Overshooting Subcellular Redox-Responses in Rett-Mouse Hippocampus during Neurotransmitter Stimulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Mouse Model for RTT
2.2. Solutions
2.3. Dissociated Cell Cultures
2.4. Culturing Media
2.5. roGFP Transduction
2.6. Immunostaining
2.7. Optical Recordings and 2-Photon Imaging
2.8. Calibration of roGFP
2.9. Calibration of Fura-2
2.10. Spectrofluorometric Quantification of H2O2
2.11. Statistics
3. Results
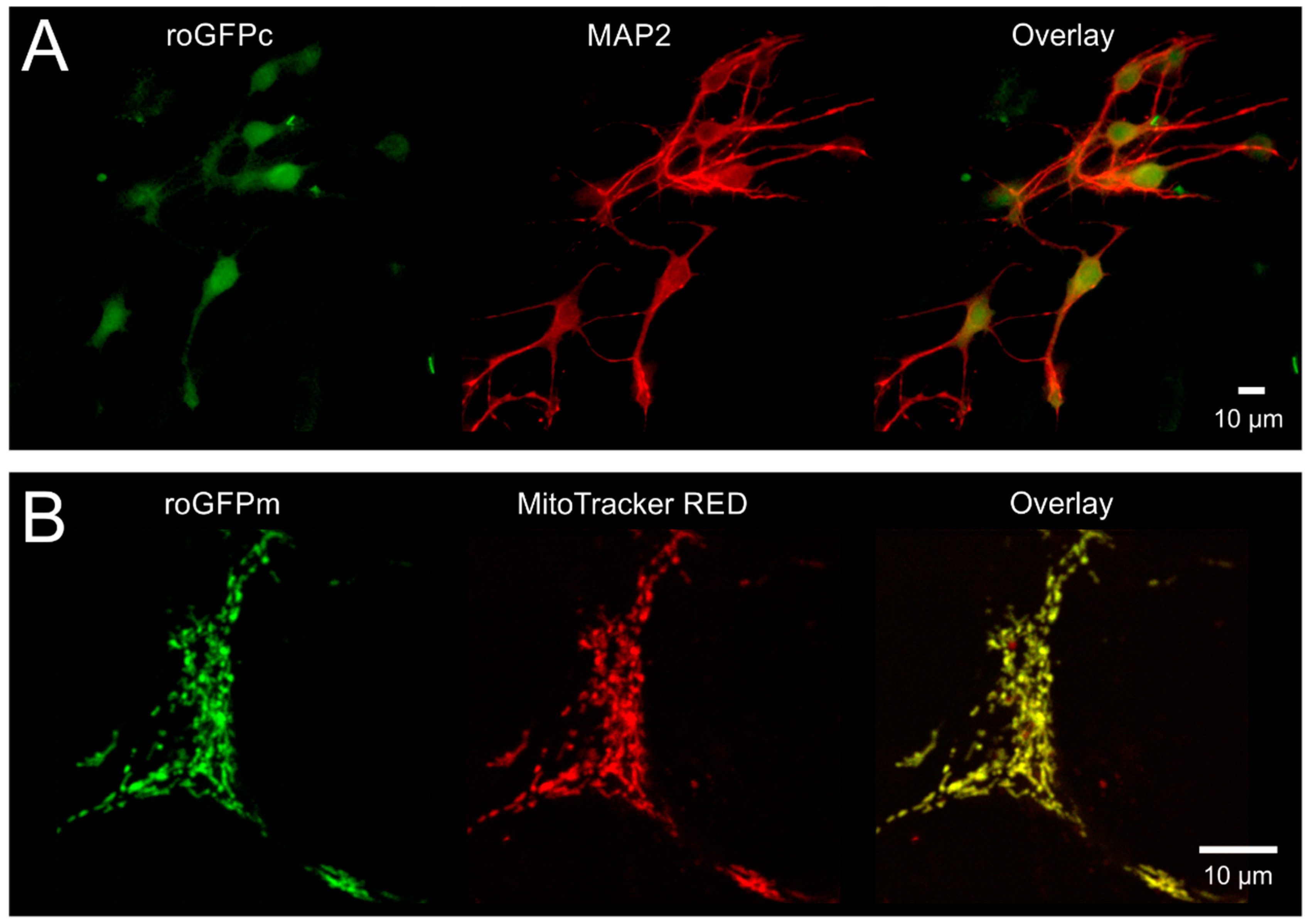
3.1. Cellular Targeting and Subcellular Expression of roGFP
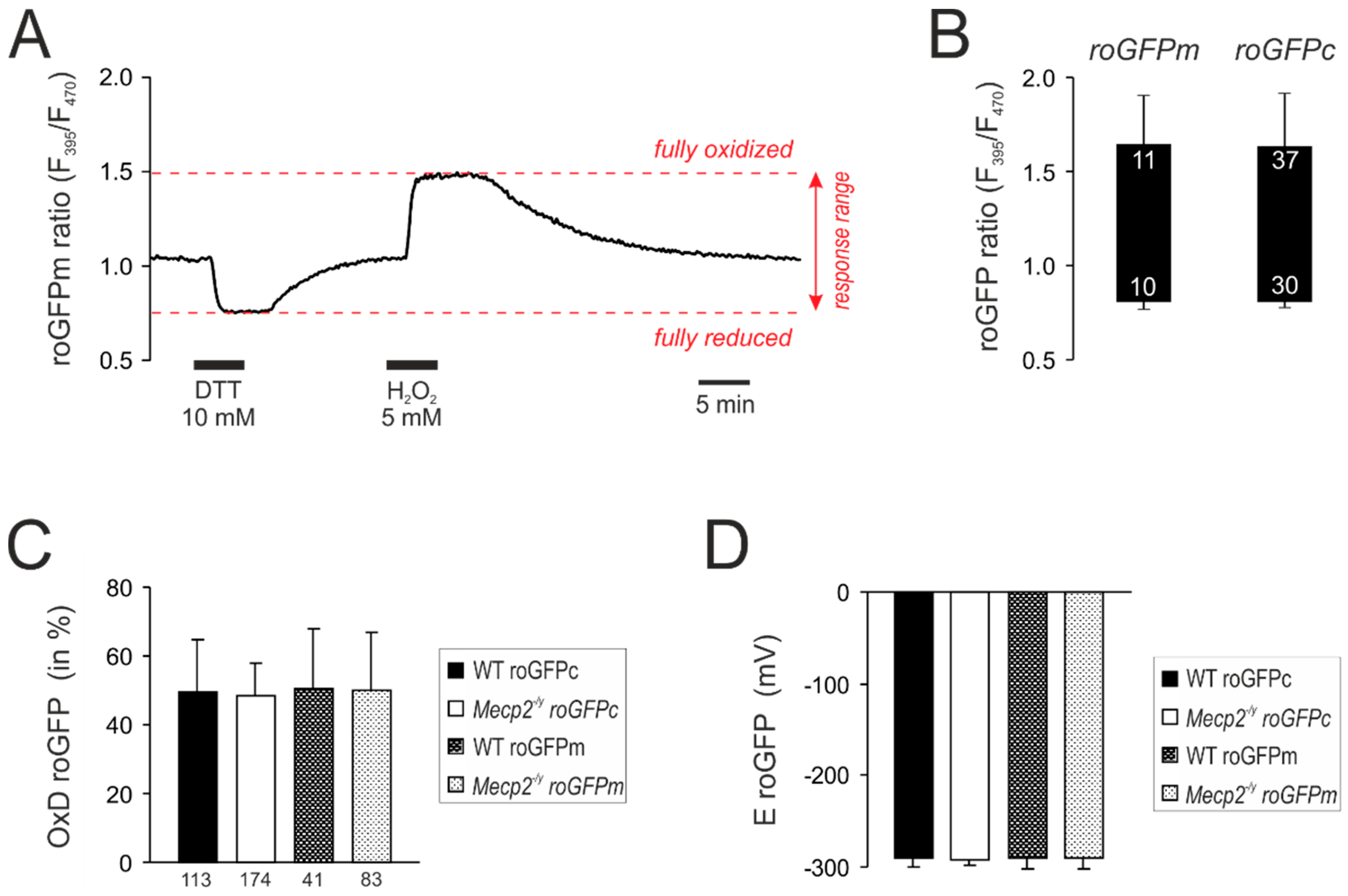
3.2. Response Calibrations of roGFPc and roGFPm
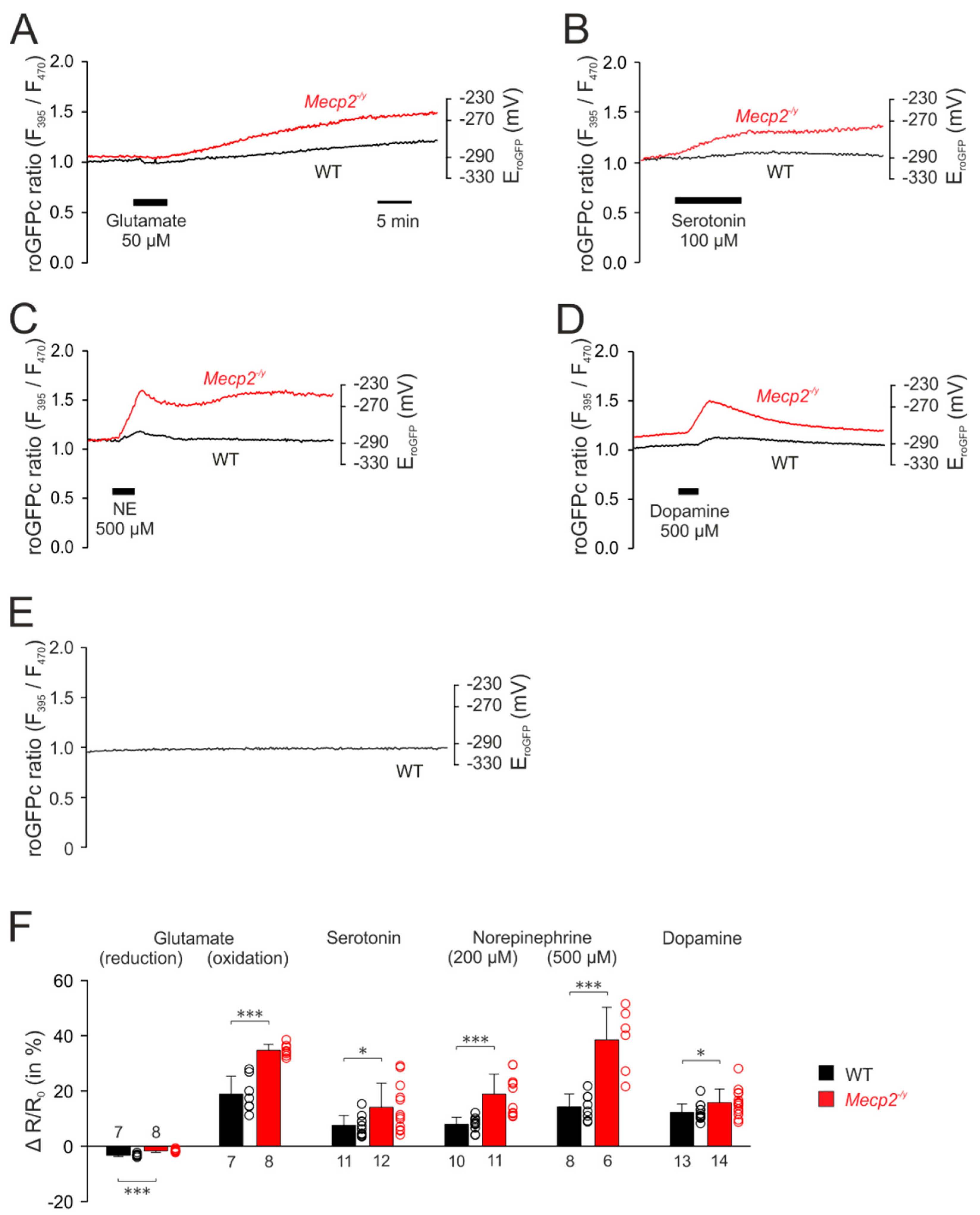
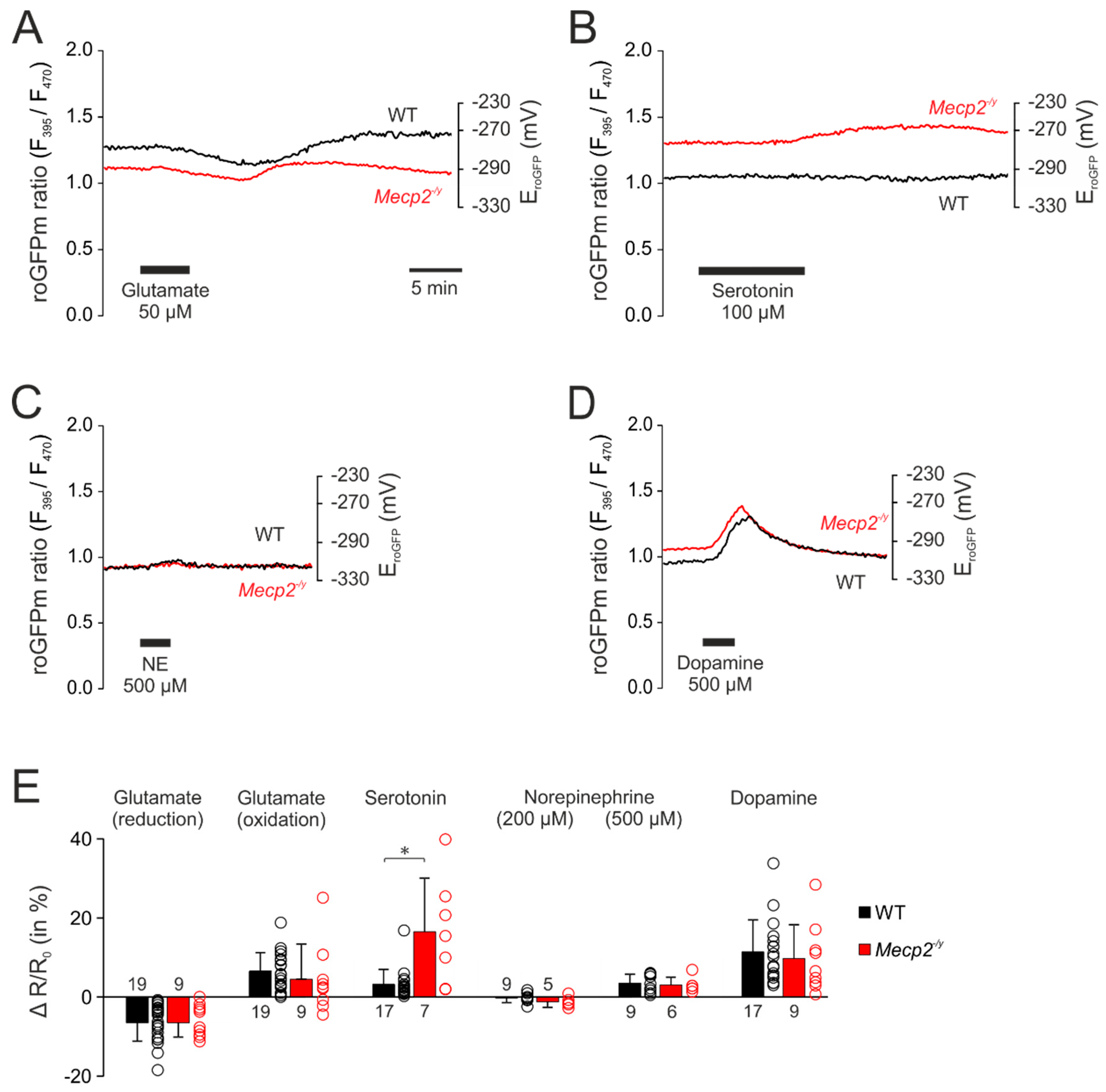
3.3. Neurotransmitter-Mediated Redox Changes
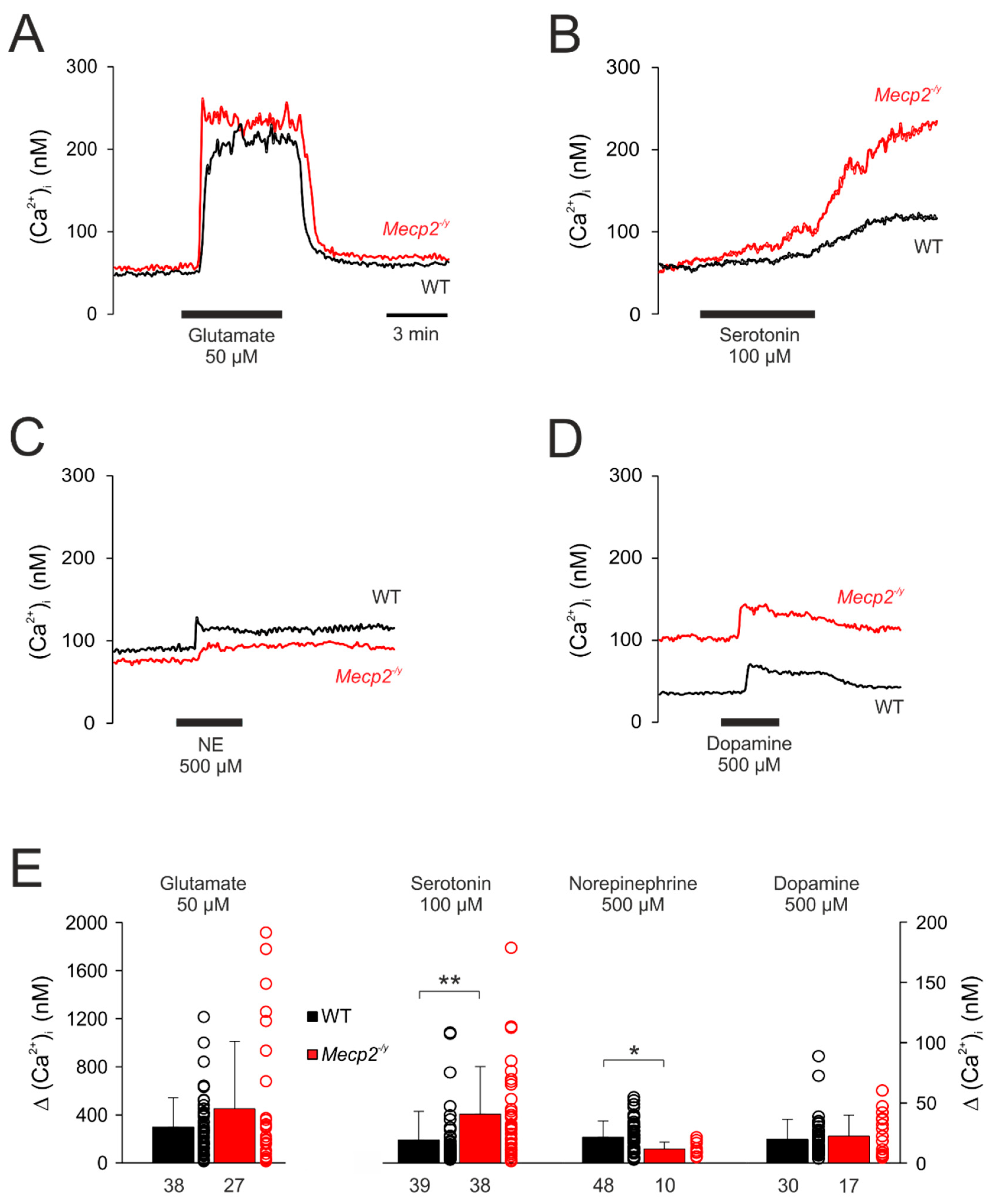
3.4. Effect of Neurotransmitters on Cytosolic Calcium Influx
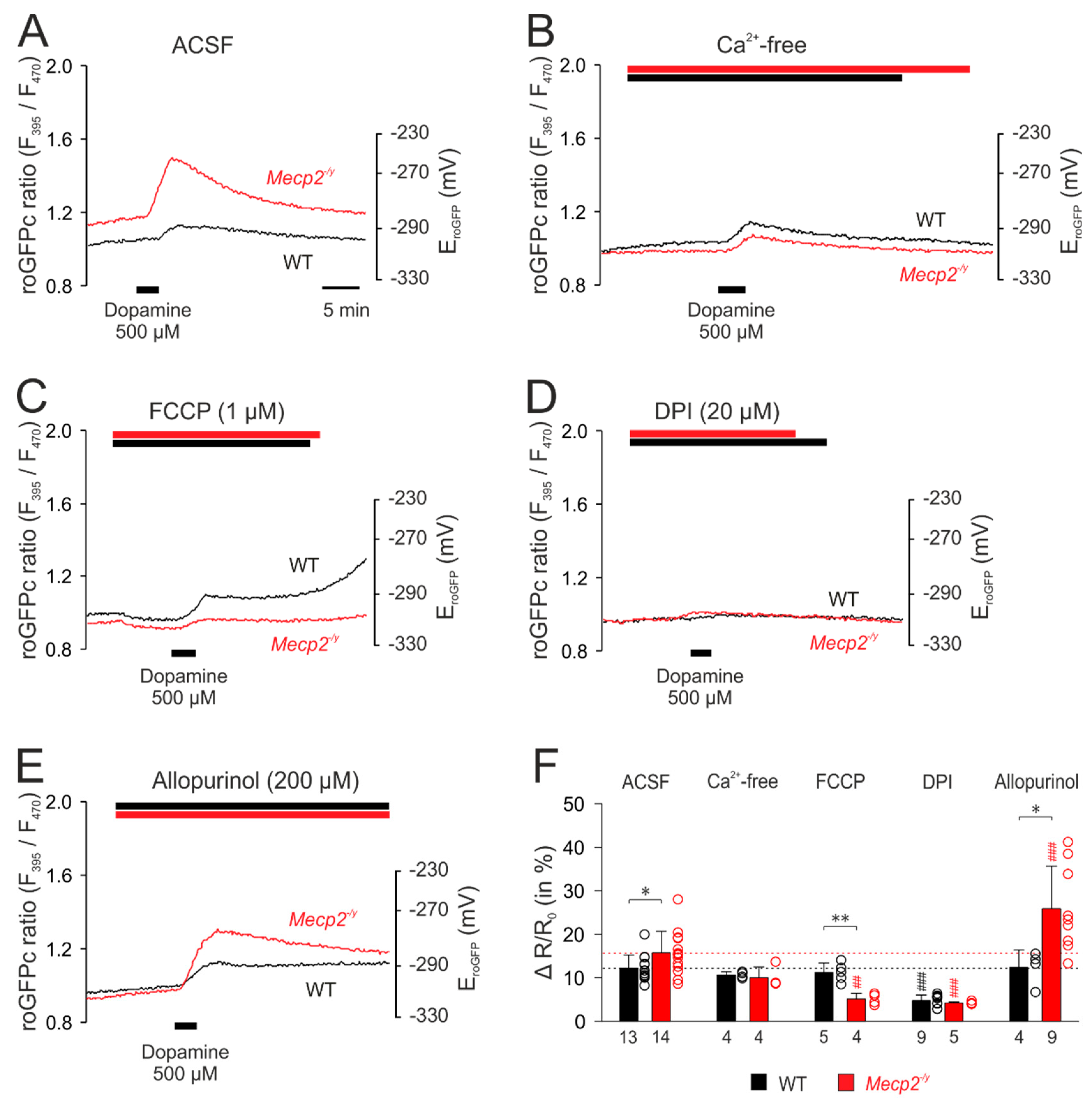
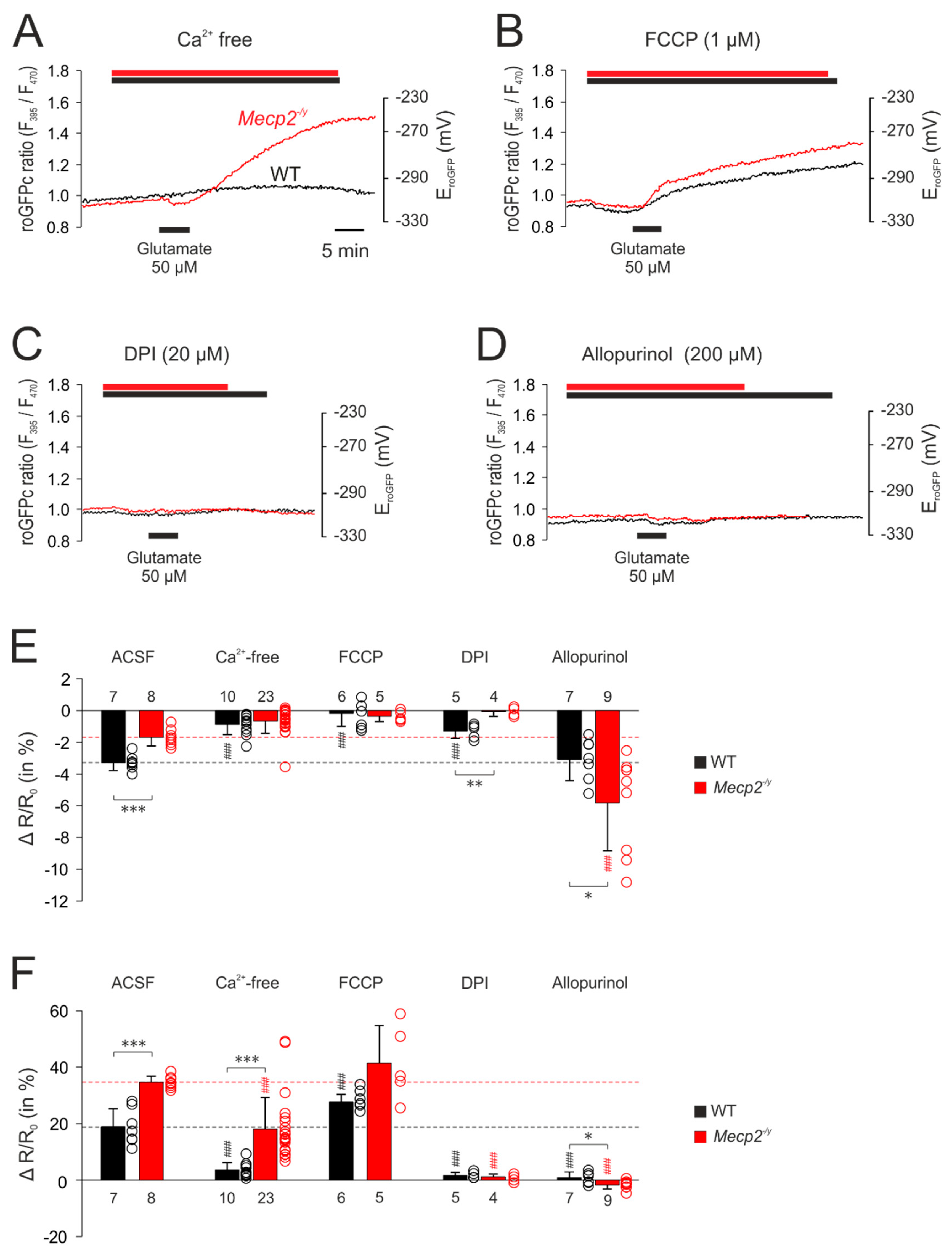
3.5. Uncovering the Redox-Signaling Pathways Involved
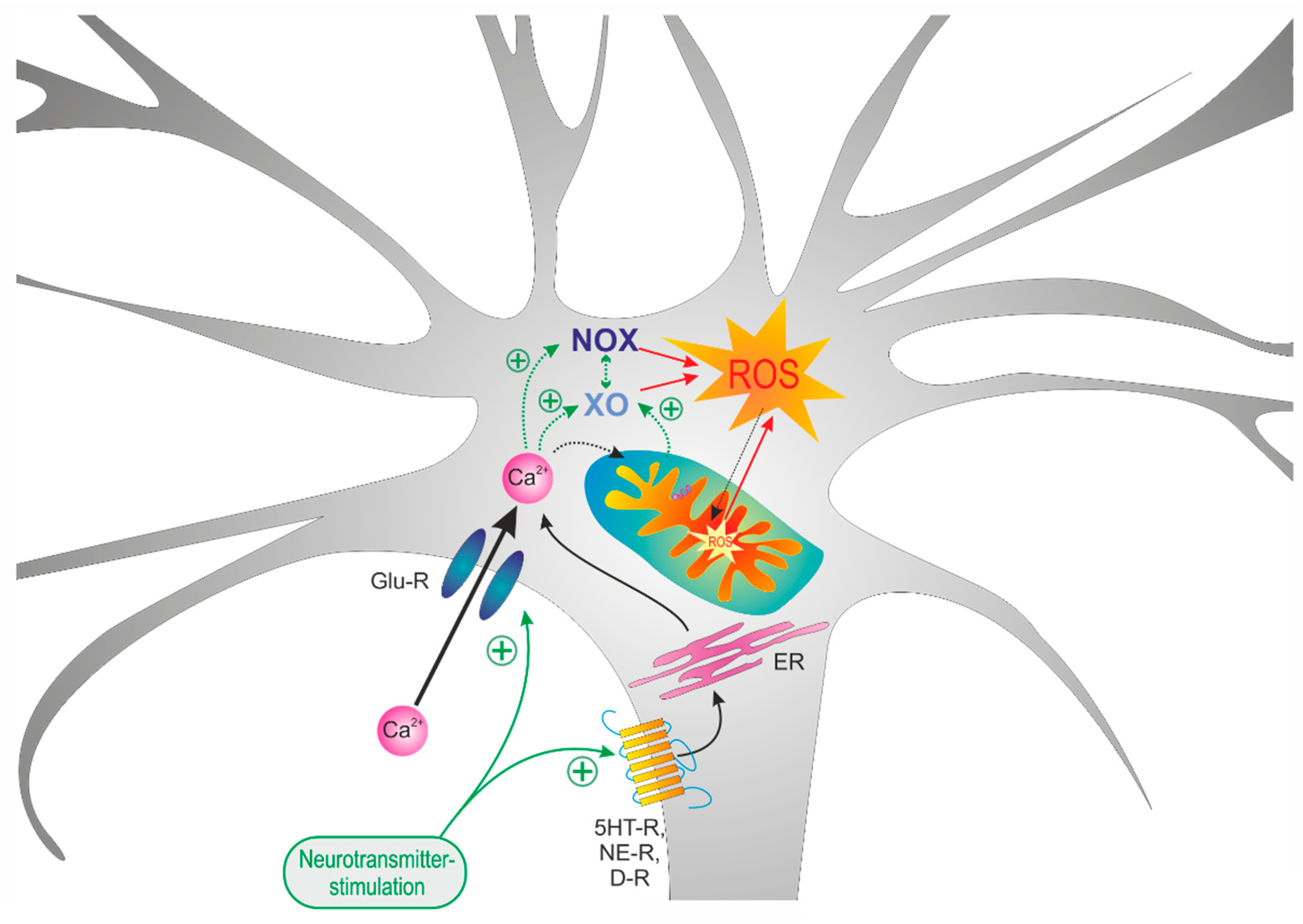
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ΔR/R0 | change in fluorescence ratio, normalized to control ratio |
| ACSF | artificial cerebrospinal fluid |
| DIV | days in vitro |
| DMSO | dimethyl sulfoxide |
| DTT | 1,4-dithio-DL-threitol |
| DPI | diphenyleneiodonium chloride |
| E0′roGFP | standard redox potential |
| EroGFP | roGFP reduction potential |
| F 380max/F 380min | fluorescence ratio at 380 nm excitation, Ca2+-free/Ca2+-saturated |
| F 395/F 470 | fluorescence ratio at 395 nm excitation and 470 nm excitation |
| F 470ox | fluorescence intensity at 470 nm excitation, fully oxidized |
| F 470ox/F 470red | fluorescence ratio at 470 nm, fully oxidized/fully reduced |
| F 470red | fluorescence intensity at 470 nm excitation, fully reduced |
| FCCP | carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| FCS | fetal calf serum |
| HBSS | Hanks’-balanced salt solution |
| HEPES | 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid |
| MAP2 | microtubule associated protein 2 |
| MeCP2 | methyl-CpG binding protein 2, protein |
| MECP2 | methyl-CpG binding protein 2, encoding gene (human) |
| Mecp2 | methyl-CpG binding protein 2, encoding gene (mouse) |
| Mecp2−/y | MeCP2-deficient male mouse (hemizygous) |
| Mecp2+/− | MeCP2-deficient female mouse (heterozygous) |
| MEM | minimal essential medium |
| NOX | NADPH oxidase |
| OxDroGFP | relative degree of roGFP oxidation |
| PBS | phosphate buffered saline |
| R | fluorescence ratio |
