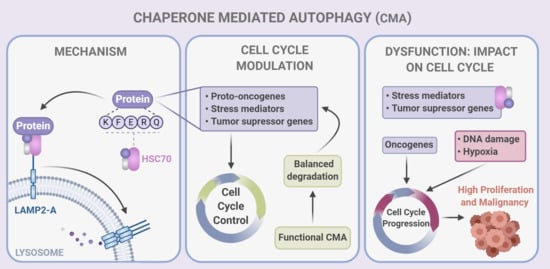
The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
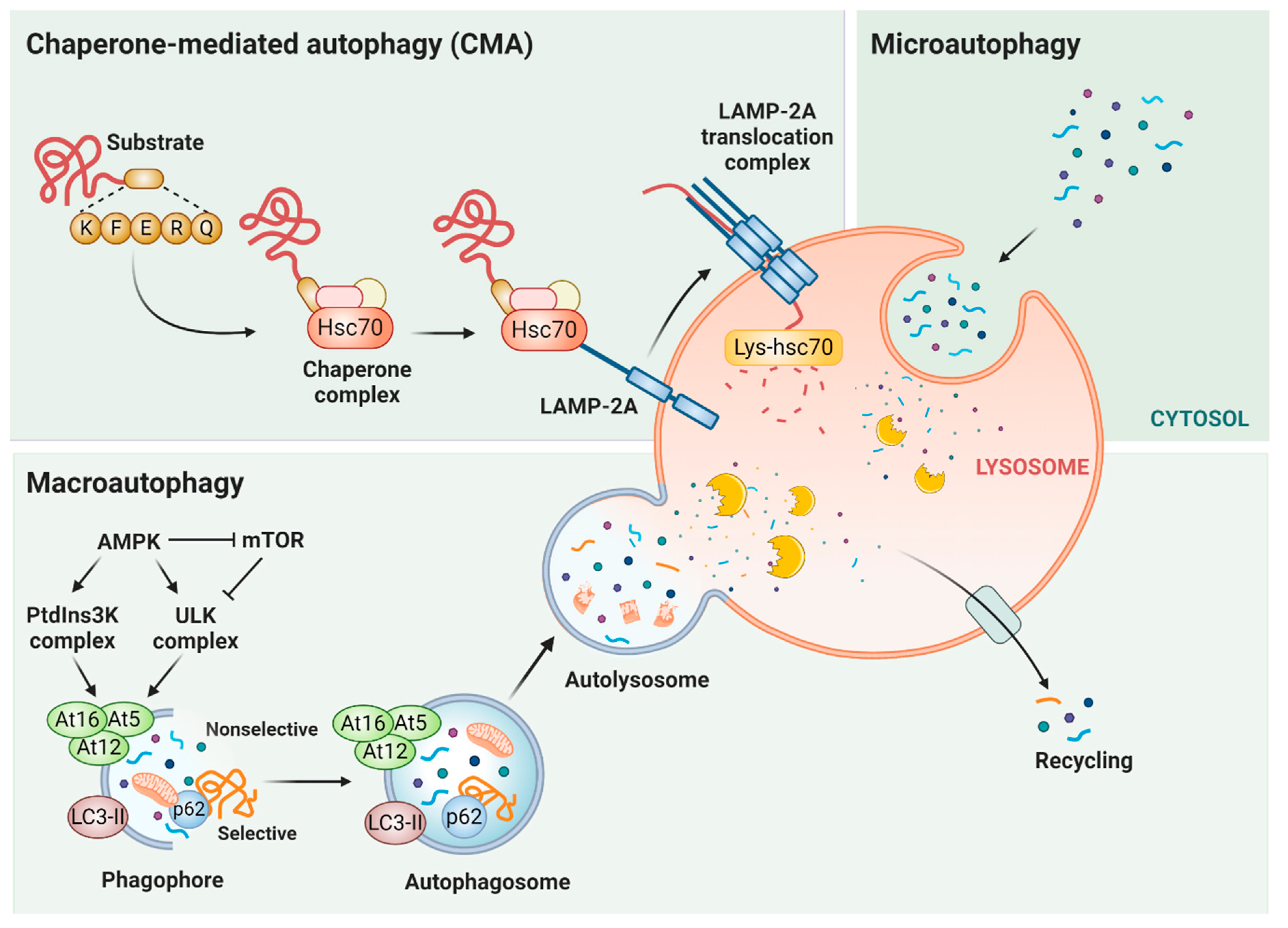
2. Chaperone-Mediated Autophagy (CMA)
3. Physiological and Pathological Roles of CMA
4. CMA’s Role in Cancer
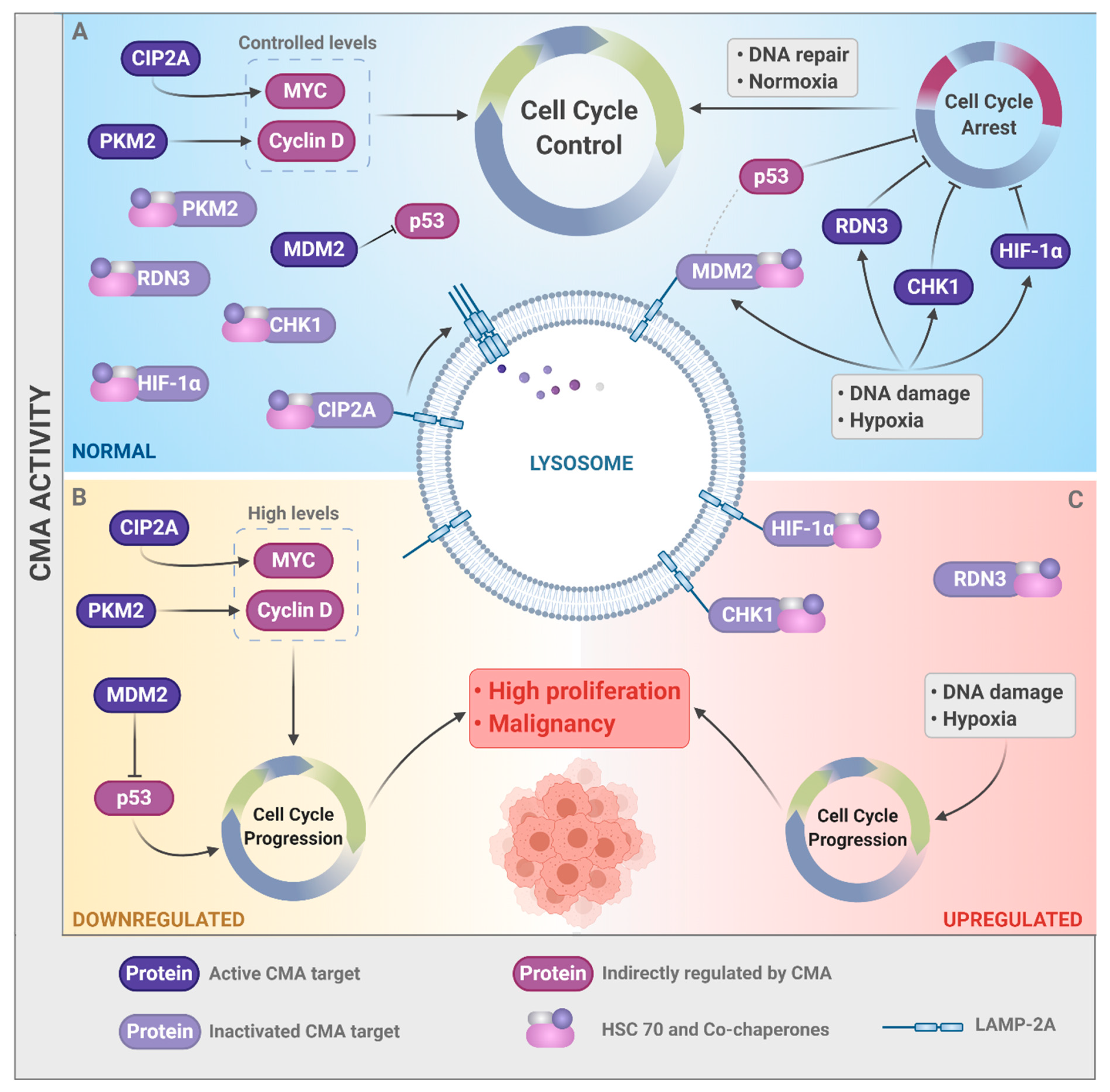
4.1. Anti-Tumor Functions of CMA
4.2. Pro-Tumor Functions of CMA
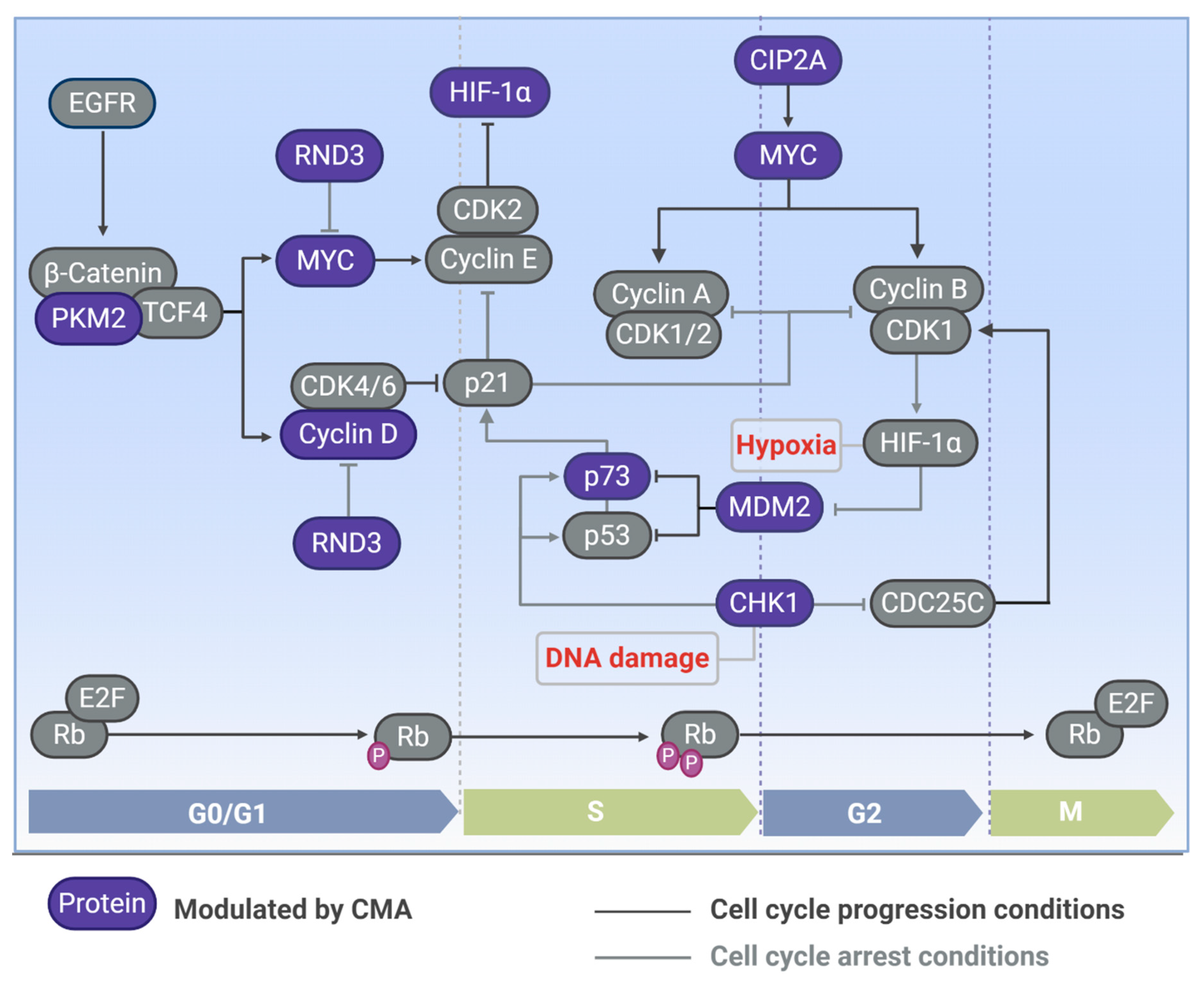
5. CMA Control of Cell Cycle in Distinct Cellular Contexts
5.1. Tumor Transformation: MYC
5.2. Hypoxia: HIF-1α
5.3. DNA Damage Response: CHK1
5.4. Different Tumor Cell Models: p73, RND3, and Cyclin D1
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, U.; Weinberg, R.A. The hallmarks of cancer. Oxf. Textb. Oncol. 2016, 100, 3–10. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Shah, M.; Schwartz, G.K. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar]
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Soussi, T.; Dehouche, K.; Béroud, C. p53 Website and analysis of p53 gene mutations in human cancer: Forging a link between epidemiology and carcinogenesis. Hum. Mutat. 2000, 15, 105–113. [Google Scholar] [CrossRef]
- Cordon-Cardo, C. Mutations of cell cycle regulators. Biological and clinical implications for human neoplasia. Am. J. Pathol. 1995, 147, 545–560. [Google Scholar]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.-L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2009, 1793, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2017, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.C. Autophagy and Stem Cells: Self-Eating for Self-Renewal. Front. Cell Dev. Boil. 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharm. 2019, 119, 109415. [Google Scholar] [CrossRef] [PubMed]
