Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy

 , and
, and
Abstract
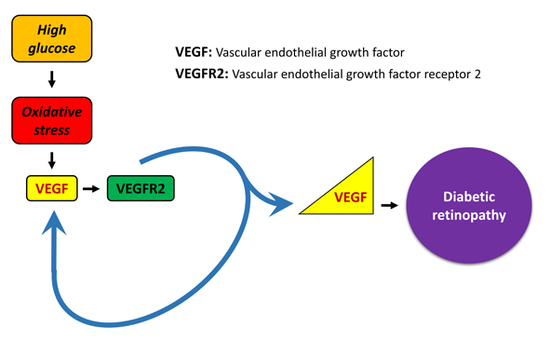
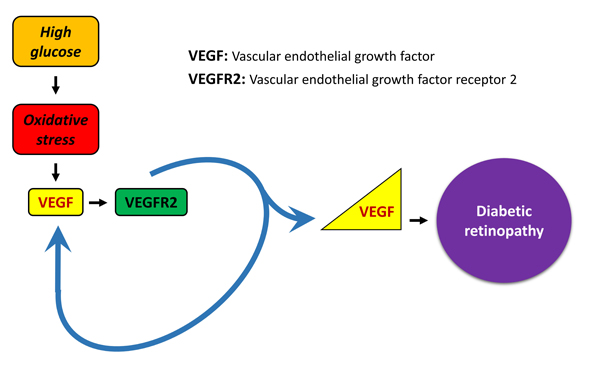
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. In Vitro Studies
2.1.1. MIO-M1 Cell Culture
2.1.2. Cell Viability/Proliferation
2.1.3. Pharmacological Treatments
2.1.4. Preparation of Conditioned Medium
2.1.5. Quantitative Real-Time PCR
2.1.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.1.7. Immunofluorescence
2.2. Ex Vivo Studies
2.2.1. Preparation of Retinal Explants
2.2.2. Pharmacological Treatments
2.2.3. Preparation of Conditioned Media
2.2.4. Quantitative Real-Time PCR
2.2.5. ELISA
2.2.6. Immunofluorescence
2.3. Statistics
3. Results
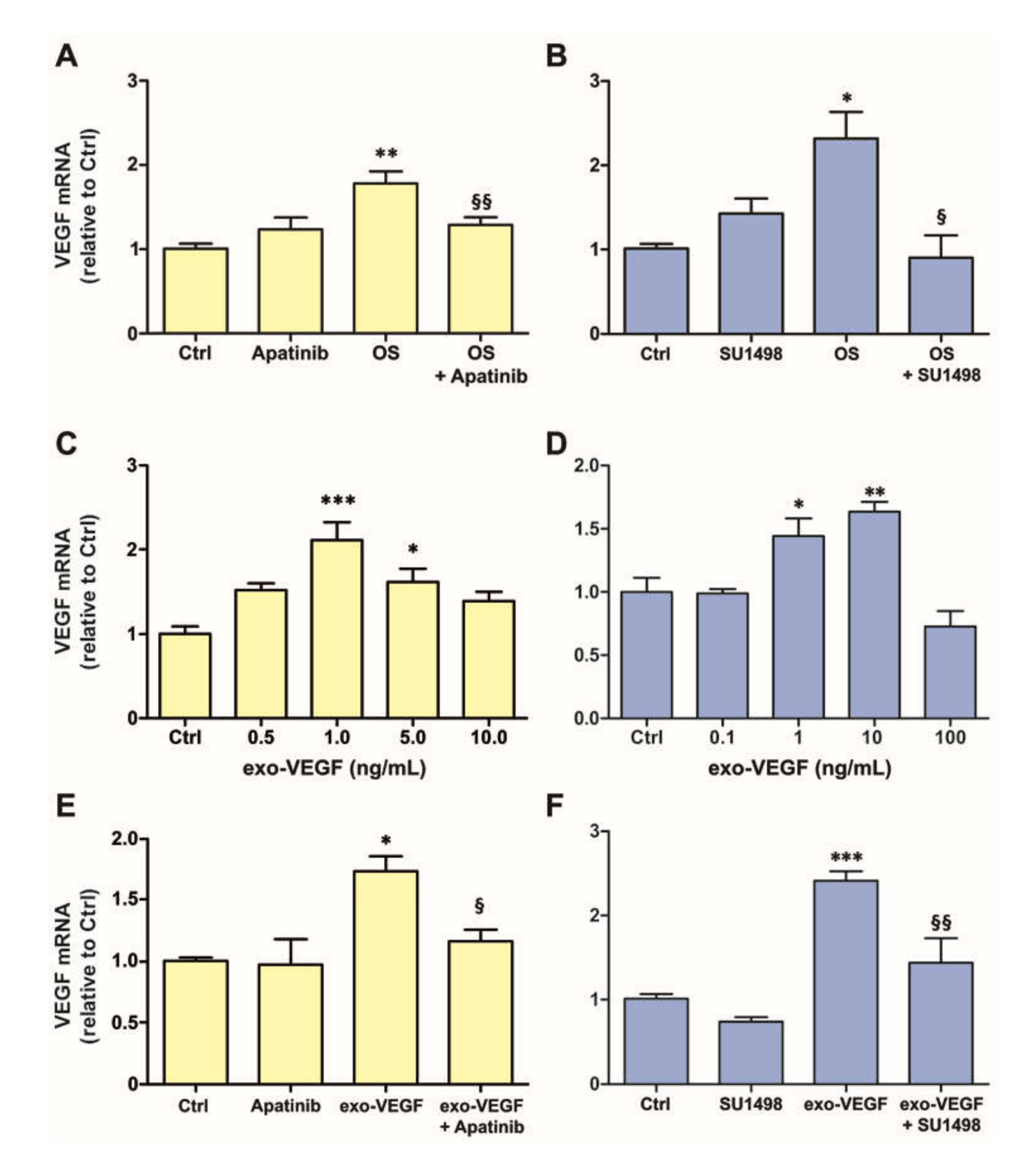
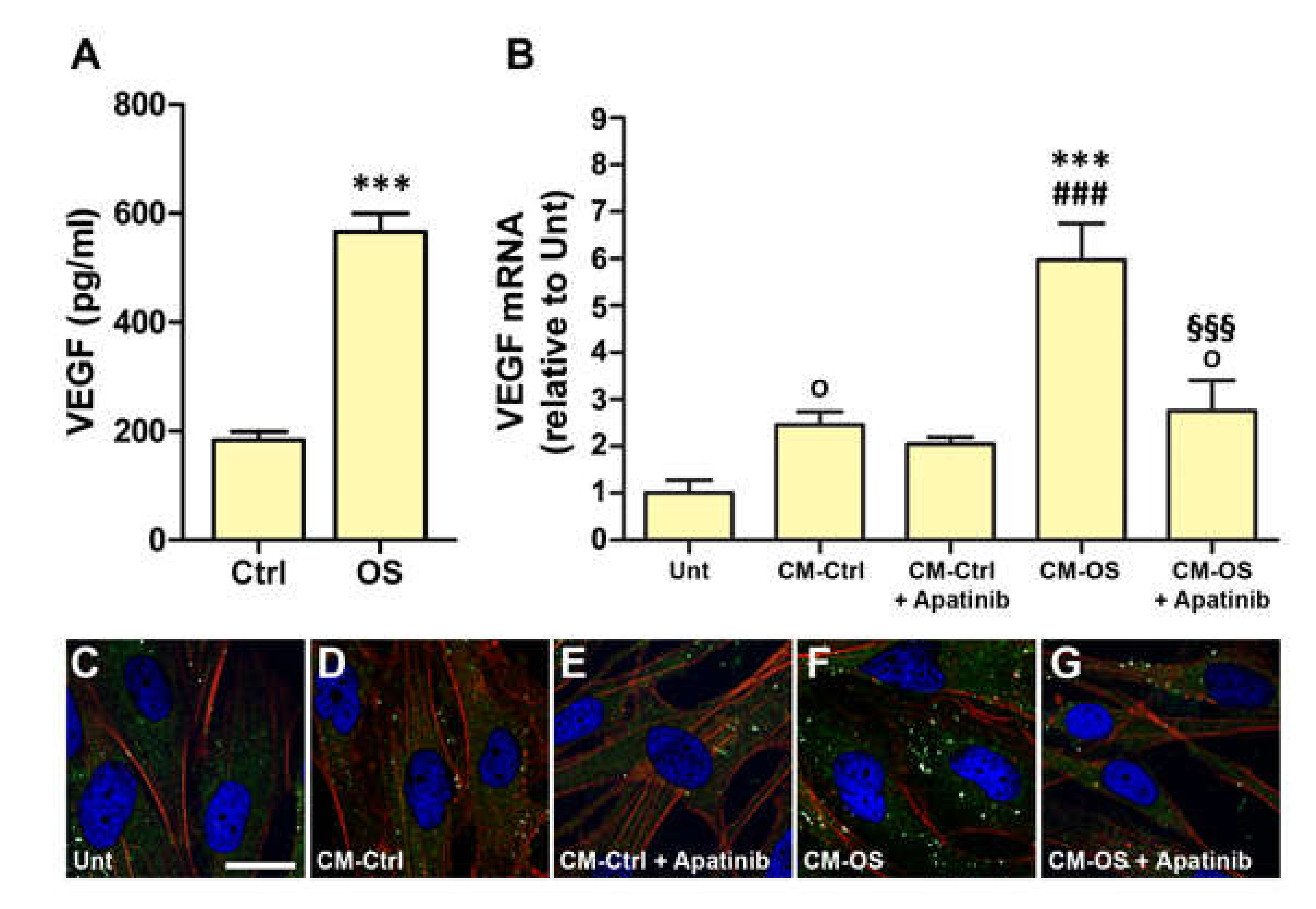
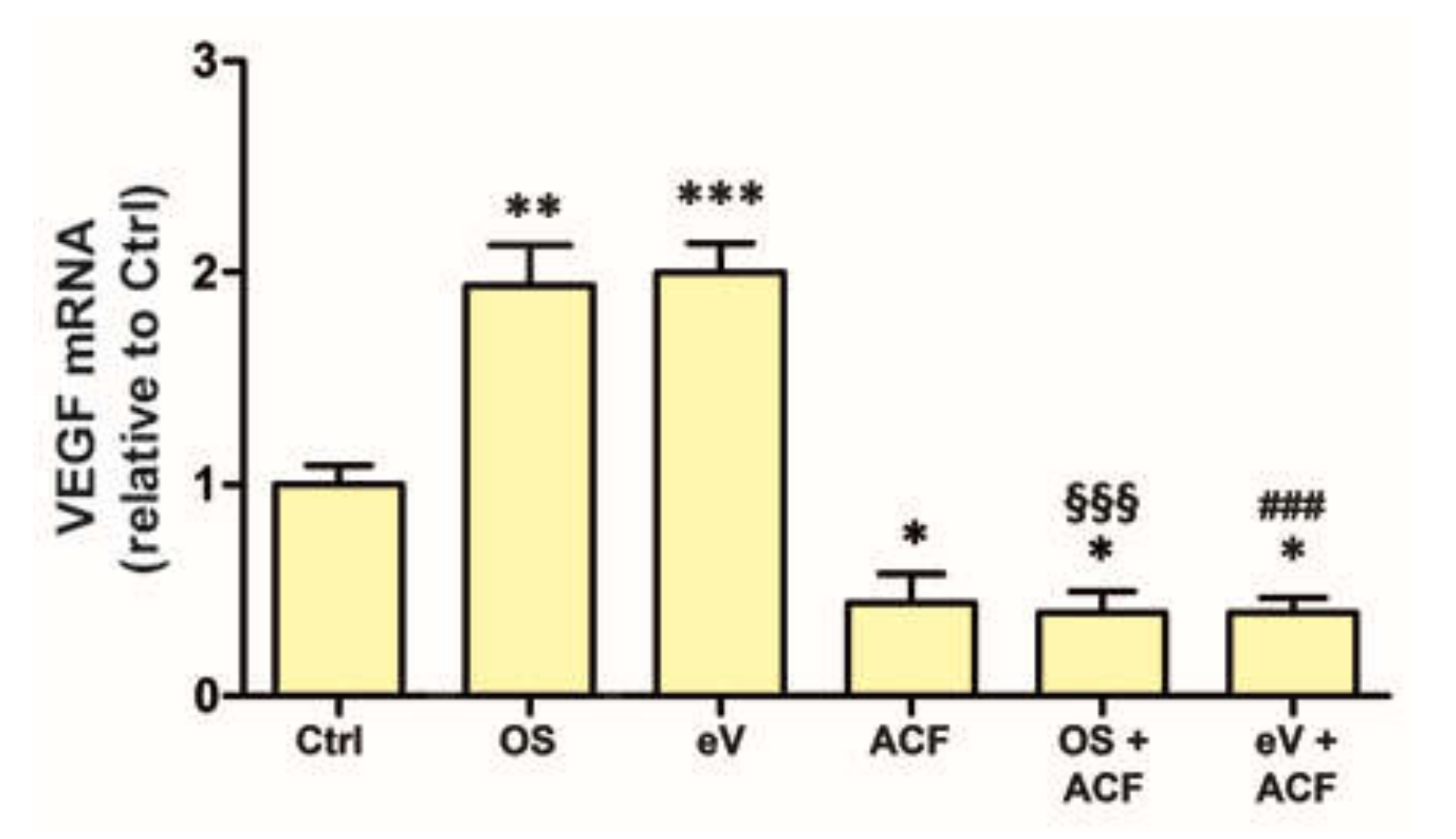
3.1. OS Induces VEGFR2-Dependent VEGF Expression Both in MIO-M1 Cells and in Retinal Explants
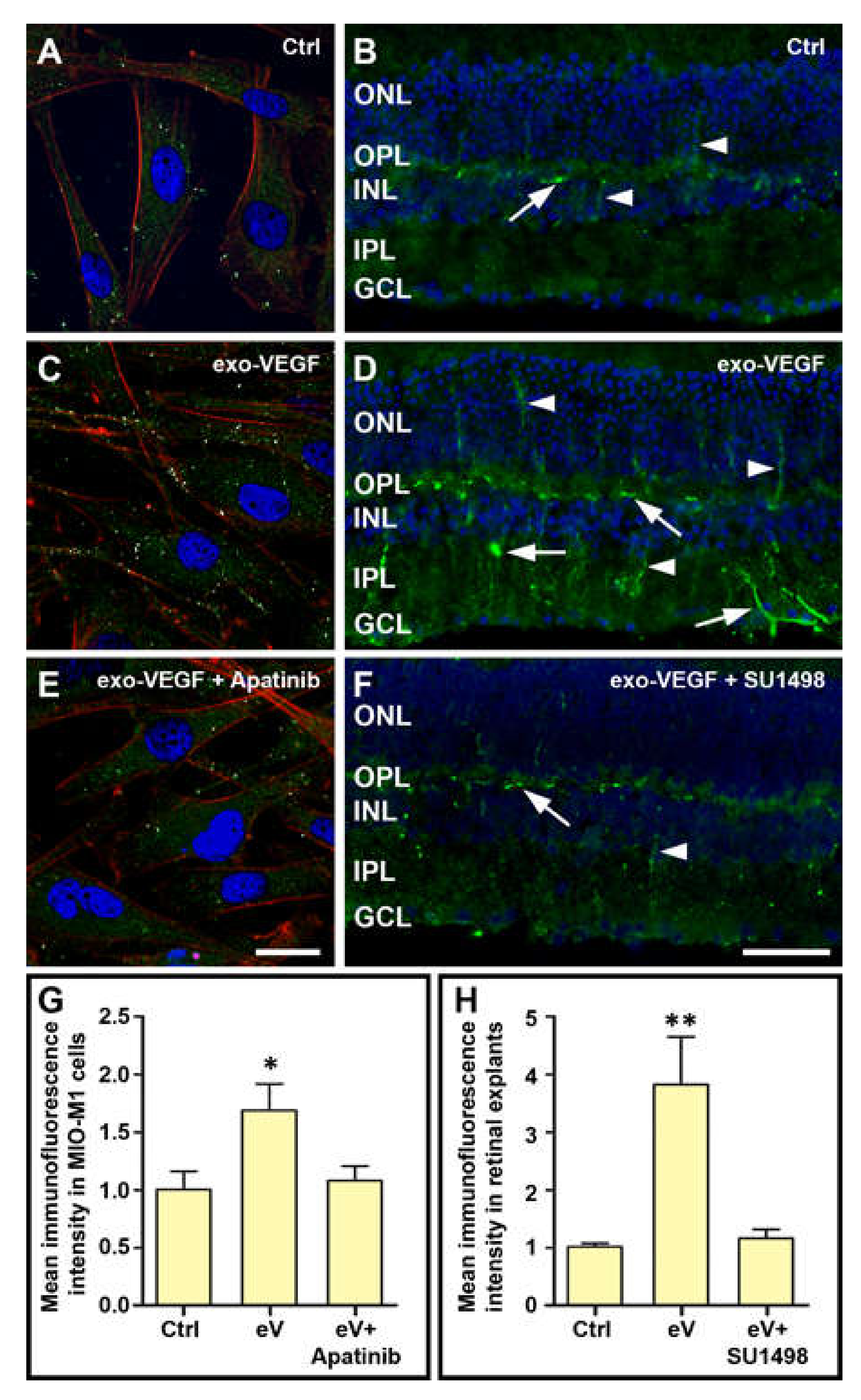
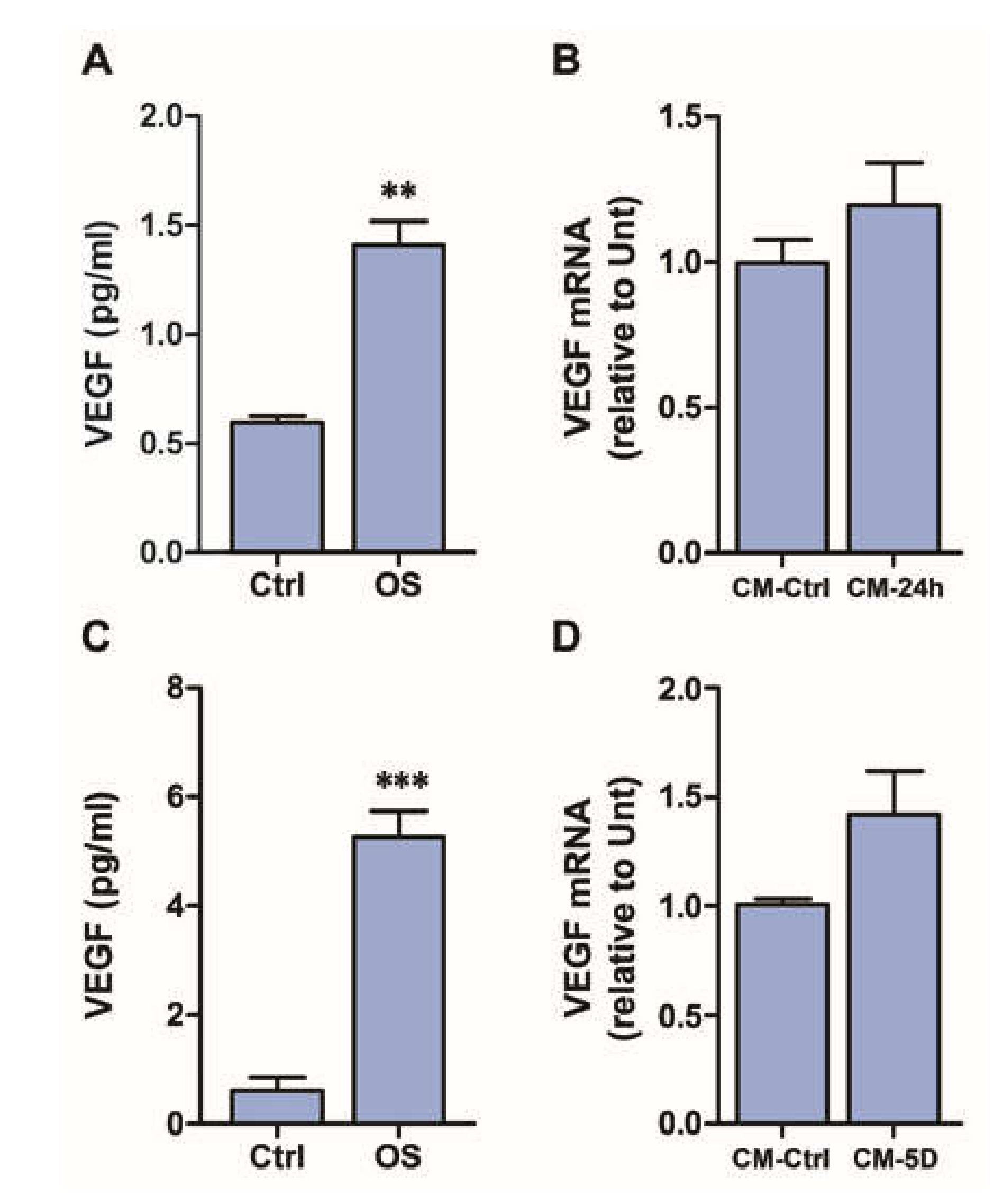
3.2. Exogenous VEGF Induces VEGFR2-Dependent VEGF Expression Both in MIO-M1 Cells and in Retinal Explants
3.3. Conditioned Medium Induces VEGF Expression in MIO-M1 Cells
3.4. Conditioned Medium Does Not Induce VEGF Expression in Retinal Explants
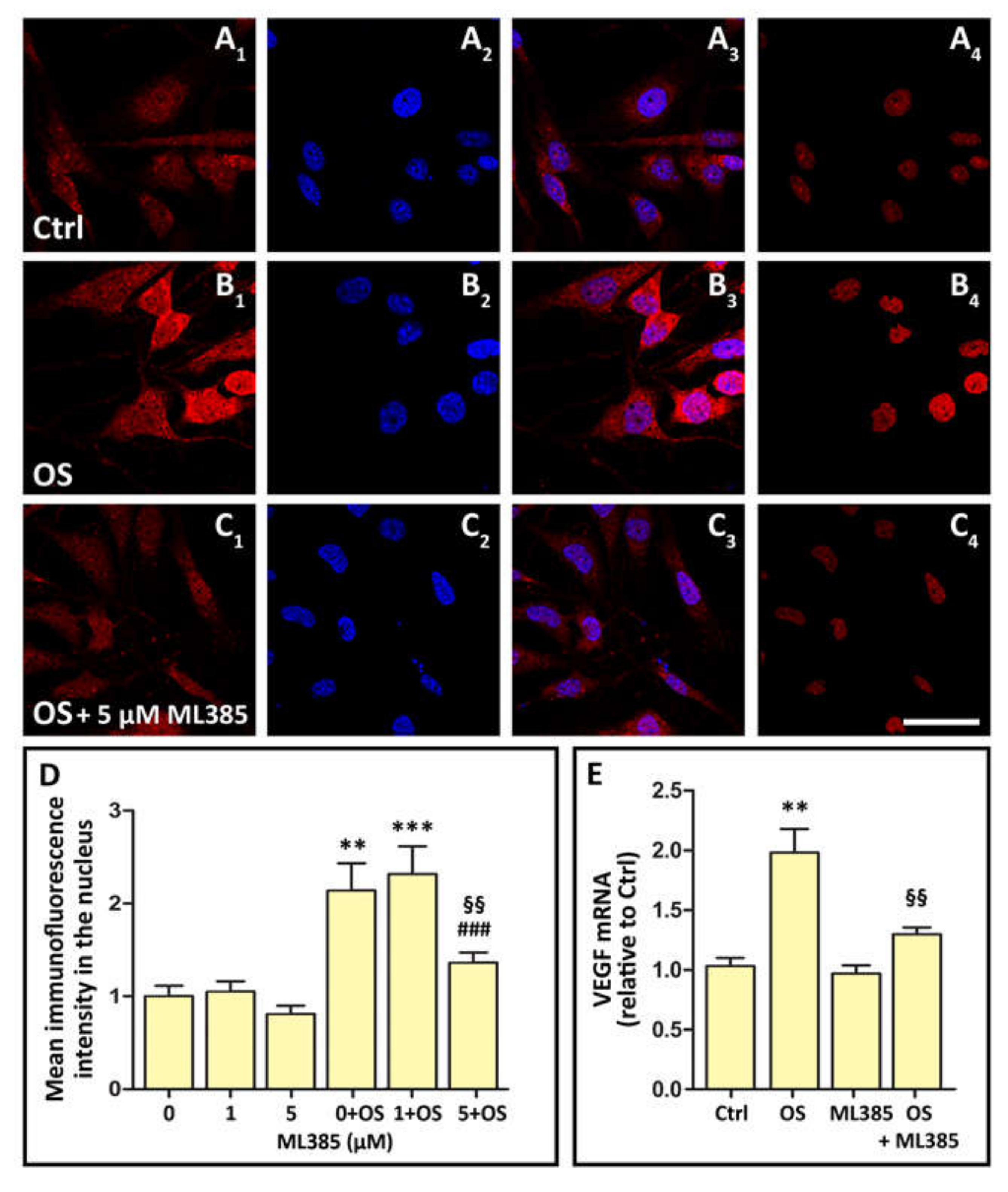
3.5. OS Triggers Nrf2 Nuclear Translocation, while OS-Induced VEGF Expression Is Inhibited by Nrf2 Blockade
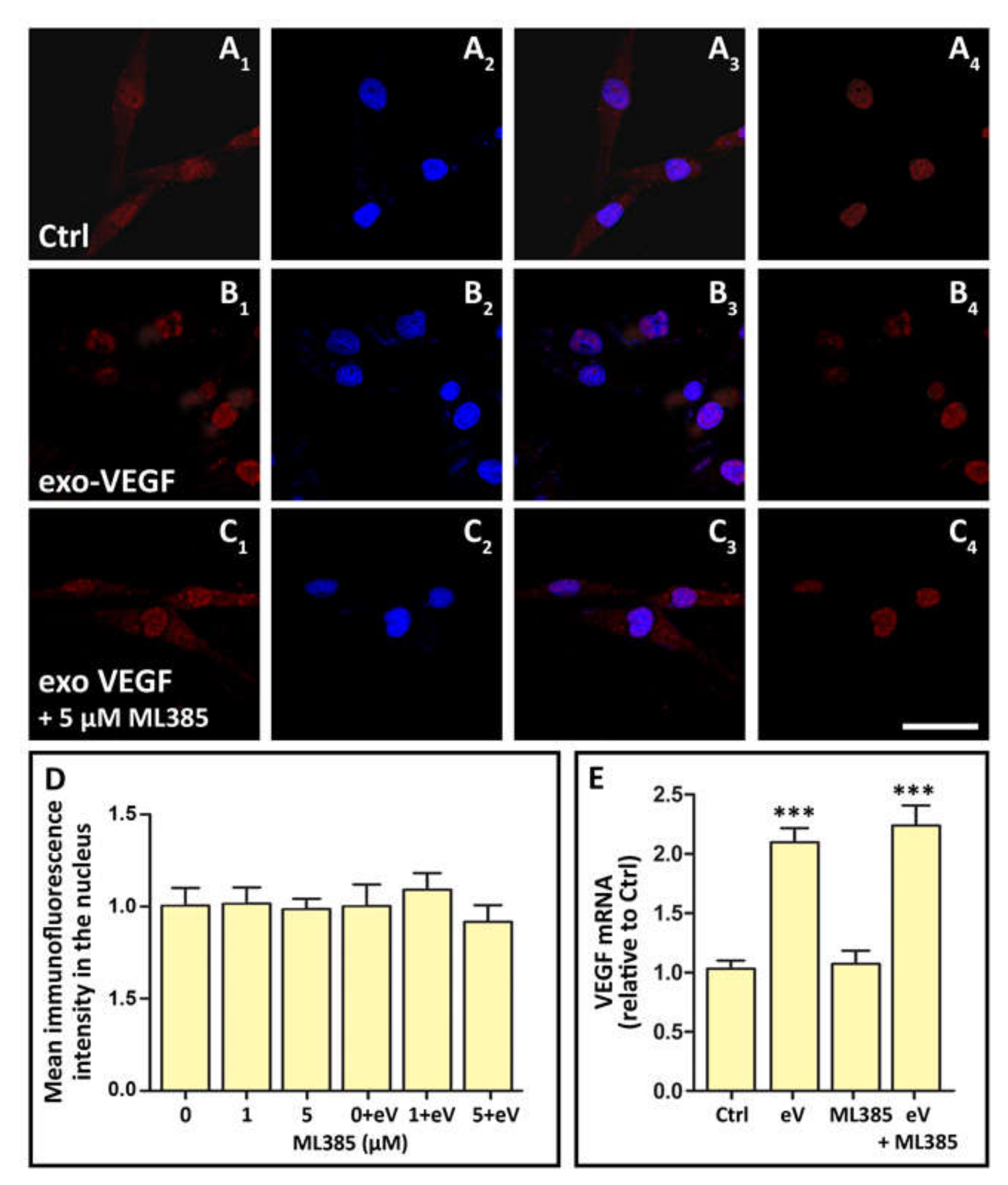
3.6. Exo-VEGF Has No Effect on Nrf2 Nuclear Translocation, while Exo-VEGF-Induced VEGF Expression Is Not Affected by Nrf2 Blockade
3.7. Blockade of HIF-1 Inhibits VEGF mRNA Expression in All Conditions
4. Discussion
4.1. The Role of OS
4.2. The Involvement of VEGFR2
4.3. The Involvement of Müller Cells
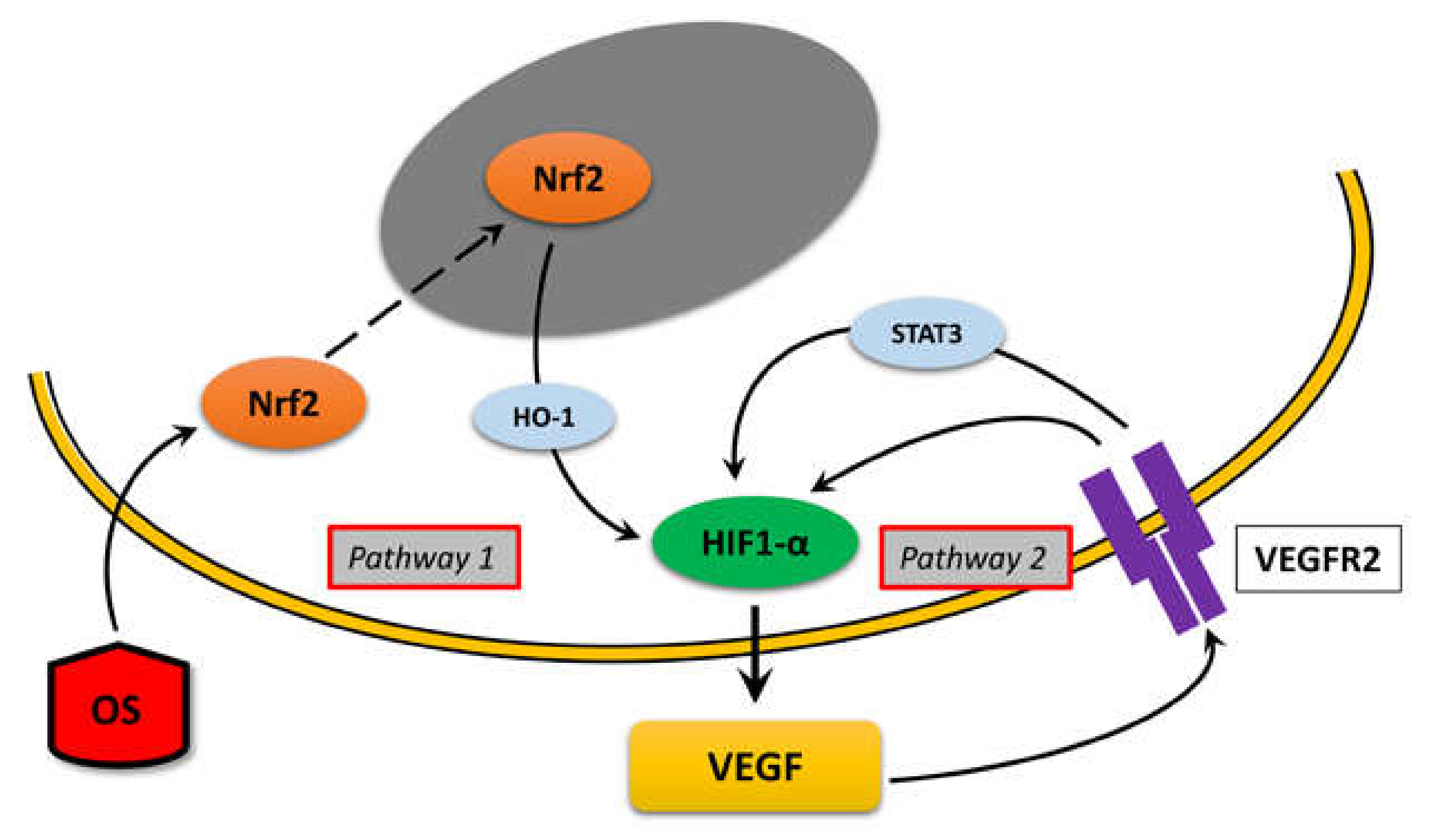