| roGFP | reduction oxidation-sensitive green fluorescent protein |
| roGFPc | roGFP expressed in cytosol |
| roGFPm | roGFP expressed in mitochondrial matrix |
| ROS | reactive oxygen species |
| Rox | ratio corresponding to full oxidation |
| Rred | ratio corresponding to full reduction |
| RTT | Rett syndrome |
| SOD1 | superoxide dismutase 1, Cu/Zn superoxide dismutase |
| WT | wildtype |
| XO | xanthine oxidase |
| XDH | xanthine dehydrogenase |
References
- Hagberg, B. Rett’s syndrome: Prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr. Scand. 1985, 74, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Rett, A. Über ein eigenartiges hirnatrophisches Syndrom bei Hyperammonämie im Kindesalter. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, R.J.; Glaze, D.G.; Motil, K.J.; Armstrong, D.D.; del Junco, D.J.; Hubbard, C.R.; Percy, A.K. The pattern of growth failure in Rett syndrome. Am. J. Dis. Child. 1993, 147, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Nagarajan, L.; de Klerk, N.; Jacoby, P.; Ellaway, C.; Leonard, H. Sleep problems in Rett syndrome. Brain Dev. 2007, 29, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, S.M.; Bailey, M.E.; Cobb, S.R. Rett syndrome: From bed to bench. Pediatr. Neonatol. 2011, 52, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marschik, P.B.; Kaufmann, W.E.; Sigafoos, J.; Wolin, T.; Zhang, D.; Bartl-Pokorny, K.D.; Pini, G.; Zappella, M.; Tager-Flusberg, H.; Einspieler, C.; et al. Changing the perspective on early development of Rett syndrome. Res. Dev. Disabil. 2013, 34, 1236–1239. [Google Scholar] [CrossRef] [Green Version]
- Marschik, P.B.; Sigafoos, J.; Kaufmann, W.E.; Wolin, T.; Talisa, V.B.; Bartl-Pokorny, K.D.; Budimirovic, D.B.; Vollmann, R.; Einspieler, C. Peculiarities in the gestural repertoire: An early marker for Rett syndrome? Res. Dev. Disabil. 2012, 33, 1715–1721. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Amir, R.E.; Zoghbi, H.Y. Rett syndrome: Methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am. J. Med. Genet. 2000, 97, 147–152. [Google Scholar] [CrossRef]
- Huppke, P.; Held, M.; Hanefeld, F.; Engel, W.; Laccone, F. Influence of mutation type and location on phenotype in 123 patients with Rett syndrome. Neuropediatrics 2002, 33, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Balmer, D.; Goldstine, J.; Rao, Y.M.; LaSalle, J.M. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J. Mol. Med. 2003, 81, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.R.; Matarazzo, V.; Palmer, A.M.; Tu, Y.; Jeon, O.H.; Pevsner, J.; Ronnett, G.V. Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol. Cell. Neurosci. 2003, 22, 417–429. [Google Scholar] [CrossRef]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, B.C.; Johnston, M.V.; Blue, M.E. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience 2004, 123, 939–949. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L.; Arin, D.M. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology 1995, 45, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D. Neuropathology of Rett syndrome. J. Child Neurol. 2005, 20, 747–753. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Oldfors, A.; Hagberg, B.; Dahlstrom, A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 1994, 5, 1509–1513. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Wright, E.E.; Belichenko, N.P.; Masliah, E.; Li, H.H.; Mobley, W.C.; Francke, U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: Evidence for disruption of neuronal networks. J. Comp. Neurol. 2009, 514, 240–258. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Percy, A.K.; Glaze, D.G.; Butler, I.J.; Riccardi, V.M. Reduction of biogenic amine levels in the Rett syndrome. N. Engl. J. Med. 1985, 313, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Milstien, S.; Butler, I.J.; Smith, E.O.; Kaufman, S.; Glaze, D.G.; Percy, A.K. Cerebrospinal fluid biogenic amines and biopterin in Rett syndrome. Ann. Neurol. 1989, 25, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Brucke, T.; Sofic, E.; Killian, W.; Rett, A.; Riederer, P. Reduced concentrations and increased metabolism of biogenic amines in a single case of Rett-syndrome: A postmortem brain study. J. Neural Transm. 1987, 68, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Lekman, A.; Witt-Engerström, I.; Holmberg, B.; Percy, A.; Svennerholm, L.; Hagberg, B. CSF and urine biogenic amine metabolites in Rett syndrome. Clin. Genet. 1990, 37, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.K. Neurochemistry of the Rett syndrome. Brain Dev. 1992, 14, S57–S62. [Google Scholar] [PubMed]
- Segawa, M. Pathophysiology of Rett syndrome from the standpoint of early catecholamine disturbance. Eur. Child Adolesc. Psychiatry 1997, 6 (Suppl. 1), 56–60. [Google Scholar]
- Wenk, G.L.; Hauss-Wegrzyniak, B. Altered cholinergic function in the basal forebrain of girls with Rett syndrome. Neuropediatrics 1999, 30, 125–129. [Google Scholar] [CrossRef]
- Wenk, G.L. Rett syndrome: Neurobiological changes underlying specific symptoms. Prog. Neurobiol. 1997, 51, 383–391. [Google Scholar] [CrossRef]
