
Cellular and Mitochondrial NAD Homeostasis in Health and Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
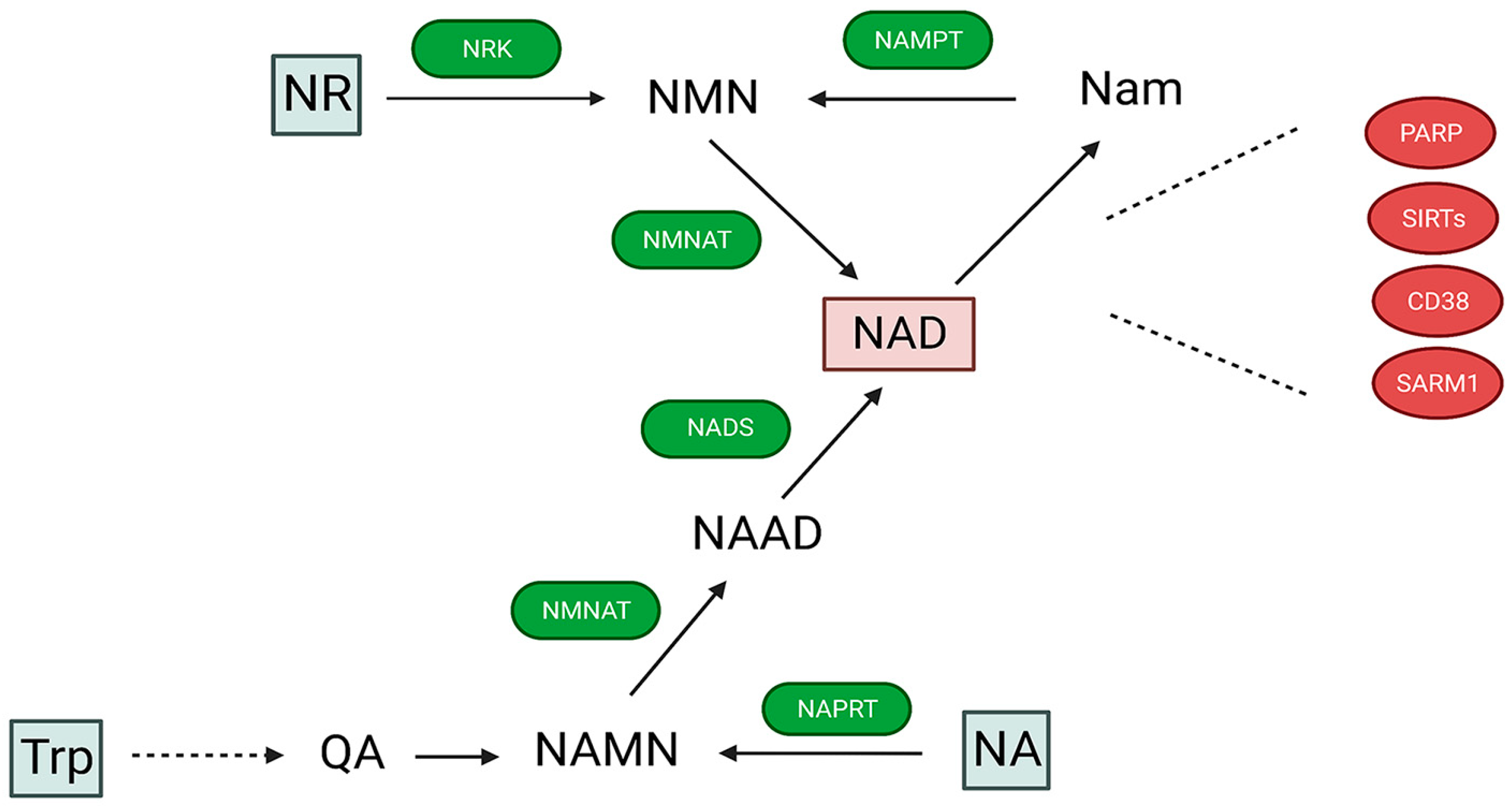
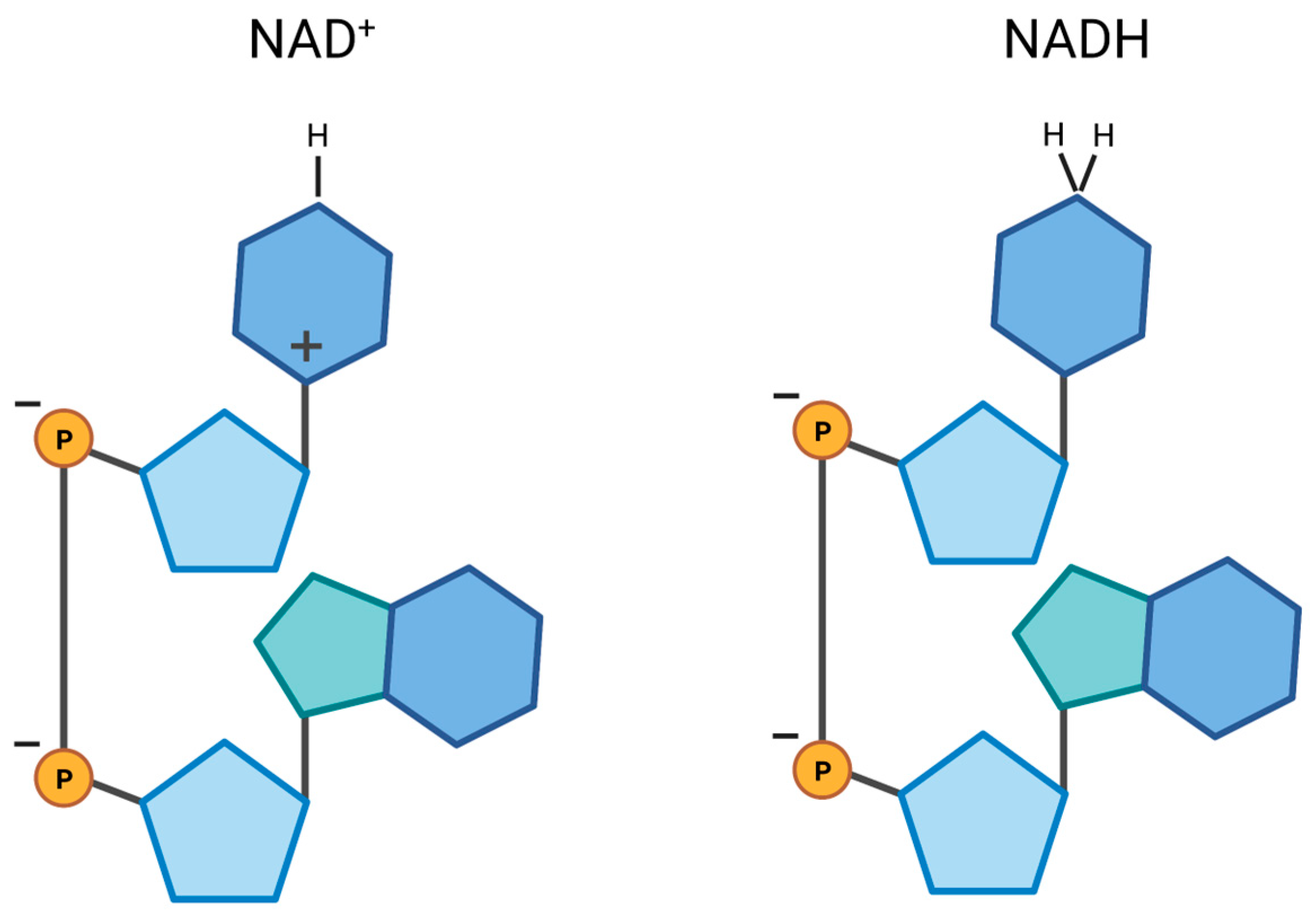
2. NAD Synthesis and Degradation in Mitochondria and Other Cellular Subcompartments
3. Intracellular NAD Degradation Enzymes and Their Compartmentation
4. Mitochondria and Intracellular NAD Exchange
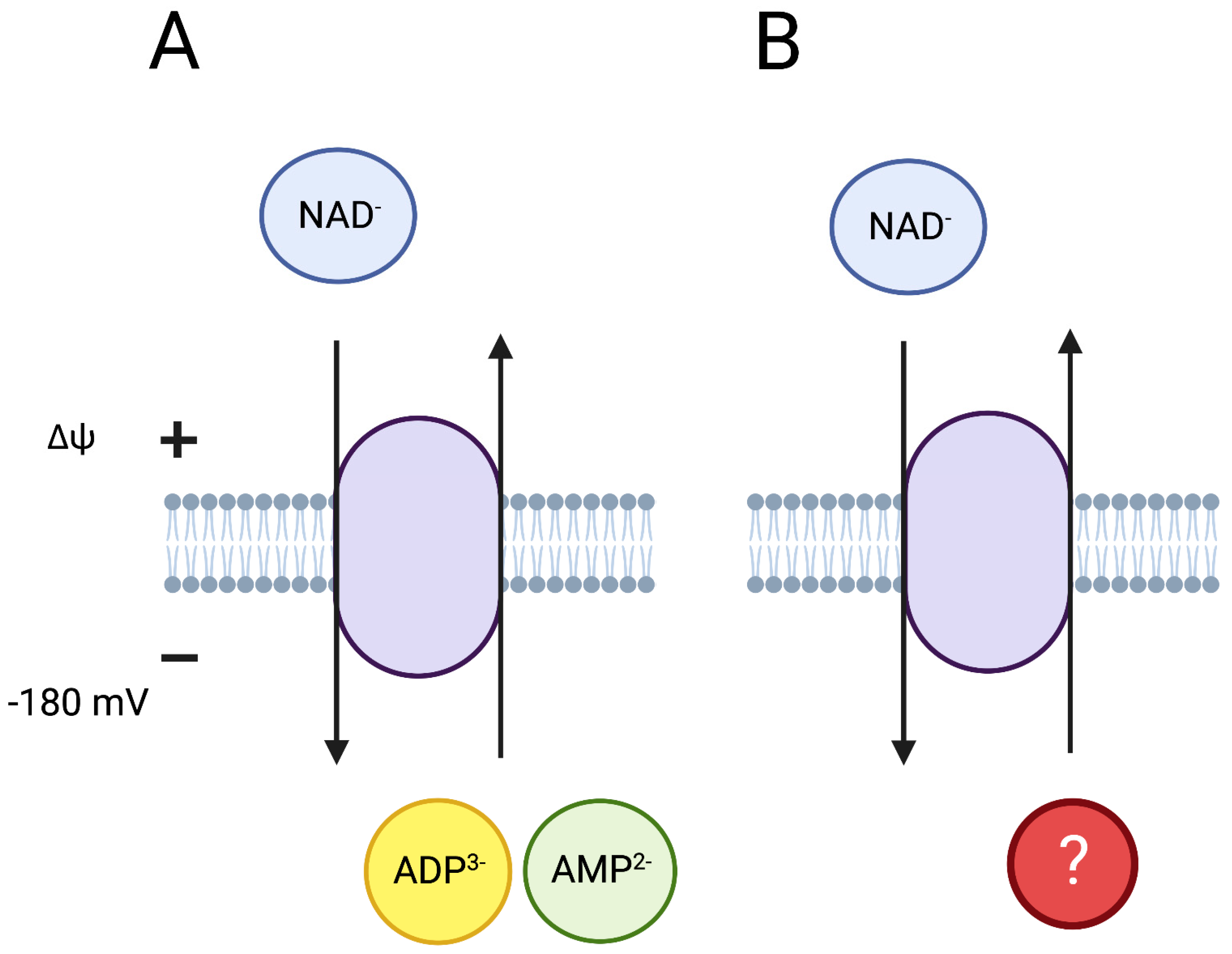
4.1. NAD and Mitochondrial Inner Membrane Permeability
4.2. Cell Membrane NAD Permeability
4.3. Mitochondria-Specific NAD Transport
5. Mitochondrial NAD Transport and Protein Acetylation
6. Pathophysiology of Perturbed NAD Homeostasis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kristian, T.; Balan, I.; Schuh, R.; Onken, M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J. Neurosci. Res. 2011, 89, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.; Park, J.H.; Schuh, R.; Kristian, T. Mitochondrial Dysfunction and NAD+ Metabolism Alterations in the Pathophysiology of Acute Brain Injury. Transl. Stroke Res. 2013, 4, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Kusumanchi, P.; Yalowitz, J.A. NMNAT expression and its relation to NAD metabolism. Curr. Med. Chem. 2011, 18, 1962–1972. [Google Scholar] [CrossRef]
- Zhai, R.G.; Zhang, F.; Hiesinger, P.R.; Cao, Y.; Haueter, C.M.; Bellen, H.J. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 2008, 452, 887–891. [Google Scholar] [CrossRef]
- Ocampo, A.; Liu, J.; Barrientos, A. NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins. Hum. Mol. Genet. 2013, 22, 1699–1708. [Google Scholar] [CrossRef]
- Ali, Y.O.; Ruan, K.; Zhai, R.G. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum. Mol. Genet. 2012, 21, 237–250. [Google Scholar] [CrossRef]
- Felici, R.; Lapucci, A.; Ramazzotti, M.; Chiarugi, A. Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PLoS ONE 2013, 8, e76938. [Google Scholar] [CrossRef]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD(+). Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef]
