FRET-Based Sorting of Live Cells Reveals Shifted Balance between PLK1 and CDK1 Activities During Checkpoint Recovery


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Flow Cytometry
2.3. FRET-Activated Cells Sorting
2.4. Protein Digestion for Mass Spectrometry Analysis
2.5. Liquid Chromatography Tandem Mass Spectrometry
2.6. Mass Spectrometry Data Analysis
2.7. Live Cell Microscopy and Quantitative Immunofluorescence (qIF)
2.8. Chemicals and Inhibitors
2.9. Western Blotting (WB)
3. Results
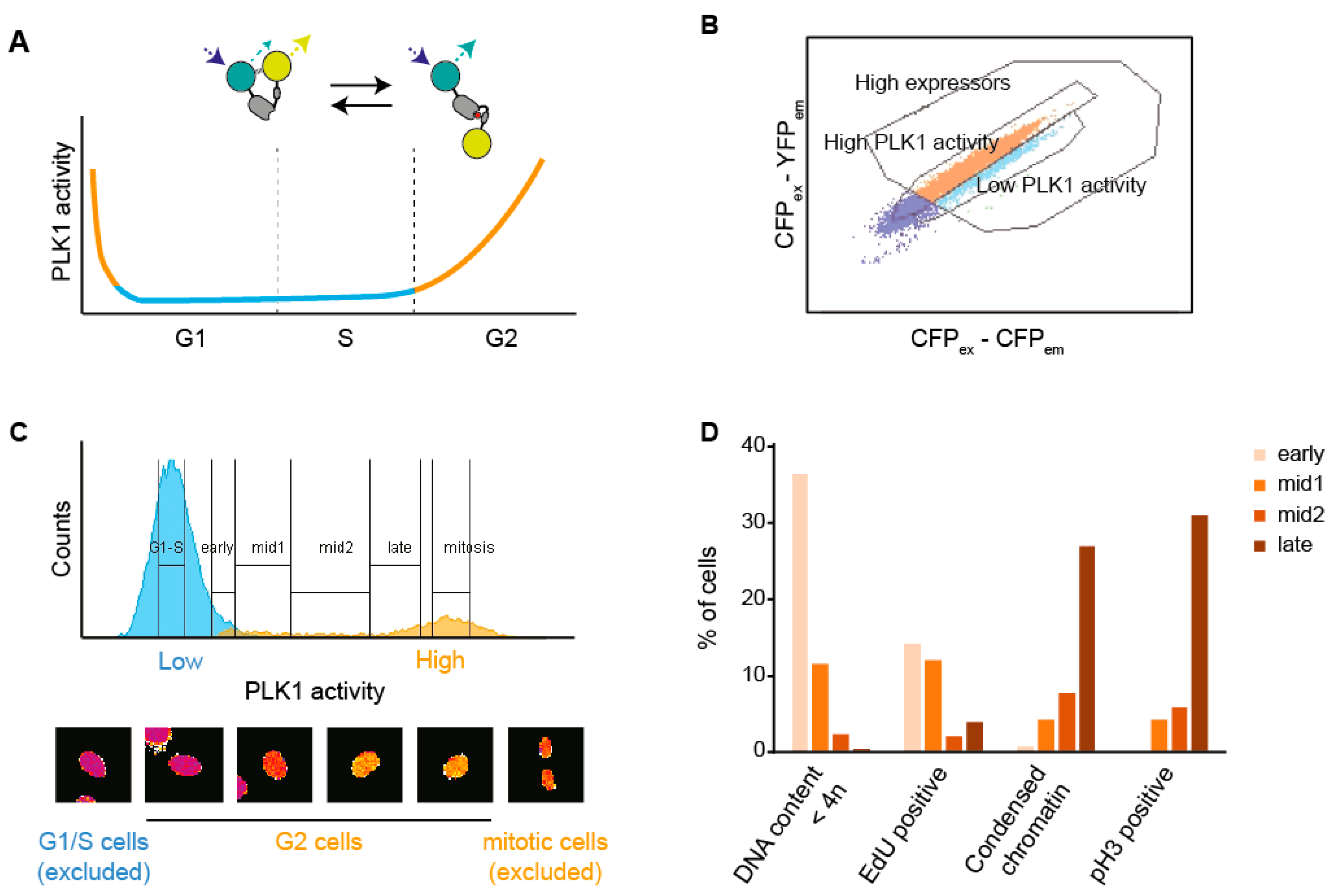
3.1. A Setup to Sort Cells Based on FRET
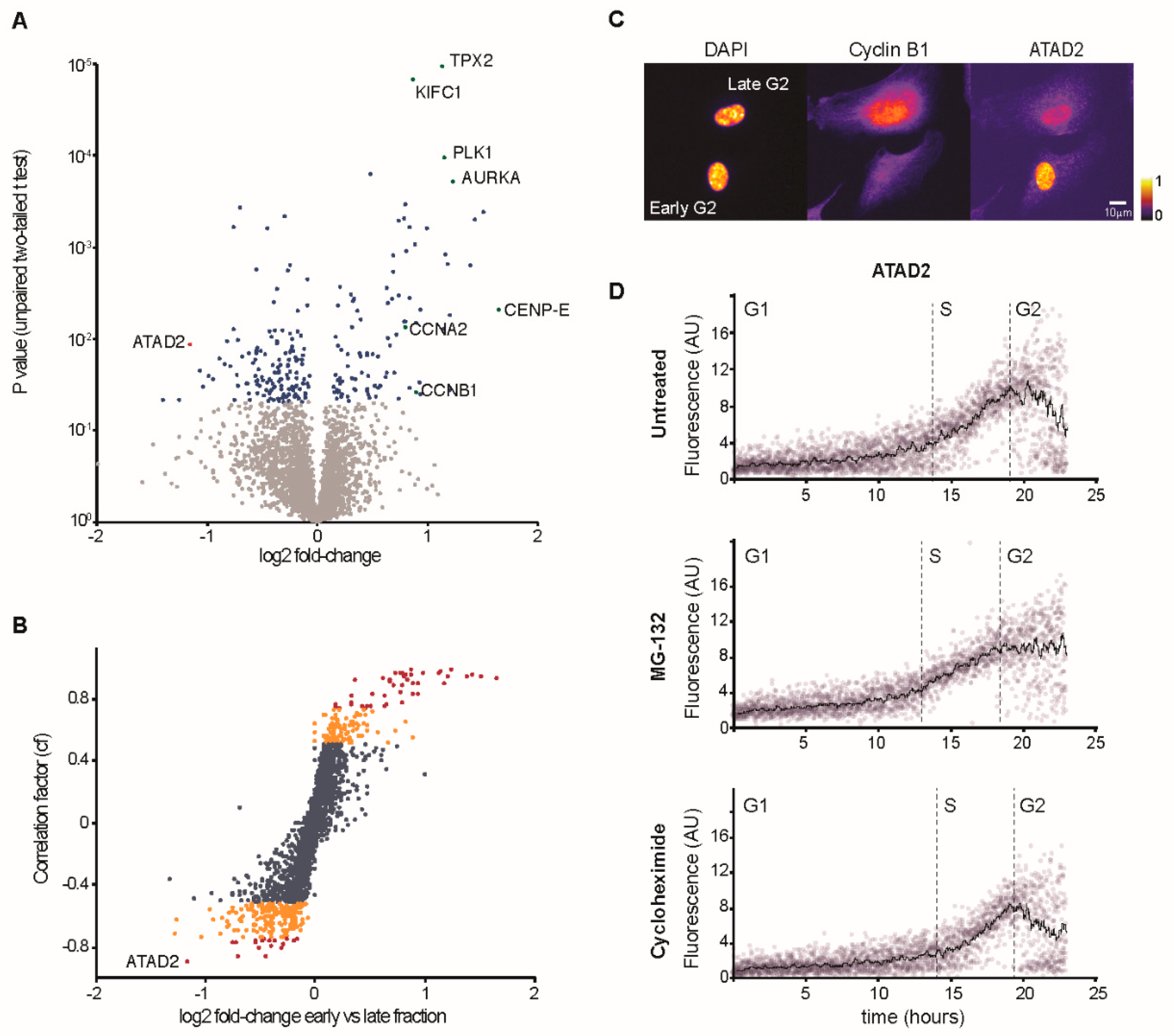
3.2. Characterization of Changes in Protein Content Through G2 Phase
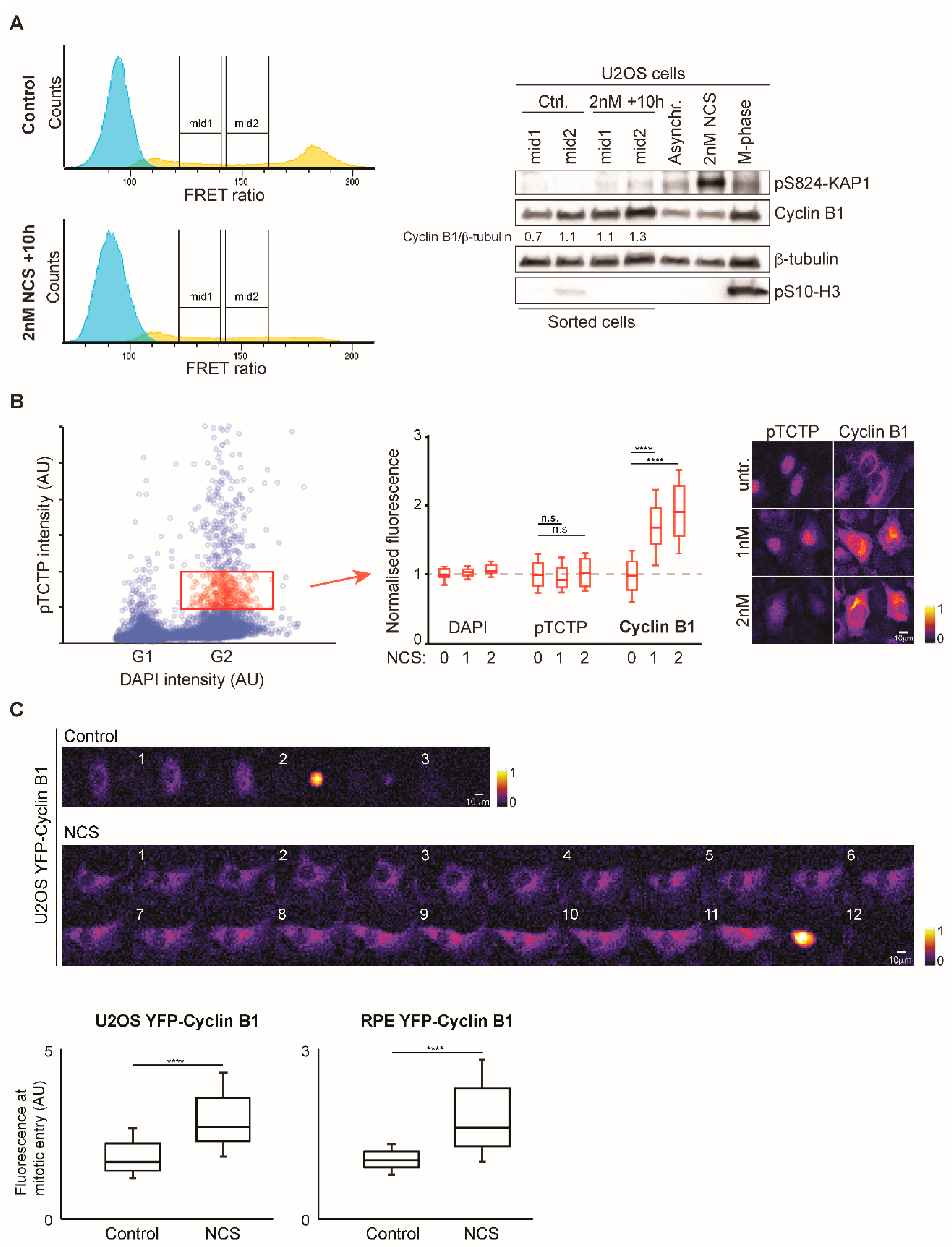
3.3. Increased Cyclin B1 Levels During DNA Damage Checkpoint Recovery
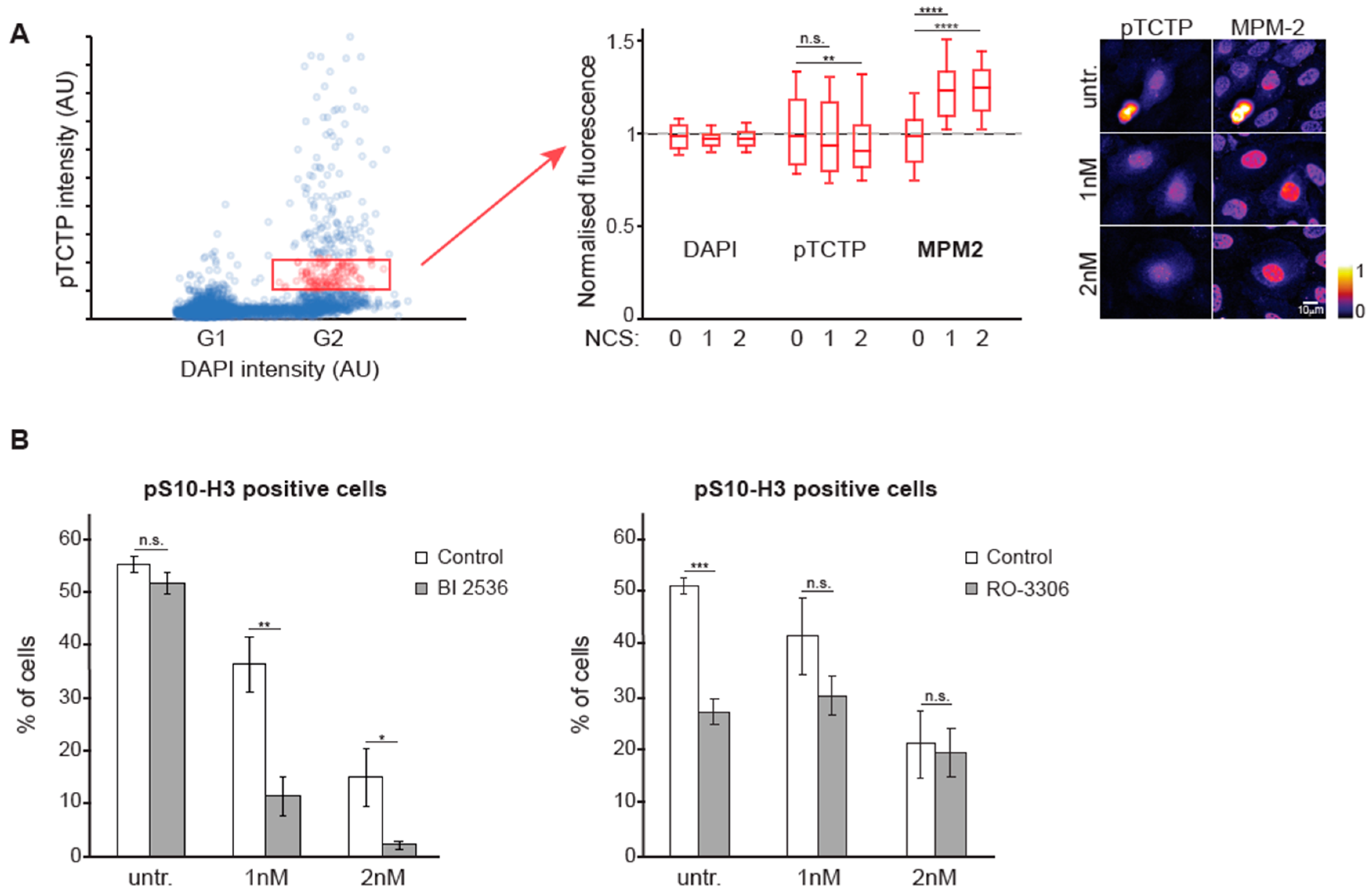
3.4. The Balance Between CDK1 and PLK1 is Altered During DNA Damage Checkpoint Recovery
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallaert, W.; Kedziora, K.M.; Chao, H.X.; Purvis, J.E. Bistable switches as integrators and actuators during cell cycle progression. FEBS Lett. 2019, 593, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Lemmens, B.; Lindqvist, A. DNA replication and mitotic entry: A brake model for cell cycle progression. J. Cell Biol. 2019, 218, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.; Medema, R.H. