; Johannsen, M.; Krause, H.; Jung, K.; Miller, K.; Willmitzer, L.; Selbig, J.; et al. Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell. Mol. Med. 2011, 15, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of Lipidomics and Transcriptomics Reveals Reprogramming of the Lipid Metabolism and Composition in Clear Cell Renal Cell Carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Wei, Q.; Mi, Q.S.; Dong, Z. The regulation and function of microRNAs in kidney diseases. IUBMB Life 2013, 65, 602–614. [Google Scholar] [CrossRef]
- Jin, L.; Zhang, Z.; Li, Y.; He, T.; Hu, J.; Liu, J.; Chen, M.; Gui, Y.; Chen, Y.; Lai, Y. miR-125b is associated with renal cell carcinoma cell migration, invasion and apoptosis. Oncol. Lett. 2017, 13, 4512–4520. [Google Scholar] [CrossRef]
- Marchionni, L.; Hayashi, M.; Guida, E.; Ooki, A.; Munari, E.; Jabboure, F.J.; Dinalankara, W.; Raza, A.; Netto, G.J.; Hoque, M.O.; et al. MicroRNA expression profiling of Xp11 renal cell carcinoma. Hum. Pathol. 2017, 67, 18–29. [Google Scholar] [CrossRef]
- Sun, J.; Yin, A.; Zhang, W.; Lv, J.; Liang, Y.; Li, H.; Li, Y.; Li, X. CircUBAP2 Inhibits Proliferation and Metastasis of Clear Cell Renal Cell Carcinoma via Targeting miR-148a-3p/FOXK2 Pathway. Cell Transpl. 2020, 29, 0963689720925751. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, H.; Gao, C.; Lai, Y.; Liang, J.; Chen, Z.; Chen, Z.; Heng, B.; Yao, N.; Lai, C. Downregulation of Circular RNA circPSD3 Promotes Metastasis by Modulating FBXW7 Expression in Clear Cell Renal Cell Carcinoma. J. Oncol. 2022, 2022, 5084631. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Chou, J.L.; Chang, J.S.; Chiu, I.J.; Chiu, H.W.; Lin, Y.F. Dysregulation of the miR-25-IMPA2 axis promotes metastatic progression in clear cell renal cell carcinoma. EBioMedicine 2019, 45, 220–230. [Google Scholar] [CrossRef]
- Heinemann, F.G.; Tolkach, Y.; Deng, M.; Schmidt, D.; Perner, S.; Kristiansen, G.; Muller, S.C.; Ellinger, J. Serum miR-122-5p and miR-206 expression: Non-invasive prognostic biomarkers for renal cell carcinoma. Clin. Epigenetics 2018, 10, 11. [Google Scholar] [CrossRef]
- Lu, J.; Chen, Z.; Zhao, H.; Dong, H.; Zhu, L.; Zhang, Y.; Wang, J.; Zhu, H.; Cui, Q.; Qi, C.; et al. ABAT and ALDH6A1, regulated by transcription factor HNF4A, suppress tumorigenic capability in clear cell renal cell carcinoma. J. Transl. Med. 2020, 18, 101. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Cho, E.; Je, Y.; Choueiri, T.K. Analgesic use and the risk of renal cell carcinoma (RCC): Results from a large up-to-date meta-analysis. J. Clin. Oncol. 2012, 30, 395. [Google Scholar] [CrossRef]
- Kang, J.; Lee, J.; Hong, D.; Lee, S.; Kim, N.; Lee, W.; Sung, T.; Gong, Y.; Kim, S. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy. Cell Death Dis. 2016, 7, e2163. [Google Scholar] [CrossRef]
- Kabel, A.M.; Atef, A.; Estfanous, R.S. Ameliorative potential of sitagliptin and/or resveratrol on experimentally-induced clear cell renal cell carcinoma. Biomed. Pharm. 2018, 97, 667–674. [Google Scholar] [CrossRef]