- Kory, N.; Uit de Bos, J.; van der Rijt, S.; Jankovic, N.; Gura, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6, eabe5310. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Stromland, O.; Niere, M.; Nikiforov, A.A.; VanLinden, M.R.; Heiland, I.; Ziegler, M. Keeping the balance in NAD metabolism. Biochem. Soc. Trans 2019, 47, 119–130. [Google Scholar] [CrossRef]
- Sauve, A.A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 384–388. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Swanson, R.A. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience 2007, 145, 1267–1272. [Google Scholar] [CrossRef]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol. Dis. 2000, 7, 225–239. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabo, C.; Clark, R.S. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem 2003, 278, 18426–18433. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M. Deadly conversations: Nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 2004, 36, 287–294. [Google Scholar] [CrossRef]
- Whalen, M.J.; Clark, R.S.; Dixon, C.E.; Robichaud, P.; Marion, D.W.; Vagni, V.; Graham, S.H.; Virag, L.; Hasko, G.; Stachlewitz, R.; et al. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J. Cereb. Blood Flow Metab. 1999, 19, 835–842. [Google Scholar] [CrossRef]
- Lai, Y.; Chen, Y.; Watkins, S.C.; Nathaniel, P.D.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Szabo, C.; Clark, R.S. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 2008, 104, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Carbone, M.; Mostocotto, C.; Mancone, C.; Tripodi, M.; Maione, R.; Amati, P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009, 284, 31616–31624. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Brunyanszki, A.; Olah, G.; Mitra, S.; Szabo, C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: Implications for the regulation of mitochondrial function. Nucleic Acids Res. 2014, 42, 13161–13173. [Google Scholar] [CrossRef] [PubMed]
- Burzio, L.O.; Saez, L.; Cornejo, R. Poly (ADP-ribose) synthetase activity in rat testis mitochondria. Biochem. Biophys. Res. Commun. 1981, 103, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef]