4.4. Intracellular Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, M.; Wickremasinghe, S.; Osborne, A.; Van Wijngaarden, P.; Martin, K.R. Diabetic retinopathy: A complex pathophysiology requiring novel therapeutic strategies. Expert Opin. Biol. Ther. 2018, 18, 1257–1270. [Google Scholar] [CrossRef]
- Hernández, C.; Monte, M.D.; Simó, R.; Casini, G. Neuroprotection as a Therapeutic Target for Diabetic Retinopathy. J. Diabetes Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, R.; Biagioni, M.; Cammalleri, M.; Monte, M.D.; Casini, G. VEGF as a Survival Factor in Ex Vivo Models of Early Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2016, 57, 3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Storkebaum, E.; De Almodóvar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Rossino, M.G.; Monte, M.D.; Casini, G. Relationships Between Neurodegeneration and Vascular Damage in Diabetic Retinopathy. Front. Mol. Neurosci. 2019, 13, 1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, K.; Morishige, K.-I.; Takahashi, T.; Hashimoto, K.; Tsutsumi, S.; Yin, L.; Ohta, T.; Kawagoe, J.; Takahashi, K.; Kurachi, H. Fasudil-induced hypoxia-inducible factor-1 degradation disrupts a hypoxia-driven vascular endothelial growth factor autocrine mechanism in endothelial cells. Mol. Cancer Ther. 2008, 7, 1551–1561. [Google Scholar] [CrossRef] [Green Version]
- Ohba, T.; Cates, J.M.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Autocrine VEGF/VEGFR1 Signaling in a Subpopulation of Cells Associates with Aggressive Osteosarcoma. Mol. Cancer Res. 2014, 12, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- González-Pacheco, F.R.; Deudero, J.J.P.; Castellanos, M.C.; Castilla, M.A.; Álvarez-Arroyo, M.V.; Yagüe, S.; Caramelo, C. Mechanisms of endothelial response to oxidative aggression: Protective role of autologous VEGF and induction of VEGFR2 by H2O2. Am. J. Physiol. Circ. Physiol. 