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marceau, A.H.; Brison, C.M.; Nerli, S.; Arsenault, H.E.; McShan, A.C.; Chen, E.; Lee, H.-W.; Benanti, J.A.; Sgourakis, N.G.; Rubin, S.M. An order-to-disorder structural switch activates the FoxM1 transcription factor. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128 e3. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, S.; Yang, Q. Incoherent Inputs Enhance the Robustness of Biological Oscillators. Cell Syst. 2017, 5, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Cooper, S. The synchronization manifesto: A critique of whole-culture synchronization. FEBS J. 2019, 286, 4650–4656. [Google Scholar] [CrossRef]
- Matson, J.P.; House, A.M.; Grant, G.D.; Wu, H.; Perez, J.; Cook, J.G. Intrinsic checkpoint deficiency during cell cycle re-entry from quiescence. J. Cell Biol. 2019, 218, 2169–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vugt, M.A.T.M.; Brás, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Macurek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mamely, I.; van Vugt, M.A.; Smits, V.A.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Medema, R.H.; Freire, R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef]
- van Vugt, M.A.T.M.; Gardino, A.K.; Linding, R.; Ostheimer, G.J.; Reinhardt, H.C.; Ong, S.-E.; Tan, C.S.; Miao, H.; Keezer, S.M.; Li, J.; et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010, 8, e1000287. [Google Scholar] [CrossRef]