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 2010, 285, 34106–34114. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef]
- Guse, A.H. Calcium mobilizing second messengers derived from NAD. Biochim. Biophys. Acta 2015, 1854, 1132–1137. [Google Scholar] [CrossRef]
- Ceni, C.; Pochon, N.; Brun, V.; Muller-Steffner, H.; Andrieux, A.; Grunwald, D.; Schuber, F.; De Waard, M.; Lund, F.; Villaz, M.; et al. CD38-dependent ADP-ribosyl cyclase activity in developing and adult mouse brain. Biochem. J. 2003, 370, 175–183. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Otsuka, N.; Sato, M.; Ishii, Y.; Kon, S.; Yamada, M.; Nishina, H.; Katada, T.; Ikeda, K. Neuronal localization of CD38 antigen in the human brain. Brain Res. 1995, 697, 235–240. [Google Scholar] [CrossRef]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.J.; Lund, F.E.; Stein, R. Dual role of CD38 in microglial activation and activation-induced cell death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef]
- Long, A.; Park, J.H.; Klimova, N.; Fowler, C.; Loane, D.J.; Kristian, T. CD38 Knockout Mice Show Significant Protection Against Ischemic Brain Damage Despite High Level Poly-ADP-Ribosylation. Neurochem. Res. 2017, 42, 283–293. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Tarrago, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Hu, J.; Dong, M.; Wang, B.S.; MacDonald, R.; Jiang, H.; Hao, Q.; Yen, A.; Lin, H. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 2014, 136, 5656–5663. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal 2012, 5, ra67. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Sasaki, Y.; Nakagawa, T.; Mao, X.; DiAntonio, A.; Milbrandt, J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD(+) depletion. Elife 2016, 5, e19749. [Google Scholar] [CrossRef] [PubMed]
- Gilley, J.; Ribchester, R.R.; Coleman, M.P. Sarm1 Deletion, but Not Wld(S), Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Rep. 2017, 21, 10–16. [Google Scholar] [CrossRef]
- Dolle, C.; Niere, M.; Lohndal, E.; Ziegler, M. Visualization of subcellular NAD pools and intra-organellar protein localization by poly-ADP-ribose formation. Cell Mol. Life Sci. 2010, 67, 433–443. [Google Scholar] [CrossRef]
