
LRP1 Deficiency Promotes Mitostasis in Response to Oxidative Stress: Implications for Mitochondrial Targeting after Traumatic Brain Injury
 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Controlled Cortical Impact
2.3. Cell Culture
2.4. Mito-Stress Test
2.5. Mitochondrial Quantification in Imaris
2.6. MitoTracker Imaging and Mitochondrial Network Analysis (MiNA)
2.7. ROS Assay in Cells
2.8. Cell Proliferation Assay
2.9. Western Blot
2.10. Immunofluorescence
2.11. Mitochondrial DNA Copy Number (mtDNA-CN) Estimation
2.12. qRT-PCR
2.13. Statistics
3. Results
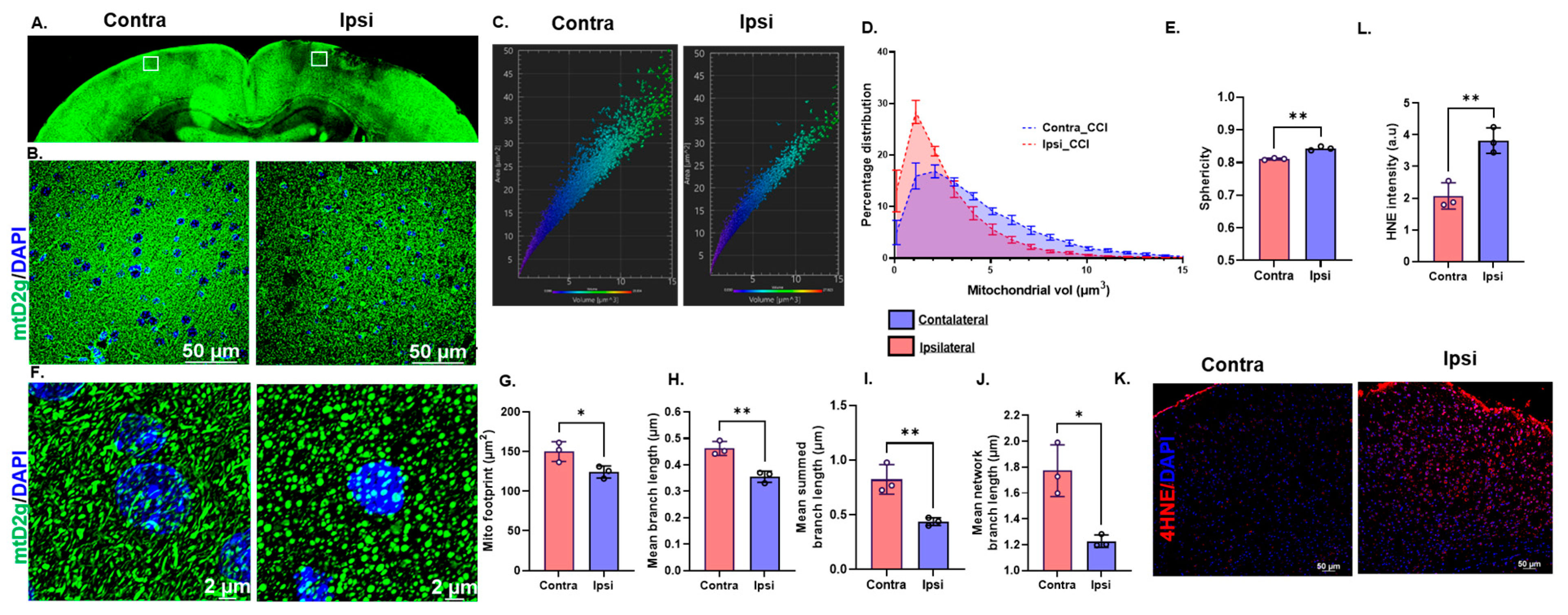
3.1. Traumatic Brain Injury Induces Oxidative Stress and Mitochondrial Fragmentation in mtD2g Mice
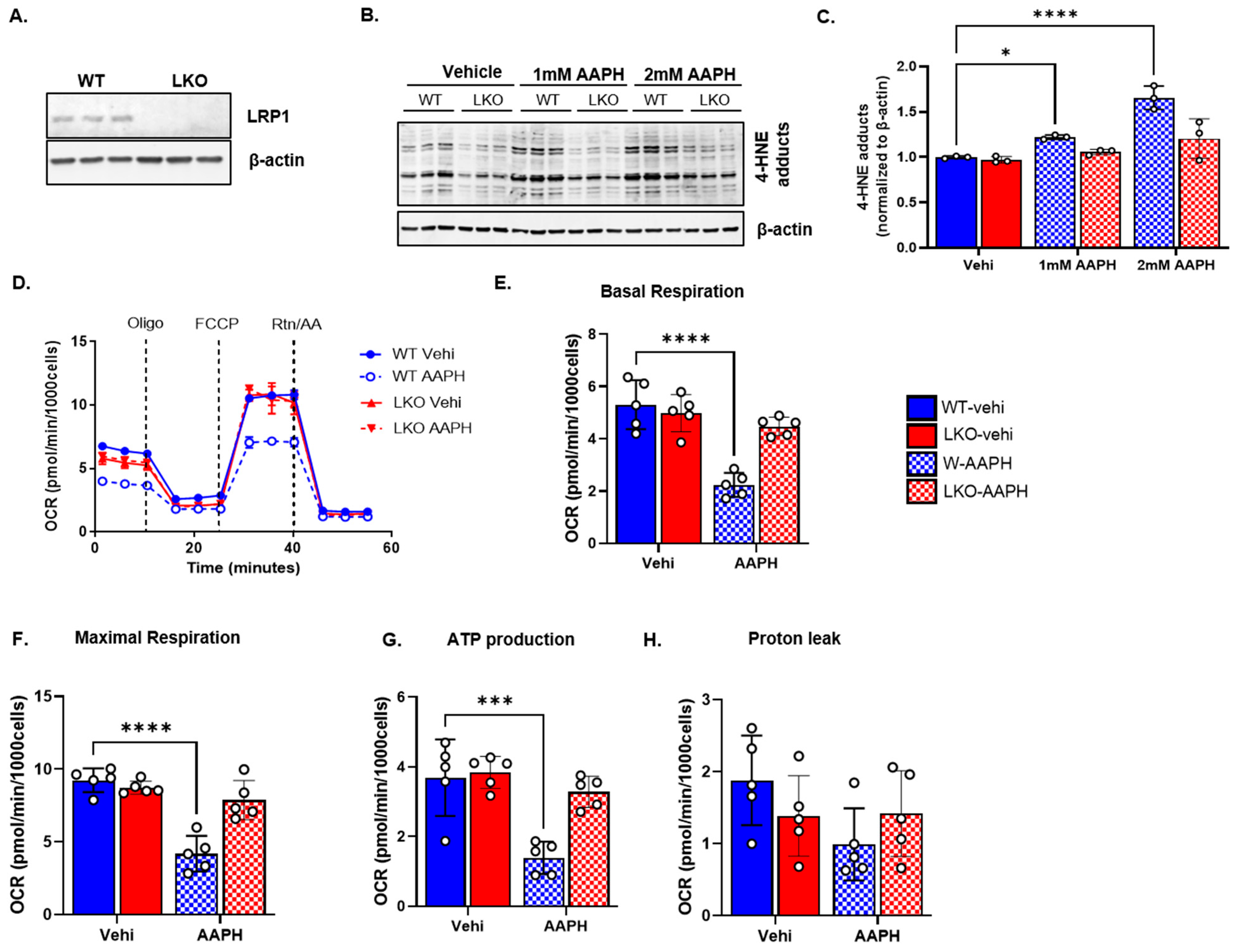
3.2. LRP1 Deficiency Improves Mitochondrial Bioenergetics following Oxidative Stress