- Lyutvinskiy, Y.; Yang, H.; Rutishauser, D.; Zubarev, R.A. In silico instrumental response correction improves precision of label-free proteomics and accuracy of proteomics-based predictive models. Mol. Cell Proteomics 2013, 12, 2324–2331. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akopyan, K.; Lindqvist, A.; Mullers, E. Cell Cycle Dynamics of Proteins and Post-translational Modifications Using Quantitative Immunofluorescence. Methods Mol. Biol. 2016, 1342, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Akopyan, K.; Silva Cascales, H.; Hukasova, E.; Saurin, A.T.; Mullers, E.; Jaiswal, H.; Hollman, D.A.; Kops, G.J.; Medema, R.H.; Lindqvist, A. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol. Cell 2014, 53, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Esposito, A.; De, S.; Ber, S.; Collin, P.; Surana, U.; Venkitaraman, A.R. Homeostatic control of polo-like kinase-1 engenders non-genetic heterogeneity in G2 checkpoint fidelity and timing. Nat. Commun. 2014, 5, 4048. [Google Scholar] [CrossRef]
- Jaiswal, H.; Benada, J.; Müllers, E.; Akopyan, K.; Burdova, K.; Koolmeister, T.; Helleday, T.; Medema, R.H.; Macurek, L.; Lindqvist, A. ATM/Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar] [CrossRef]
- Cucchi, U.; Gianellini, L.M.; De Ponti, A.; Sola, F.; Alzani, R.; Patton, V.; Pezzoni, A.; Troiani, S.; Saccardo, M.B.; Rizzi, S.; et al. Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res. 2010, 30, 4973–4985. [Google Scholar]