- Leite, M.F.; Thrower, E.C.; Echevarria, W.; Koulen, P.; Hirata, K.; Bennett, A.M.; Ehrlich, B.E.; Nathanson, M.H. Nuclear and cytosolic calcium are regulated independently. Proc. Natl. Acad. Sci. USA 2003, 100, 2975–2980. [Google Scholar] [CrossRef]
- Wright, R.H.; Lioutas, A.; Le Dily, F.; Soronellas, D.; Pohl, A.; Bonet, J.; Nacht, A.S.; Samino, S.; Font-Mateu, J.; Vicent, G.P.; et al. ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 2016, 352, 1221–1225. [Google Scholar] [CrossRef]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef]
- Knockenhauer, K.E.; Schwartz, T.U. The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell 2016, 164, 1162–1171. [Google Scholar] [CrossRef]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Kraus, W.L. Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 2018, 360, eaan5780. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflug. Arch. Eur. J. Physiol. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Ali, Y.O.; Li-Kroeger, D.; Bellen, H.J.; Zhai, R.G.; Lu, H.C. NMNATs, evolutionarily conserved neuronal maintenance factors. Trends Neurosci. 2013, 36, 632–640. [Google Scholar] [CrossRef]
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Kristian, T.; Fiskum, G. A fluorescence-based technique for screening compounds that protect against damage to brain mitochondria. Brain Res. Brain Res. Protoc. 2004, 13, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [CrossRef]
- Kristian, T.; Bernardi, P.; Siesjo, B.K. Acidosis promotes the permeability transition in energized mitochondria: Implications for reperfusion injury. J. Neurotrauma 2001, 18, 1059–1074. [Google Scholar] [CrossRef]
- Robertson, C.L.; Scafidi, S.; McKenna, M.C.; Fiskum, G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 2009, 218, 371–380. [Google Scholar] [CrossRef]
- Siesjo, B.K.; Elmer, E.; Janelidze, S.; Keep, M.; Kristian, T.; Ouyang, Y.B.; Uchino, H. Role and mechanisms of secondary mitochondrial failure. In Current Progress in the Understanding of Secondary Brain Damage from Trauma and Ischemia; Springer: Berlin/Heidelberg, Germany, 1999; Volume 73, pp. 7–13. [Google Scholar]
