
An Atypical Presentation of Upper Motor Neuron Predominant Juvenile Amyotrophic Lateral Sclerosis Associated with TARDBP Gene: A Case Report and Review of the Literature

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Patient and Method
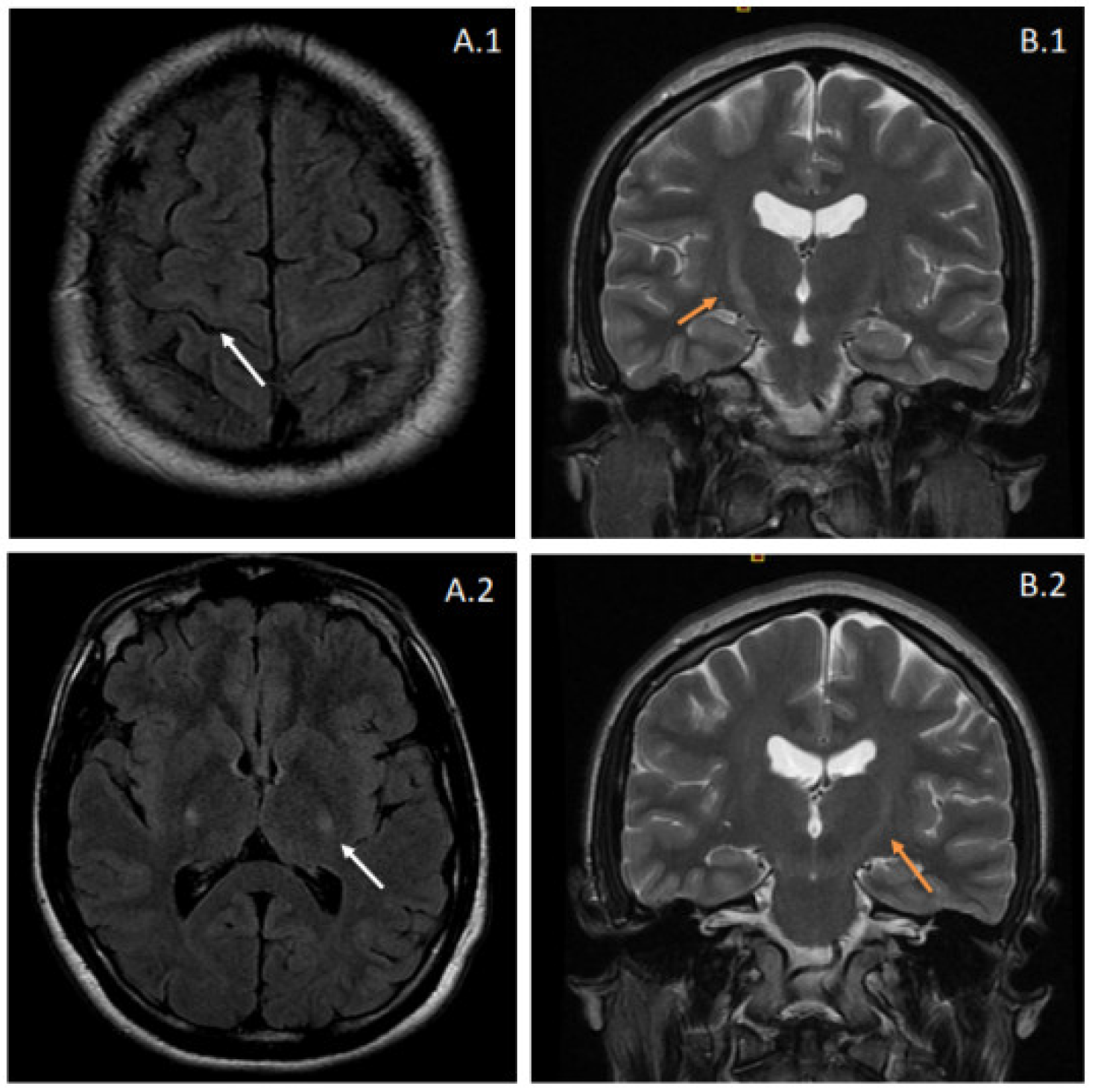
2.1. Clinical Presentation
2.2. Genetic Results
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G.L. Genetics of Amyotrophic Lateral Sclerosis: A Review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Orban, P.; Devon, R.S.; Hayden, M.R.; Leavitt, B.R. Chapter 15 Juvenile amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 82, pp. 301–312. ISBN 978-0-444-51894-1. [Google Scholar]
- Sreedharan, J.; Brown, R.H. Juvenile Amyotrophic Lateral Sclerosis. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence; Elsevier: Amsterdam, The Netherlands, 2015; pp. 146–159. ISBN 978-0-12-417044-5. [Google Scholar]
- Kacem, I.; Sghaier, I.; Bougatef, S.; Nasri, A.; Gargouri, A.; Ajroud-Driss, S.; Gouider, R. Epidemiological and Clinical Features of Amyotrophic Lateral Sclerosis in a Tunisian Cohort. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Lehky, T.; Grunseich, C. Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes 2021, 12, 1935. [Google Scholar] [CrossRef] [PubMed]
- Esselin, F.; Mouzat, K.; Polge, A.; Juntas-Morales, R.; Pageot, N.; De la Cruz, E.; Bernard, E.; Lagrange, E.; Danel, V.; Alphandery, S.; et al. Clinical Phenotype and Inheritance in Patients with C9ORF72 Hexanucleotide Repeat Expansion: Results from a Large French Cohort. Front. Neurosci. 2020, 14, 316. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hara, K.; Ishihara, T.; Onodera, O.; Ishiguro, H. A New Japanese Amyotrophic Lateral Sclerosis Family with TARDBP (TDP-43) Mutation. Neurol. Clin. Neurosci. 2019, 7, 101–102. [Google Scholar] [CrossRef]
- Takeda, T.; Iijima, M.; Shimizu, Y.; Yoshizawa, H.; Miyashiro, M.; Onizuka, H.; Yamamoto, T.; Nishiyama, A.; Suzuki, N.; Aoki, M.; et al. P.N345K Mutation in TARDBP in a Patient with Familial Amyotrophic Lateral Sclerosis: An Autopsy Case. Neuropathology 2019, 39, 286–293. [Google Scholar] [CrossRef]
- Leventoux, N.; Morimoto, S.; Hara, K.; Nakamura, S.; Ozawa, F.; Mitsuzawa, S.; Akiyama, T.; Nishiyama, A.; Suzuki, N.; Warita, H.; et al. Generation of an ALS Human IPSC Line KEIOi001-A from Peripheral Blood of a Charcot Disease-Affected Patient Carrying TARDBP p.N345K Heterozygous SNP Mutation. Stem Cell Res. 2020, 47, 101896. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 Proteinopathies: A New Wave of Neurodegenerative Diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 86–95. [Google Scholar] [CrossRef] [PubMed]