2006, 291, H1395–H1401. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Xu, X.; Zhang, L.; Fu, Y.; Wang, M.; Gu, H.; Xie, X. Blocking autocrine VEGF signaling by sunitinib, an anti-cancer drug, promotes embryonic stem cell self-renewal and somatic cell reprogramming. Cell Res. 2014, 24, 1121–1136. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Chen, S.; Li, K.; Xiao, X.; Xu, T.; Zheng, S. Upregulated autocrine vascular endothelial growth factor (VEGF)/VEGF receptor-2 loop prevents apoptosis in haemangioma-derived endothelial cells. Br. J. Dermatol. 2014, 170, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; Maharaj, A.S.R.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous VEGF Is Required for Visual Function: Evidence for a Survival Role on Müller Cells and Photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byeon, S.H.; Lee, S.C.; Choi, S.H.; Lee, H.K.; Lee, J.H.; Chu, Y.K.; Kwon, O.W. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Chen, T.T.; Barber, C.L.; Jordan, M.C.; Murdock, J.; Desai, S.; Ferrara, N.; Nagy, A.; Roos, K.P.; Iruela-Arispe, M.L. Autocrine VEGF Signaling Is Required for Vascular Homeostasis. Cell 2007, 130, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, M.; Platt, D.H.; Lemtalsi, T.; Gu, X.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. VEGF differentially activates STAT3 in microvascular endothelial cells. FASEB J. 2003, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Monte, M.D.; Martini, D.; Ristori, C.; Azara, D.; Armani, C.; Balbarini, A.; Bagnoli, P. Hypoxia effects on proangiogenic factors in human umbilical vein endothelial cells: Functional role of the peptide somatostatin. Naunyn Schmiedeberg’s Arch. Pharmacol. 2011, 383, 593–612. [Google Scholar] [CrossRef]