- Brustovetsky, N.; Dubinsky, J.M. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J. Neurosci. 2000, 20, 8229–8237. [Google Scholar] [CrossRef]
- Ayoub, I.A.; Lee, E.J.; Ogilvy, C.S.; Beal, M.F.; Maynard, K.I. Nicotinamide reduces infarction up to two hours after the onset of permanent focal cerebral ischemia in Wistar rats. Neurosci. Lett. 1999, 259, 21–24. [Google Scholar] [CrossRef]
- Yang, J.; Klaidman, L.K.; Chang, M.L.; Kem, S.; Sugawara, T.; Chan, P.; Adams, J.D. Nicotinamide therapy protects against both necrosis and apoptosis in a stroke model. Pharmacol. Biochem. Behav. 2002, 73, 901–910. [Google Scholar] [CrossRef]
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009, 11, 28–42. [Google Scholar] [CrossRef]
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004, 279, 18895–18902. [Google Scholar] [CrossRef]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- Wang, S.; Xing, Z.; Vosler, P.S.; Yin, H.; Li, W.; Zhang, F.; Signore, A.P.; Stetler, R.A.; Gao, Y.; Chen, J. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: Role of enhanced DNA repair. Stroke 2008, 39, 2587–2595. [Google Scholar] [CrossRef]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef]
- Okuda, H.; Higashi, Y.; Nishida, K.; Fujimoto, S.; Nagasawa, K. Contribution of P2X7 receptors to adenosine uptake by cultured mouse astrocytes. Glia 2010, 58, 1757–1765. [Google Scholar] [CrossRef]
- Alano, C.C.; Tran, A.; Tao, R.; Ying, W.; Karliner, J.S.; Swanson, R.A. Differences among cell types in NAD(+) compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 2007, 85, 3378–3385. [Google Scholar] [CrossRef]
- Todisco, S.; Agrimi, G.; Castegna, A.; Palmieri, F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 1524–1531. [Google Scholar] [CrossRef]