- Wenk, G.L.; Mobley, S.L. Choline acetyltransferase activity and vesamicol binding in Rett syndrome and in rats with nucleus basalis lesions. Neuroscience 1996, 73, 79–84. [Google Scholar] [CrossRef]
- Hamberger, A.; Gillberg, C.; Palm, A.; Hagberg, B. Elevated CSF glutamate in Rett syndrome. Neuropediatrics 1992, 23, 212–213. [Google Scholar] [CrossRef]
- Lappalainen, R.; Riikonen, R.S. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr. Neurol. 1996, 15, 213–216. [Google Scholar] [CrossRef]
- Lappalainen, R.; Lindholm, D.; Riikonen, R. Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J. Child Neurol. 1996, 11, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, R.; Vanhala, R. Levels of cerebrospinal fluid nerve-growth factor differ in infantile autism and Rett syndrome. Dev. Med. Child Neurol. 1999, 41, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Matsuishi, T.; Nagamitsu, S.; Yamashita, Y.; Murakami, Y.; Kimura, A.; Sakai, T.; Shoji, H.; Kato, H.; Percy, A.K. Decreased cerebrospinal fluid levels of substance P in patients with Rett syndrome. Ann. Neurol. 1997, 42, 978–981. [Google Scholar] [CrossRef]
- Deguchi, K.; Antalffy, B.A.; Twohill, L.J.; Chakraborty, S.; Glaze, D.G.; Armstrong, D.D. Substance P immunoreactivity in Rett syndrome. Pediatr. Neurol. 2000, 22, 259–266. [Google Scholar] [CrossRef]
- Budden, S.S.; Myer, E.C.; Butler, I.J. Cerebrospinal fluid studies in the Rett syndrome: Biogenic amines and beta-endorphins. Brain Dev. 1990, 12, 81–84. [Google Scholar] [CrossRef]
- Ogier, M.; Katz, D.M. Breathing dysfunction in Rett syndrome: Understanding epigenetic regulation of the respiratory network. Respir. Physiol. Neurobiol. 2008, 164, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Jian, L.; Nagarajan, L.; de Klerk, N.; Ravine, D.; Bower, C.; Anderson, A.; Williamson, S.; Christodoulou, J.; Leonard, H. Predictors of seizure onset in Rett syndrome. J. Pediatr. 2006, 149, 542–547. [Google Scholar] [CrossRef]
- Goffin, D.; Zhou, Z.J. The neural circuit basis of Rett syndrome. Front. Biol. (Beijing) 2012, 7, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Calfa, G.; Hablitz, J.J.; Pozzo-Miller, L. Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J. Neurophysiol. 2011, 105, 1768–1784. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Reuter, J.; Gerich, F.J.; Hildebrandt, B.; Hägele, S.; Katschinski, D.; Müller, M. Enhanced hypoxia susceptibility in hippocampal slices from a mouse model of Rett syndrome. J. Neurophysiol. 2009, 101, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Partin, J. Evidence for mitochondrial control of neuronal polarity. J. Neurosci. Res. 1999, 56, 8–20. [Google Scholar] [CrossRef]