- Deudero, J.J.P.; Caramelo, C.; Castellanos, M.C.; Neria, F.; Fernández-Sánchez, R.; Calabia, O.; Peñate, S.; González-Pacheco, F.R. Induction of Hypoxia-inducible Factor 1α Gene Expression by Vascular Endothelial Growth Factor. J. Biol. Chem. 2008, 283, 11435–11444. [Google Scholar] [CrossRef] [Green Version]
- Klettner, A.; Westhues, D.; Lassen, J.; Bartsch, S.; Roider, J. Regulation of constitutive vascular endothelial growth factor secretion in retinal pigment epithelium/choroid organ cultures: p38, nuclear factor kappaB, and the vascular endothelial growth factor receptor-2/phosphatidylinositol 3 kinase pathway. Mol. Vis. 2013, 19, 281–291. [Google Scholar]
- Mei, S.; Cammalleri, M.; Azara, D.; Casini, G.; Bagnoli, P.; Monte, M.D. Mechanisms underlying somatostatin receptor 2 down-regulation of vascular endothelial growth factor expression in response to hypoxia in mouse retinal explants. J. Pathol. 2011, 226, 519–533. [Google Scholar] [CrossRef]
- Grigsby, J.; Allen, D.; Ferrigno, A.; Vellanki, S.; Pouw, C.; Hejny, W.; Tsin, A.T. Autocrine and Paracrine Secretion of Vascular Endothelial Growth Factor in the Pre-Hypoxic Diabetic Retina. Curr. Diabetes Rev. 2017, 13, 161–174. [Google Scholar] [CrossRef]
- Matsuda, M.; Krempel, P.G.; Marquezini, M.V.; Sholl-Franco, A.; Lameu, A.; Monteiro, M.L.R.; Miguel, N.C.D.O. Cellular stress response in human Müller cells (MIO-M1) after bevacizumab treatment. Exp. Eye Res. 2017, 160, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.-Z. VEGF production and signaling in Müller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vis. Res. 2017, 139, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2016, 103, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, T.; Westenskow, P.D.; Friedlander, M. Hypoxia-inducible factor (HIF)/vascular endothelial growth factor (VEGF) signaling in the retina. Adv. Exp. Med. Biol. 2014, 801, 275–281. [Google Scholar]