- Palmieri, F.; Rieder, B.; Ventrella, A.; Blanco, E.; Do, P.T.; Nunes-Nesi, A.; Trauth, A.U.; Fiermonte, G.; Tjaden, J.; Agrimi, G.; et al. Molecular identification and functional characterization of Arabidopsis thaliana mitochondrial and chloroplastic NAD+ carrier proteins. J. Biol. Chem. 2009, 284, 31249–31259. [Google Scholar] [CrossRef]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gurtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 2020, 11, 6145. [Google Scholar] [CrossRef]
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1alpha and PGC-1beta regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Kim, H.; Morse, P.T.; Lu, M.J.; Huttemann, M.; Cambronne, X.A.; Zhang, K.; Zhang, R. The mitochondrial NAD(+) transporter SLC25A51 is a fasting-induced gene affecting SIRT3 functions. Metabolism 2022, 135, 155275. [Google Scholar] [CrossRef]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Kristian, T.; Karimi, A.J.; Fearnow, A.; Waddell, J.; McKenna, M.C. Perturbed Brain Glucose Metabolism Caused by Absent SIRT3 Activity. Cells 2021, 10, 2348. [Google Scholar] [CrossRef]
- Klimova, N.; Kristian, T. Multi-targeted Effect of Nicotinamide Mononucleotide on Brain Bioenergetic Metabolism. Neurochem. Res. 2019, 44, 2280–2287. [Google Scholar] [CrossRef]
- Mokudai, T.; Ayoub, I.A.; Sakakibara, Y.; Lee, E.J.; Ogilvy, C.S.; Maynard, K.I. Delayed treatment with nicotinamide (Vitamin B(3)) improves neurological outcome and reduces infarct volume after transient focal cerebral ischemia in Wistar rats. Stroke 2000, 31, 1679–1685. [Google Scholar] [CrossRef]
- Lo, E.H.; Bosque-Hamilton, P.; Meng, W. Inhibition of poly(ADP-ribose) polymerase: Reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke 1998, 29, 830–836. [Google Scholar] [CrossRef]
- Szabo, C.; Dawson, V.L. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol. Sci. 1998, 19, 287–298. [Google Scholar] [CrossRef]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146–148, 53–64. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef]
- Chiarugi, A. Poly(ADP-ribosyl)ation and stroke. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2005, 52, 15–24. [Google Scholar] [CrossRef]