- François-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-Translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L.; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T Mutation in Familial Motor Neuron Disease. Ann. Neurol. 2008, 63, 535–538. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP Mutations in Individuals with Sporadic and Familial Amyotrophic Lateral Sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Budini, M.; Romano, V.; Avendaño-Vázquez, S.E.; Bembich, S.; Buratti, E.; Baralle, F.E. Role of Selected Mutations in the Q/N Rich Region of TDP-43 in EGFP-12xQ/N-Induced Aggregate Formation. Brain Res. 2012, 1462, 139–150. [Google Scholar] [CrossRef]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and Genotype Analysis in Amyotrophic Lateral Sclerosis with TARDBP Gene Mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Wang, H.; Liu, J.; Wang, Z.; Xu, B.; Zhao, K.; Tao, X.; He, Z.; Yang, F.; Huang, X. Genetic and Clinical Features of Chinese Sporadic Amyotrophic Lateral Sclerosis Patients with TARDBP Mutations. Brain Behav. 2021, 11, e2312. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High Frequency of TARDBP Gene Mutations in Italian Patients with Amyotrophic Lateral Sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Lin, H.-X.; Liu, G.-L.; Tao, Q.-Q.; Ni, W.; Xiao, B.-G.; Wu, Z.-Y. The Investigation of Genetic and Clinical Features in Chinese Patients with Juvenile Amyotrophic Lateral Sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Newell, K.; Paron, F.; Mompean, M.; Murrell, J.; Salis, E.; Stuani, C.; Pattee, G.; Romano, M.; Laurents, D.; Ghetti, B.; et al. Dysregulation of TDP-43 Intracellular Localization and Early Onset ALS Are Associated with a TARDBP S375G Variant. Brain Pathol. 2019, 29, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fu, S.; Lei, J.; Wu, H.; Shi, S.; Chen, K.; Hu, J.; Xu, X. Identification of Novel FUS and TARDBP Gene Mutations in Chinese Amyotrophic Lateral Sclerosis Patients with HRM Analysis. Aging 2020, 12, 22859. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Peng, Y.; Wang, X.-N.; Liu, M.-S.; Li, X.-G.; Cui, L.-Y. Screening of the TARDBP Gene in Familial and Sporadic Amyotrophic Lateral Sclerosis Patients of Chinese Origin. Neurobiol. Aging 2012, 33, 2229.e11–2229.e18. [Google Scholar] [CrossRef] [PubMed]
- Black, H.A.; Leighton, D.J.; Cleary, E.M.; Rose, E.; Stephenson, L.; Colville, S.; Ross, D.; Warner, J.; Porteous, M.; Gorrie, G.H.; et al. Genetic Epidemiology of Motor Neuron Disease-Associated Variants in the Scottish Population. Neurobiol. Aging 2017, 51, 178.e11–178.e20. [Google Scholar] [CrossRef]