- Cervia, D.; Catalani, E.; Monte, M.D.; Casini, G. Vascular endothelial growth factor in the ischemic retina and its regulation by somatostatin. J. Neurochem. 2012, 120, 818–829. [Google Scholar] [CrossRef]
- Behl, T.; Kotwani, A. Exploring the various aspects of the pathological role of vascular endothelial growth factor (VEGF) in diabetic retinopathy. Pharmacol. Res. 2015, 99, 137–148. [Google Scholar] [CrossRef]
- Klettner, A.; Roider, J. Mechanisms of Pathological VEGF Production in the Retina and Modification with VEGF-Antagonists. Stud. Retinal Choroidal Disord. 2012, 277–305. [Google Scholar]
- Penn, J.S.; Madan, A.; Caldwell, R.; Bartoli, M.; Hartnett, M.; Caldwell, R. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [Green Version]
- Amato, R.; Monte, M.D.; Lulli, M.; Raffa, V.; Casini, G. Nanoparticle-Mediated Delivery of Neuroprotective Substances for the Treatment of Diabetic Retinopathy. Curr. Neuropharmacol. 2018, 16, 993–1003. [Google Scholar] [CrossRef]
- Semeraro, F.; Cancarini, A.; Dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Canning, P.; Sorrell, F.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Pan, H.; Wang, H.; Li, X.; Bu, X.; Wang, Q.; Gao, Y.; Wen, G.; Zhou, Y.; Cong, Z.; et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci. Rep. 2016, 6, 37338. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [Green Version]