- Yaffe, M.B.; Schutkowski, M.; Shen, M.; Zhou, X.Z.; Stukenberg, P.T.; Rahfeld, J.U.; Xu, J.; Kuang, J.; Kirschner, M.W.; Fischer, G.; et al. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science 1997, 278, 1957–1960. [Google Scholar] [CrossRef]
- Lin, W.; Mehta, S.; Zhang, J. Genetically encoded fluorescent biosensors illuminate kinase signaling in cancer. J. Biol. Chem. 2019, 294, 14814–14822. [Google Scholar] [CrossRef] [Green Version]
- Terai, K.; Imanishi, A.; Li, C.; Matsuda, M. Two Decades of Genetically Encoded Biosensors Based on Förster Resonance Energy Transfer. Cell Struct. Funct. 2019, 44, 153–169. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Hu, Y.; Yang, X.; Tang, Y.; Han, S.; Kang, A.; Deng, H.; Chi, Y.; Zhu, D.; Lu, Y. FÖrster resonance energy transfer (FRET)-based biosensors for biological applications. Biosens. Bioelectron. 2019, 138, 111314. [Google Scholar] [CrossRef]
- Boström, J.; Sramkova, Z.; Salašová, A.; Johard, H.; Mahdessian, D.; Fedr, R.; Marks, C.; Medalová, J.; Souček, K.; Lundberg, E.; et al. Comparative cell cycle transcriptomics reveals synchronization of developmental transcription factor networks in cancer cells. PLoS ONE 2017, 12, e0188772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giotti, B.; Chen, S.