- Kirby, J.; Goodall, E.F.; Smith, W.; Highley, J.R.; Masanzu, R.; Hartley, J.A.; Hibberd, R.; Hollinger, H.C.; Wharton, S.B.; Morrison, K.E.; et al. Broad Clinical Phenotypes Associated with TAR-DNA Binding Protein (TARDBP) Mutations in Amyotrophic Lateral Sclerosis. Neurogenetics 2010, 11, 217–225. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic Lateral Sclerosis Onset Is Influenced by the Burden of Rare Variants in Known Amyotrophic Lateral Sclerosis Genes: Rare Variants in ALS Genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef]
- Chen, W.; Xie, Y.; Zheng, M.; Lin, J.; Huang, P.; Pei, Z.; Yao, X. Clinical and Genetic Features of Patients with Amyotrophic Lateral Sclerosis in Southern China. Eur. J. Neurol. 2020, 27, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, R.-L.; Zhang, W.; Che, C.-H.; Feng, S.-Y.; Huang, H.-P.; Liu, C.-Y.; Zou, Z.-Y. Novel TARDBP Missense Mutation Caused Familial Amyotrophic Lateral Sclerosis with Frontotemporal Dementia and Parkinsonism. Neurobiol. Aging 2021, 107, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Min, J.; Staropoli, J.F.; Collin, E.; Bi, S.; Feng, X.; Barone, R.; Cao, Y.; O’Malley, L.; Xin, W.; et al. SOD1, ANG, TARDBP and FUS Mutations in Amyotrophic Lateral Sclerosis: A United States Clinical Testing Lab Experience. Amyotroph. Lateral Scler. 2012, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Caroppo, P.; Camuzat, A.; Guillot-Noel, L.; Thomas-Antérion, C.; Couratier, P.; Wong, T.H.; Teichmann, M.; Golfier, V.; Auriacombe, S.; Belliard, S.; et al. Defining the Spectrum of Frontotemporal Dementias Associated with TARDBP Mutations. Neurol. Genet. 2016, 2, e80. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Occhineri, P.; Cau, T.B.; et al. Genetic Architecture of ALS in Sardinia. Neurobiol. Aging 2014, 35, 2882.e7–2882.e12. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome Sequencing in Amyotrophic Lateral Sclerosis Identifies Risk Genes and Pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 Mutations in Familial Amyotrophic Lateral Sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 Variants Confer Susceptibility to Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Mossevelde, S.; Dillen, L.; De Bleecker, J.L.; Moisse, M.; Van Damme, P.; Van Broeckhoven, C.; van der Zee, J.; Engelborghs, S.; Crols, R.; et al. NEK1 Genetic Variability in a Belgian Cohort of ALS and ALS-FTD Patients. Neurobiol. Aging 2018, 61, 255.e1–255.e7. [Google Scholar] [CrossRef]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Yoshimura, J.; Doi, K.; Morishita, S.; Goto, J.; Toda, T.; et al. Loss-of-Function Variants in NEK1 Are Associated with an Increased Risk of Sporadic ALS in the Japanese Population. J. Hum. Genet. 2021, 66, 237–241. [Google Scholar] [CrossRef]