- Paramasivan, P.; Kankia, I.H.; Langdon, S.P.; Deeni, Y.Y. Emerging role of nuclear factor erythroid 2-related factor 2 in the mechanism of action and resistance to anticancer therapies. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Dong, F.; Zhou, X.; Li, C.; Yan, S.; Deng, X.; Cao, Z.; Li, L.; Tang, B.; Allen, T.; Liu, J. Dihydroartemisinin targets VEGFR2 via the NF-κB pathway in endothelial cells to inhibit angiogenesis. Cancer Biol. Ther. 2014, 15, 1479–1488. [Google Scholar] [CrossRef] [Green Version]
- Basagiannis, D.; Zografou, S.; Murphy, C.; Fotsis, T.; Morbidelli, L.; Ziche, M.; Bleck, C.; Mercer, J.; Christoforidis, S. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation via macropinocytosis. J. Cell Sci. 2016, 129, 4091–4104. [Google Scholar] [CrossRef] [Green Version]
- Dougher, M.; I Terman, B. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene 1999, 18, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 174, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Huang, W.; Yang, S.F.; Zhang, X.P.; Yu, Q.; Zhang, Z.Q.; Yao, J.; Li, K.R.; Jiang, Q.; Cao, C. Galphai1 and Galphai3mediate VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis. Theranostics 2018, 8, 4695–4709. [Google Scholar] [CrossRef]
- Devoldere, J.; Peynshaert, K.; De Smedt, S.C.; Remaut, K. Müller cells as a target for retinal therapy. Drug Discov. Today 2019, 24, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Cáceres-Del-Carpio, J.; Moustafa, M.T.; Toledo-Corral, J.; Hamid, M.A.; Atilano, S.R.; Schneider, K.; Fukuhara, P.S.; Costa, R.D.; Norman, J.L.; Malik, D.; et al. In vitro response and gene expression of human retinal Müller cells treated with different anti-VEGF drugs. Exp. Eye Res. 2020, 191, 107903. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Dong, S.; Zhu, M.; Le, Y.-Z. VEGF as a Trophic Factor for Müller Glia in Hypoxic Retinal Diseases. In Plant Promoters and Transcription Factors; Springer: Cham, Switzerland, 2018; pp. 473–478. [Google Scholar] [CrossRef]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.; Le, Y.