- Belaid, A.; Cerezo, M.; Chargui, A.; Corcelle–Termeau, E.; Pedeutour, F.; Giuliano, S.; Ilié, M.; Rubera, I.; Tauc, M.; Barale, S.; et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013, 73, 4311–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; He, M.-X.; McLeod, I.X.; Guo, J.; Ji, D.; He, Y.-W. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip. Autophagy 2015, 11, 2335–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H.; et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of Gata. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Boil. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient Chaperone-Mediated Autophagy in Liver Leads to Metabolic Dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-Mediated Autophagy Is Required for Tumor Growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Hu, H.; Kshitiz Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated Autophagy Targets Hypoxia-inducible Factor-1α (HIF-1α) for Lysosomal Degradation. J. Boil. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.; Yang, K.; Chiao, L.; Deng, Y.; Zhou, X.; Zhang, Z.; Zeng, S.X.; Lu, H. Inhibition of tumor suppressor p73 by nerve growth factor receptor via chaperone-mediated autophagy. J. Mol. Cell Boil. 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martínez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-mediated autophagy at a glance. J. Cell Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Boil. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Boil. 2017, 217, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Cuervo, A.M. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol. Metab. 2019, 31, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Boil. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M.; Dice, J.F. Covalent linkage of ribonuclease S-peptide to microinjected proteins causes their intracellular degradation to be enhanced during serum withdrawal. Proc. Natl. Acad. Sci. USA 1986, 83, 5830–5834. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef] [Green Version]
- Aniento, F.; Roche, E.; Cuervo, A.M.; Knecht, E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Boil. Chem. 1993, 268, 10463–10470. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Farese, R.V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef]
- Franch, H.A.; Sooparb, S.; Du, J.; Brown, N.S.; Shen, W.T.T.A.O.L.L. A Mechanism Regulating Proteolysis of Specific Proteins during Renal Tubular Cell Growth. J. Boil. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdor, R.; Mocholí, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U. Cellular Homeostasis and Aging. Annu. Rev. Biochem. 2016, 85, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant -Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Carasa, I.F.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild Type α-Synuclein Is Degraded by Chaperone-mediated Autophagy and Macroautophagy in Neuronal Cells. J. Boil. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Martínez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, P.; Song, W.; Sun, X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009, 23, 3383–3392. [Google Scholar] [CrossRef]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martínez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.-B.; Fu, X.-T.; Shi, Y.; Zhou, J.; Peng, Y.-F.; Liu, W.-R.; Shi, G.