- Lattante, S.; Doronzio, P.N.; Conte, A.; Marangi, G.; Martello, F.; Bisogni, G.; Meleo, E.; Colavito, D.; Del Giudice, E.; Patanella, A.K.; et al. Novel Variants and Cellular Studies on Patients’ Primary Fibroblasts Support a Role for NEK1 Missense Variants in ALS Pathogenesis. Hum. Mol. Genet. 2021, 30, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; He, X.; Cui, B.; Zhao, F.; Zhou, C. NEK1 Mutations and the Risk of Amyotrophic Lateral Sclerosis (ALS): A Meta-Analysis. Neurol. Sci. 2021, 42, 1277–1285. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Case No. | Nucleotide Change | Amino Acid Change | Exon | FALS | Gender | Age of Onset (Years) | Region of Onset | Spread to Bulbar Region | Phenotype: Motor Neuron Predominance | CI/ Dementia | Disease Duration (Months) | Major Source of Disability | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Onset | Last Follow-Up | |||||||||||||
| 1 | c.1004G > A | p.Gly335Asp | 6 | No | Male | 20 | Spinal(UL) | No | LMN | LMN | No | 24 | LMN | Corrado et al. [25] |
| 2 | c.1043G > T | p.Gly348Val | 6 | Yes | Male | 24 | Spinal (UL) | Yes | LMN | Both | No | 46 | LMN | Liu et al. [26] |
| 3 | c.1123A > G | p.Ser375Gly | 6 | No | Female | 22 | Spinal (LL) | Yes | LMN | Both | No | 48 | LMN | Newell et al. [27] |
| 4 | c.1043G > T | p.Gly348Val | 6 | No | Male | 24 | Spinal(UL) | Yes | LMN | LMN | No | >120 (Alive at the time of publication) | LMN | Wang et al. [28] |
| 5 | c.1035C > G | p.Asn345Lys | 6 | No | Male | 24 | Spinal(LL) | Yes | UMN | UMN>>LMN | No | Alive at the time of writing | UMN | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Tejerina, D.; Restrepo-Vera, J.L.; Rovira-Moreno, E.; Codina-Sola, M.; Llauradó, A.; Sotoca, J.; Salvado, M.; Raguer, N.; García-Arumí, E.; Juntas-Morales, R. An Atypical Presentation of Upper Motor Neuron Predominant Juvenile Amyotrophic Lateral Sclerosis Associated with TARDBP Gene: A Case Report and Review of the Literature. Genes 2022, 13, 1483. https://doi.org/10.3390/genes13081483
Sánchez-Tejerina D, Restrepo-Vera JL, Rovira-Moreno E, Codina-Sola M, Llauradó A, Sotoca J, Salvado M, Raguer N, García-Arumí E, Juntas-Morales R. An Atypical Presentation of Upper Motor Neuron Predominant Juvenile Amyotrophic Lateral Sclerosis Associated with TARDBP Gene: A Case Report and Review of the Literature. Genes. 2022; 13(8):1483. https://doi.org/10.3390/genes13081483
Chicago/Turabian StyleSánchez-Tejerina, Daniel, Juan Luis Restrepo-Vera, Eulalia Rovira-Moreno, Marta Codina-Sola, Arnau Llauradó, Javier Sotoca, Maria Salvado, Núria Raguer, Elena García-Arumí, and Raúl Juntas-Morales. 2022. "An Atypical Presentation of Upper Motor Neuron Predominant Juvenile Amyotrophic Lateral Sclerosis Associated with TARDBP Gene: A Case Report and Review of the Literature" Genes 13, no. 8: 1483. https://doi.org/10.3390/genes13081483