-Z. Müller Glia Are a Major Cellular Source of Survival Signals for Retinal Neurons in Diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, H.J.; Chiang, C.W.; Chen, J.; Song, S.K. Vitreous volume of the mouse measured by quantitative high-resolution MRI. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4414. [Google Scholar]
- Arjamaa, O.; Nikinmaa, M. Oxygen-dependent diseases in the retina: Role of hypoxia-inducible factors. Exp. Eye Res. 2006, 83, 473–483. [Google Scholar] [CrossRef]
- Agani, F.; Jiang, B.-H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 2013, 13, 245–251. [Google Scholar] [CrossRef]
- Borsi, E.; Terragna, C.; Brioli, A.; Tacchetti, P.; Martello, M.; Cavo, M. Therapeutic targeting of hypoxia and hypoxia-inducible factor 1 alpha in multiple myeloma. Transl. Res. 2015, 165, 641–650. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kim, M.S.; Park, J.W. Oxygen-Dependent and -Independent Regulation of HIF-1alpha. J. Korean Med. Sci. 2002, 17, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Di Gesualdo, F.; Capaccioli, S.; Lulli, M. A pathophysiological view of the long non-coding RNA world. Oncotarget 2014, 5, 10976–10996. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.K.; Diamond, P.; Gronthos, S.; Peet, D.J.; Zannettino, A.C. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia 2011, 25, 1533–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacher, S.E.; Levings, D.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, B.; Ren, Q. Baicalin relieves hypoxia-aroused H9c2 cell apoptosis by activating Nrf2/HO-1-mediated HIF1alpha/BNIP3 pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3657–3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.K.; Park, J.H.; Yun, J.A.; Cha, J.H.; Kim, Y.; Won, M.H.; Kim, K.W.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Heme oxygenase metabolites improve astrocytic mitochondrial function via a Ca2 + -dependent HIF-1alpha/ERRalpha circuit. PloS ONE 2018, 13, e0202039. [Google Scholar]