- Vayssiere, J.L.; Cordeau-Lossouarn, L.; Larcher, J.C.; Basseville, M.; Gros, F.; Croizat, B. Participation of the mitochondrial genome in the differentiation of neuroblastoma cells. In Vitro Cell. Dev. Biol. 1992, 28A, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Chada, S.R.; Hollenbeck, P.J. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol. 2004, 14, 1272–1276. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Peng, H.B. Mitochondrial clustering at the vertebrate neuromuscular junction during presynaptic differentiation. J. Neurobiol. 2006, 66, 522–536. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J. Neurosci. 2004, 24, 10878–10887. [Google Scholar] [CrossRef] [Green Version]
- Kamsler, A.; Segal, M. Paradoxical actions of hydrogen peroxide on long-term potentiation in transgenic superoxide dismutase-1 mice. J. Neurosci. 2003, 23, 10359–10367. [Google Scholar] [CrossRef] [Green Version]
- Cornford, M.E.; Philippart, M.; Jacobs, B.; Scheibel, A.B.; Vinters, H.V. Neuropathology of Rett syndrome: Case report with neuronal and mitochondrial abnormalities in the brain. J. Child Neurol. 1994, 9, 424–431. [Google Scholar] [CrossRef]
- Ruch, A.; Kurczynski, T.W.; Velasco, M.E. Mitochondrial alterations in Rett syndrome. Pediatr. Neurol. 1989, 5, 320–323. [Google Scholar] [CrossRef]
- Eeg-Olofsson, O.; al-Zuhair, A.G.; Teebi, A.S.; Daoud, A.S.; Zaki, M.; Besisso, M.S.; Al-Essa, M.M. Rett syndrome: A mitochondrial disease? J. Child Neurol. 1990, 5, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Coker, S.B.; Melnyk, A.R. Rett syndrome and mitochondrial enzyme deficiencies. J. Child Neurol. 1991, 6, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.H.; Slobedman, B.; Harikrishnan, K.N.; Williamson, S.L.; Minchenko, D.; El-Osta, A.; Stern, J.L.; Christodoulou, J. Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci. 2010, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriaucionis, S.; Paterson, A.; Curtis, J.; Guy, J.; Macleod, N.; Bird, A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell. Biol. 2006, 26, 5033–5042. [Google Scholar] [CrossRef] [Green Version]
- Can, K.; Menzfeld, C.; Rinne, L.; Rehling, P.; Kügler, S.; Golubiani, G.; Dudek, J.; Müller, M. Neuronal redox-imbalance in Rett syndrome affects mitochondria as well as cytosol, and is accompanied by intensified mitochondrial O2 consumption and ROS release. Front. Physiol. 2019, 10, 479. [Google Scholar] [CrossRef]