-H.; Barnett, M.W.; Regan, T.; Ly, T.; Wiemann, S.; Hume, D.A.; Freeman, T.C. Assembly of a parts list of the human mitotic cell cycle machinery. J. Mol. Cell Biol. 2019, 11, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.R.; Moreno, M.V.; Olshen, A.B.; Taylor, B.S.; Ruggero, D. The translational landscape of the mammalian cell cycle. Mol. Cell 2013, 52, 574–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, T.; Whigham, A.; Clarke, R.; Brenes-Murillo, A.J.; Estes, B.; Madhessian, D.; Lundberg, E.; Wadsworth, P.; Lamond, A.I. Proteomic analysis of cell cycle progression in asynchronous cultures, including mitotic subphases, using PRIMMUS. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Kelly, V.; al-Rawi, A.; Lewis, D.; Ly, T. Cell cycle state proteomics and classification using in-cell protease digests and mass spectrometry. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mahdessian, D.; Cesnik, A.J.; Gnann, C.; Danielsson, F.; Stenström, L.; Arif, M.; Zhang, C.; Shutten, R.; Bäckström, A.; Thul, P.; et al. Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. bioRxiv 2020, 543231. [Google Scholar] [CrossRef]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.; Cole, R.W.; Rieder, C.L. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 2002, 12, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Deckbar, D.; Birraux, J.; Krempler, A.; Tchouandong, L.; Beucher, A.; Walker, S.; Stiff, T.; Jeggo, P.; Löbrich, M. Chromosome breakage after G2 checkpoint release. J. Cell Biol. 2007, 176, 749–755. [Google Scholar] [CrossRef]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell. Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Target | Species | Supplier/Reference | Application |