- Kweider, N.; Fragoulis, A.; Rosen, C.; Pecks, U.; Rath, W.; Pufe, T.; Wruck, C.J. Interplay between vascular endothelial growth factor (VEGF) and nuclear factor erythroid 2-related factor-2 (Nrf2): Implications for preeclampsia. J. Biol. Chem. 2011, 286, 42863–42872. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Niu, N.; Wei, T.; Tozawa, H.; Chen, X.; Zhang, C.; Zhang, J.; Wada, Y.; Kapron, C.M.; Liu, J. The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget 2017, 8, 69139–69161. [Google Scholar] [CrossRef] [Green Version]
- Rossino, M.G.; Casini, G. Nutraceuticals for the Treatment of Diabetic Retinopathy. Nutrition 2019, 11, 771. [Google Scholar] [CrossRef] [Green Version]
- Amato, R.; Rossino, M.G.; Cammalleri, M.; Locri, F.; Pucci, L.; Monte, M.D.; Casini, G. Lisosan G Protects the Retina from Neurovascular Damage in Experimental Diabetic Retinopathy. Nutrition 2018, 10, 1932. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossino, M.G.; Lulli, M.; Amato, R.; Cammalleri, M.; Dal Monte, M.; Casini, G. Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy. Cells 2020, 9, 1452. https://doi.org/10.3390/cells9061452
Rossino MG, Lulli M, Amato R, Cammalleri M, Dal Monte M, Casini G. Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy. Cells. 2020; 9(6):1452. https://doi.org/10.3390/cells9061452
Chicago/Turabian StyleRossino, Maria Grazia, Matteo Lulli, Rosario Amato, Maurizio Cammalleri, Massimo Dal Monte, and Giovanni Casini. 2020. "Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy" Cells 9, no. 6: 1452. https://doi.org/10.3390/cells9061452