-M.; Gao, Q.; Wang, X.-Y.; Song, K.; et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Ali, A.; Nin, D.S.; Tam, J.; Khan, M. Role of Chaperone Mediated Autophagy (CMA) in the Degradation of Misfolded N-CoR Protein in Non-Small Cell Lung Cancer (NSCLC) Cells. PLoS ONE 2011, 6, e25268. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.-L.; Huang, G.-J.; Wang, H.-J.; Chen, J.-L.; Hsu, H.-P.; Lu, T.-J. Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem. Biophys. Res. Commun. 2010, 398, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Bonhoure, A.; Vallentin, A.; Martin, M.; Senff-Ribeiro, A.; Amson, R.; Telerman, A.; Vidal, M. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur. J. Cell Boil. 2017, 96, 83–98. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lv, L.; Li, N.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.-Y.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martínez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Boil. 2019, 17, e3000301. [Google Scholar] [CrossRef] [PubMed]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Jã³ÅºWiak, P.; Forma, E.; Bryå›, M.; Krzeå›Lak, A.; Jóźwiak, P.; Krześlak, A. O-GlcNAcylation and Metabolic Reprograming in Cancer. Front. Endocrinol. 2014, 5, 145. [Google Scholar] [CrossRef] [Green Version]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1 to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef] [Green Version]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-Inducible Factor 1α Is Essential for Cell Cycle Arrest during Hypoxia. Mol. Cell. Boil. 2003, 23, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Hackenbeck, T.; Knaup, K.X.; Schietke, R.E.; Schödel, J.; Willam, C.; Wu, X.; Warnecke, C.; Eckardt, K.