|---|---|---|---|
| 53BP1 | Rabbit | Abcam, ab21083 | IF |
| ATAD2 | Rabbit | ATLAS antibodies HPA029424 | IF, WB |
| Cyclin B1 (V152) | Mouse | Cell signaling #4135 | IF, WB |
| Cyclin B1 (D-1) | Mouse | Santa Cruz, 166210 | IF |
| pSer10-Histone H3 (D2C8) | Rabbit | Cell signaling #3377 | IF, WB |
| pSer824-KAP1 | Rabbit | Bethyl laboratories, A300-767A | WB |
| pSer/Thr-Pro MPM-2 | Mouse | Merck Millipore, 05-368 | IF |
| β-tubulin (93F) | Rabbit | Cell signaling, #2128 | WB |
| pSer46-TCTP | Rabbit | Cell signaling, #5251 | IF |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lafranchi, L.; Müllers, E.; Rutishauser, D.; Lindqvist, A. FRET-Based Sorting of Live Cells Reveals Shifted Balance between PLK1 and CDK1 Activities During Checkpoint Recovery. Cells 2020, 9, 2126. https://doi.org/10.3390/cells9092126
Lafranchi L, Müllers E, Rutishauser D, Lindqvist A. FRET-Based Sorting of Live Cells Reveals Shifted Balance between PLK1 and CDK1 Activities During Checkpoint Recovery. Cells. 2020; 9(9):2126. https://doi.org/10.3390/cells9092126
Chicago/Turabian StyleLafranchi, Lorenzo, Erik Müllers, Dorothea Rutishauser, and Arne Lindqvist. 2020. "FRET-Based Sorting of Live Cells Reveals Shifted Balance between PLK1 and CDK1 Activities During Checkpoint Recovery" Cells 9, no. 9: 2126. https://doi.org/10.3390/cells9092126