- Xu, R.; Wu, Q.; Gong, Y.; Wu, Y.; Chi, Q.; Sun, D. A novel prognostic target-gene signature and nomogram based on an integrated bioinformatics analysis in hepatocellular carcinoma. Biocell 2022, 46, 1261–1288. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, E.; Long, J.; Hu, Z.; Peng, J.; Liu, L.; Tang, F.; Li, L.; Ouyang, Y.; Zeng, Z. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019, 110, 1564–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, M.; Righetto, M.; Noessner, E. Checkpoint Inhibition in Bladder Cancer: Clinical Expectations, Current Evidence, and Proposal of Future Strategies Based on a Tumor-Specific Immunobiological Approach. Cancers 2021, 13, 6016. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, Q.; Zhen, Q.; Zhao, Y.; Liu, N.; Li, T.; Hao, Y.; Zhang, Y.; Luo, C.; Wu, X. Negative regulation of tumor-infiltrating NK cell in clear cell renal cell carcinoma patients through the exosomal pathway. Oncotarget 2017, 8, 37783. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Linlin, M.; Ye, T.; Yuhai, Z. Alterations of dendritic cell subsets and TH1/TH2 cytokines in the peripheral circulation of patients with superficial transitional cell carcinoma of the bladder. J. Clin. Lab. Anal. 2012, 26, 365–371. [Google Scholar] [CrossRef]
- Li, L.; Yang, C.; Zhao, Z.; Xu, B.; Zheng, M.; Zhang, C.; Min, Z.; Guo, J.; Rong, R. Skewed T-helper (Th) 1/2-and Th17/T regulatory-cell balances in patients with renal cell carcinoma. Mol. Med. Rep. 2015, 11, 947–953. [Google Scholar] [CrossRef]
- Cavalcanti, E.; Gigante, M.; Mancini, V.; Battaglia, M.; Ditonno, P.; Capobianco, C.; Cincione, R.I.; Selvaggi, F.P.; Herr, W.; Storkus, W.J.; et al. JAK3/STAT5/6 pathway alterations are associated with immune deviation in CD8 T cells in renal cell carcinoma patients. J. Biomed. Biotechnol. 2010, 2010, 935764. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | TCGA Training Set (n = 424) | TCGA Test Set (n = 106) | ||
|---|---|---|---|---|
| NO. | % | NO. | % | |
| Sex | ||||
| Men | 278 | 65.6 | 66 | 62.3 |
| Women | 146 | 34.4 | 40 | 37.7 |
| Age, years | ||||
| Median | 61 | ― | 60 | ― |
| Range | 26–90 | ― | 36–90 | ― |
| T | ||||
| T1&T2 | 277 | 65.4 | 63 | 59.4 |
| T3&T4 | 147 | 34.6 | 43 | 40.6 |
| M | ||||
| M0 | 335 | 79.0 | 85 | 80.2 |
| M1 | 61 | 14.4 | 17 | 16.0 |
| MX | 28 | 6.6 | 4 | 3.8 |
| N | ||||
| N0 | 185 | 43.6 | 54 | 50.9 |
| N1 | 14 | 3.3 | 2 | 1.9 |
| NX | 225 | 53.0 | 50 | 47.2 |
| Grade | ||||
| G1&G2 | 197 | 46.5 | 42 | 39.6 |
| G3&G4 | 217 | 53.5 | 64 | 60.4 |
| Overall survival, years | ||||
| Median | 3.51 | ― | 3.69 | ― |
| No. | Features | Prognostic Model Construction Methods | Accuracy (Training Set/Test Set) | Source |
|---|---|---|---|---|
| 1 | 2 risk groups | LASSO Cox and multivariate Cox regression | 0.808/0.863 | Our method |
| 2 | 5 Hypoxic-Genes | LASSO Cox and multivariate Cox regression | 0.782/0.806 | [33] |
| 3 | PRAS40 (mRNA) | Univariate Cox regression | 0.635/0.627 | [34] |
| 4 | 9 DERRlncRNAs | LASSO Cox regression | 0.762/0.729 | [35] |
| 5 | 3 DEGs | LASSO Cox regression | 0.668/0.665 | [36] |
| 6 | C19orf10 (mRNA) | Univariate Cox regression | 0.644/0.692 | [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Hu, F.; Huang, D.; Chi, Q.; Tang, N.L.S. Nonsense-Mediated Decay Targeted RNA (ntRNA): Proposal of a ntRNA–miRNA–lncRNA Triple Regulatory Network Usable as Biomarker of Prognostic Risk in Patients with Kidney Cancer. Genes 2022, 13, 1656. https://doi.org/10.3390/genes13091656
Zhou Z, Hu F, Huang D, Chi Q, Tang NLS. Nonsense-Mediated Decay Targeted RNA (ntRNA): Proposal of a ntRNA–miRNA–lncRNA Triple Regulatory Network Usable as Biomarker of Prognostic Risk in Patients with Kidney Cancer. Genes. 2022; 13(9):1656. https://doi.org/10.3390/genes13091656
Chicago/Turabian StyleZhou, Zhiyue, Fuyan Hu, Dan Huang, Qingjia Chi, and Nelson L. S. Tang. 2022. "Nonsense-Mediated Decay Targeted RNA (ntRNA): Proposal of a ntRNA–miRNA–lncRNA Triple Regulatory Network Usable as Biomarker of Prognostic Risk in Patients with Kidney Cancer" Genes 13, no. 9: 1656. https://doi.org/10.3390/genes13091656