- Großer, E.; Hirt, U.; Janc, O.A.; Menzfeld, C.; Fischer, M.; Kempkes, B.; Vogelgesang, S.; Manzke, T.U.; Opitz, L.; Salinas-Riester, G.; et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis. 2012, 48, 102–114. [Google Scholar] [CrossRef]
- Bebensee, D.F.; Can, K.; Müller, M. Increased Mitochondrial Mass and Cytosolic Redox Imbalance in Hippocampal Astrocytes of a Mouse Model of Rett Syndrome: Subcellular Changes Revealed by Ratiometric Imaging of JC-1 and roGFP1 Fluorescence. Oxid. Med. Cell. Longev. 2017, 2017, 3064016. [Google Scholar] [CrossRef] [Green Version]
- Cervellati, C.; Sticozzi, C.; Romani, A.; Belmonte, G.; De Rasmo, D.; Signorile, A.; Cervellati, F.; Milanese, C.; Mastroberardino, P.G.; Pecorelli, A.; et al. Impaired enzymatic defensive activity, mitochondrial dysfunction and proteasome activation are involved in RTT cell oxidative damage. Biochim. Biophys. Acta 2015, 1852, 2066–2074. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef]
- Müller, M.; Can, K. Aberrant redox homoeostasis and mitochondrial dysfunction in Rett syndrome. Biochem. Soc. Trans. 2014, 42, 959–964. [Google Scholar] [CrossRef]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial dysfunction in the pathogenesis of Rett syndrome: Implications for mitochondria-targeted therapies. Front. Cell. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.; Dunn, J.K.; Antalffy, B.; Trivedi, R. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 1995, 54, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Boggio, E.M.; Lonetti, G.; Pizzorusso, T.; Giustetto, M. Synaptic determinants of Rett syndrome. Front. Synaptic Neurosci. 2010, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Clement, M.V.; Long, L.H.; Ramalingam, J.; Halliwell, B. The cytotoxicity of dopamine may be an artefact of cell culture. J. Neurochem. 2002, 81, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [Green Version]
- Can, K.; Kügler, S.; Müller, M. Live imaging of mitochondrial ROS production and dynamic redox balance in neurons. In Techniques to Investigate Mitochondrial Function in Neurons; Strack, S., Usachev, Y.M., Eds.; Springer Science+Business Media: Berlin, Germany, 2017; Volume 123, pp. 179–197. [Google Scholar]
- Funke, F.; Gerich, F.; Müller, M. Dynamic, semi-quantitative imaging of intracellular ROS levels and redox status in rat hippocampal neurons. Neuroimage 2011, 54, 2590–2602. [Google Scholar] [CrossRef]