-U.; Wiesener, M. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle 2009, 8, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Villalonga, P.; De Mattos, S.F.; Ridley, A.J. RhoE Inhibits 4E-BP1 Phosphorylation and eIF4E Function Impairing Cap-dependent Translation. J. Boil. Chem. 2009, 284, 35287–35296. [Google Scholar] [CrossRef] [Green Version]
- Poch, E.; Miñambres, R.; Mocholí, E.; Ivorra, C.; Pérez-Aragó, A.; Guerri, C.; Pérez-Roger, I.; Guasch, R.M. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp. Cell Res. 2007, 313, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, P.; Guasch, R.M.; Riento, K.; Ridley, A.J.; Huamani, J.; Mcmahan, C.A.; Herbert, D.C.; Reddick, R.; McCarrey, J.R.; MacInnes, M.I.; et al. RhoE Inhibits Cell Cycle Progression and Ras-Induced Transformation. Mol. Cell. Boil. 2004, 24, 8145–8153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Li, L.; Guo, J.; Liu, A.; Wu, J.; Wang, H.; Shi, J.; Pang, D.; Cao, Q. M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 2017, 8, 44465–44476. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade-Tomaz, M.; de Souza, I.; Rocha, C.R.R.; Gomes, L.R. The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer. Cells 2020, 9, 2140. https://doi.org/10.3390/cells9092140
Andrade-Tomaz M, de Souza I, Rocha CRR, Gomes LR. The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer. Cells. 2020; 9(9):2140. https://doi.org/10.3390/cells9092140
Chicago/Turabian StyleAndrade-Tomaz, Marina, Izadora de Souza, Clarissa Ribeiro Reily Rocha, and Luciana Rodrigues Gomes. 2020. "The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer" Cells 9, no. 9: 2140. https://doi.org/10.3390/cells9092140