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Voorhees, J.R.; Remy, M.T.; Cintron-Perez, C.J.; El Rassi, E.; Khan, M.Z.; Dutca, L.M.; Yin, T.C.; McDaniel, L.N.; Williams, N.S.; Brat, D.J.; et al. (−)-P7C3-S243 Protects a Rat Model of Alzheimer’s Disease from Neuropsychiatric Deficits and Neurodegeneration Without Altering Amyloid Deposition or Reactive Glia. Biol. Psychiatry 2018, 84, 488–498. [Google Scholar] [CrossRef]
- Yang, J.; Klaidman, L.K.; Nalbandian, A.; Oliver, J.; Chang, M.L.; Chan, P.H.; Adams, J.D., Jr. The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci. Lett. 2002, 333, 91–94. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Zhao, J.H.; Zong, W.F.; Wei, X.J.; Xu, Z.; Yuan, Y.; Jiang, Y.F.; Luo, X.; Wang, W.; Qu, W.S. Acute Treatment with Nicotinamide Riboside Chloride Reduces Hippocampal Damage and Preserves the Cognitive Function of Mice with Ischemic Injury. Neurochem. Res. 2022, 47, 2244–2253. [Google Scholar] [CrossRef]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef]
- Harlan, B.A.; Killoy, K.M.; Pehar, M.; Liu, L.; Auwerx, J.; Vargas, M.R. Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1-linked ALS mouse models. Exp. Neurol. 2020, 327, 113219. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Nakamura, T.; Cho, D.H.; Lipton, S.A. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp. Neurol 2012, 238, 12–21. [Google Scholar] [CrossRef]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, L.; Wang, P.; Li, X.; Qiu, D.; Li, L.; Zhang, J.; Hou, X.; Han, L.; Ge, J.; et al. Sirt3-dependent deacetylation of SOD2 plays a protective role against oxidative stress in oocytes from diabetic mice. Cell Cycle 2017, 16, 1302–1308. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waddell, J.; Khatoon, R.; Kristian, T. Cellular and Mitochondrial NAD Homeostasis in Health and Disease. Cells 2023, 12, 1329. https://doi.org/10.3390/cells12091329
Waddell J, Khatoon R, Kristian T. Cellular and Mitochondrial NAD Homeostasis in Health and Disease. Cells. 2023; 12(9):1329. https://doi.org/10.3390/cells12091329
Chicago/Turabian StyleWaddell, Jaylyn, Rehana Khatoon, and Tibor Kristian. 2023. "Cellular and Mitochondrial NAD Homeostasis in Health and Disease" Cells 12, no. 9: 1329. https://doi.org/10.3390/cells12091329