- Meyer, A.J.; Dick, T.P. Fluorescent protein-based redox probes. Antioxid. Redox Signal. 2010, 13, 621–650. [Google Scholar] [CrossRef]
- Müller, M.; Ballanyi, K. Dynamic recording of cell death in the in vitro dorsal vagal nucleus of rats in response to metabolic arrest. J. Neurophysiol. 2003, 89, 551–561. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar]
- Adebayo, O.L.; Dewenter, I.; Rinne, L.; Golubiani, G.; Solomonia, R.; Müller, M. Intensified mitochondrial hydrogen peroxide release occurs in all brain regions, affects male as well as female Rett mice, and constitutes a life-long burden. Arch Biochem. Biophys. 2020, 696, 108666. [Google Scholar] [CrossRef] [PubMed]
- Smialowski, A.; Bijak, M. Excitatory and inhibitory action of dopamine on hippocampal neurons in vitro. Involvement of D2 and D1 receptors. Neuroscience 1987, 23, 95–101. [Google Scholar] [CrossRef]
- Ropert, N. Inhibitory action of serotonin in CA1 hippocampal neurons in vitro. Neuroscience 1988, 26, 69–81. [Google Scholar] [CrossRef]
- Madison, D.V.; Nicoll, R.A. Norepinephrine decreases synaptic inhibition in the rat hippocampus. Brain Res. 1988, 442, 131–138. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Li, V.; Brustovetsky, N. Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium 2009, 46, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Choi, D.W.; Koh, J.Y.; Peters, S. Pharmacology of glutamate neurotoxicity in cortical cell culture: Attenuation by NMDA antagonists. J. Neurosci. 1988, 8, 185–196. [Google Scholar] [CrossRef]
- Savolainen, K.M.; Loikkanen, J.; Naarala, J. Amplification of glutamate-induced oxidative stress. Toxicol. Lett. 1995, 82–83, 399–405. [Google Scholar] [CrossRef]
- Sapoznik, S.; Ivenshitz, M.; Segal, M. Age-dependent glutamate induction of synaptic plasticity in cultured hippocampal neurons. Learn. Mem. 2006, 13, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S413–S426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kron, M.; Müller, M. Impaired hippocampal Ca2+ homeostasis and concomitant K+ channel dysfunction in a mouse model of Rett syndrome during anoxia. Neuroscience 2010, 171, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, S.; Mironov, S.L. Regenerative glutamate release in the hippocampus of Rett syndrome model mice. PLoS ONE 2018, 13, e0202802. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Liu, Q.; Li, R.; Wang, A.; Bu, Q.; Wang, K.H.; Chang, Q. Mechanism and consequence of abnormal calcium homeostasis in Rett syndrome astrocytes. Elife 2018, 7, e33417. [Google Scholar] [CrossRef]
- Mironov, S.L.; Skorova, E.; Hartelt, N.; Mironova, L.A.; Hasan, M.T.; Kügler, S. Remodelling of the respiratory network in a mouse model of Rett syndrome depends on brain-derived neurotrophic factor regulated slow calcium buffering. J. Physiol. 2009, 587, 2473–2485. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Tejada-Simon, M.V.; Serrano, F.; Villasana, L.E.; Kanterewicz, B.I.; Wu, G.Y.; Quinn, M.T.; Klann, E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell. Neurosci. 2005, 29, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makarenko, V.; Semenza, G.L.; Prabhakar, N.R. HIF-1alpha activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS ONE 2015, 10, e0119762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Sun, A.Y. Oxidative mechanisms involved in kainate-induced cytotoxicity in cortical neurons. Neurochem. Res. 1994, 19, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Stern, A.; Trenkner, E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J. Neurochem. 1987, 49, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Saksela, M.; Lapatto, R.; Raivio, K.O. Irreversible conversion of xanthine dehydrogenase into xanthine oxidase by a mitochondrial protease. FEBS Lett. 1999, 443, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [Green Version]
- Kamsler, A.; Segal, M. Hydrogen peroxide modulation of synaptic plasticity. J. Neurosci. 2003, 23, 269–276. [Google Scholar] [CrossRef]
- Pellmar, T.C. Peroxide alters neuronal excitability in the CA1 region of guinea-pig hippocampus in vitro. Neuroscience 1987, 23, 447–456. [Google Scholar] [CrossRef]
- Calfa, G.; Percy, A.K.; Pozzo-Miller, L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. 2011, 236, 3–19. [Google Scholar] [CrossRef] [Green Version]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Festerling, K.; Can, K.; Kügler, S.; Müller, M. Overshooting Subcellular Redox-Responses in Rett-Mouse Hippocampus during Neurotransmitter Stimulation. Cells 2020, 9, 2539. https://doi.org/10.3390/cells9122539
Festerling K, Can K, Kügler S, Müller M. Overshooting Subcellular Redox-Responses in Rett-Mouse Hippocampus during Neurotransmitter Stimulation. Cells. 2020; 9(12):2539. https://doi.org/10.3390/cells9122539
Chicago/Turabian StyleFesterling, Karina, Karolina Can, Sebastian Kügler, and Michael Müller. 2020. "Overshooting Subcellular Redox-Responses in Rett-Mouse Hippocampus during Neurotransmitter Stimulation" Cells 9, no. 12: 2539. https://doi.org/10.3390/cells9122539