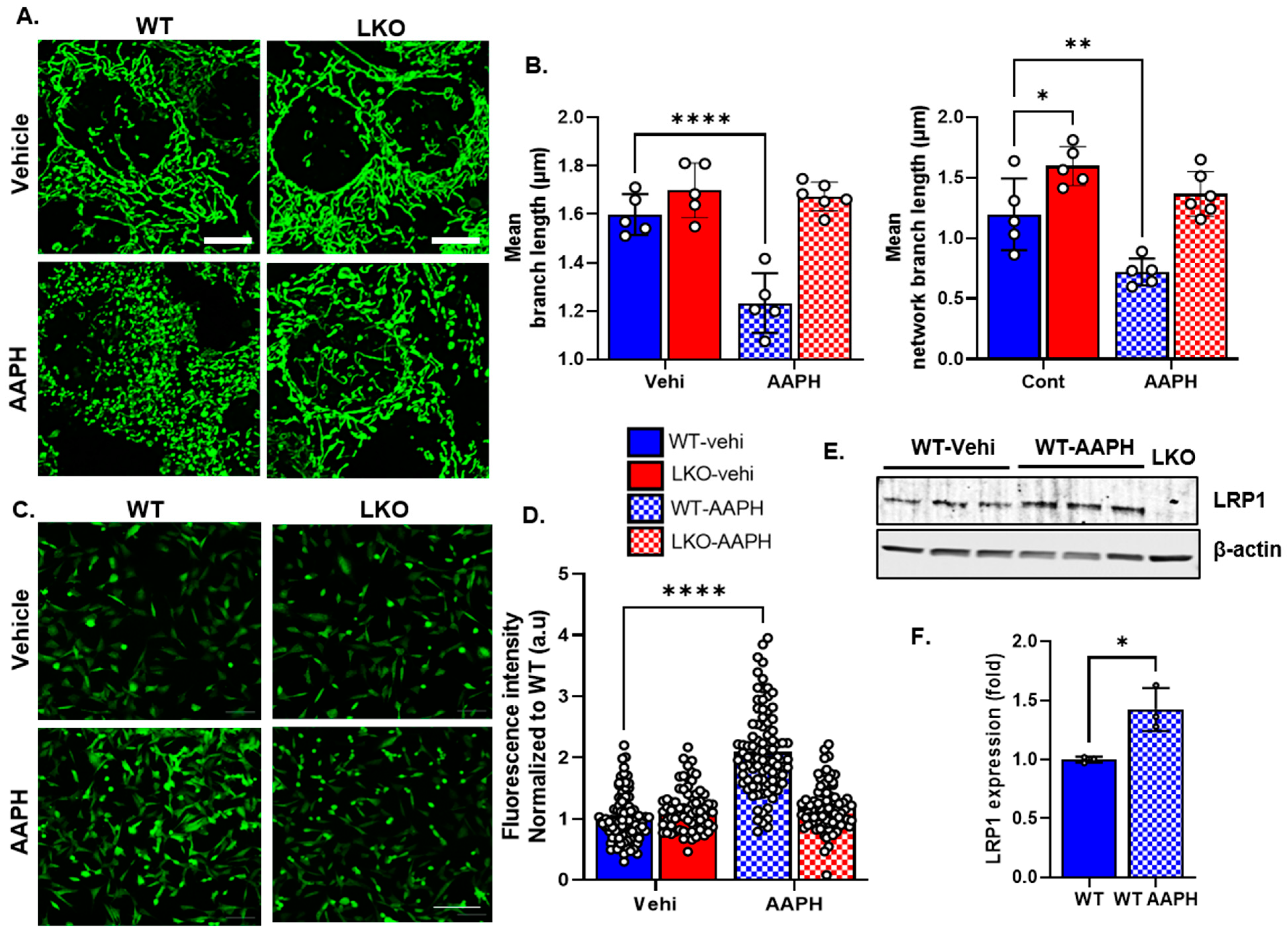
3.3. LRP1 Deficiency Protects the Cell from Oxidative Stress and Mitochondrial Fragmentation
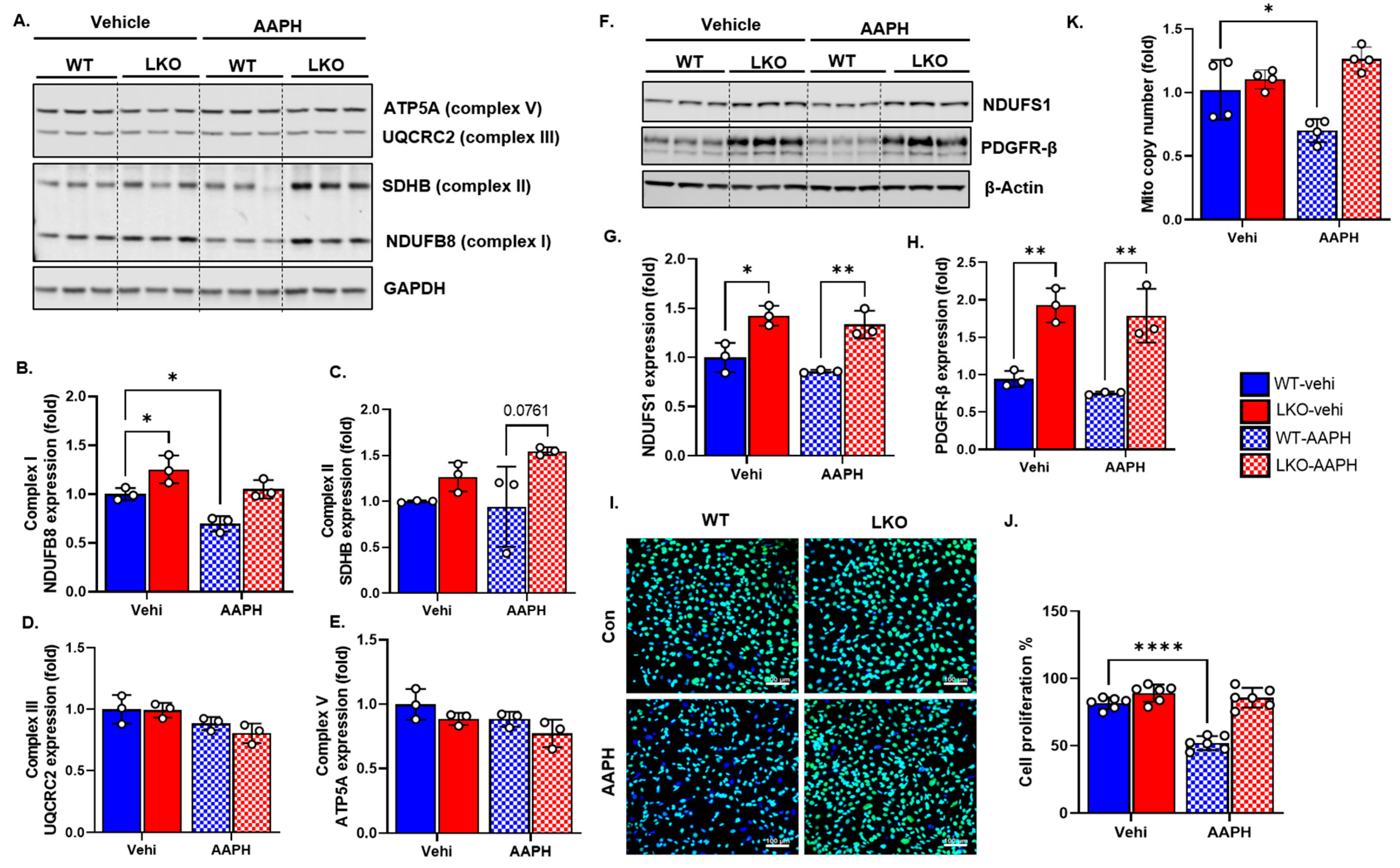
3.4. LRP1 Deficiency Protects Mitochondrial Complex Integrity, DNA Copy Number, and Cell Growth following Oxidative Stress
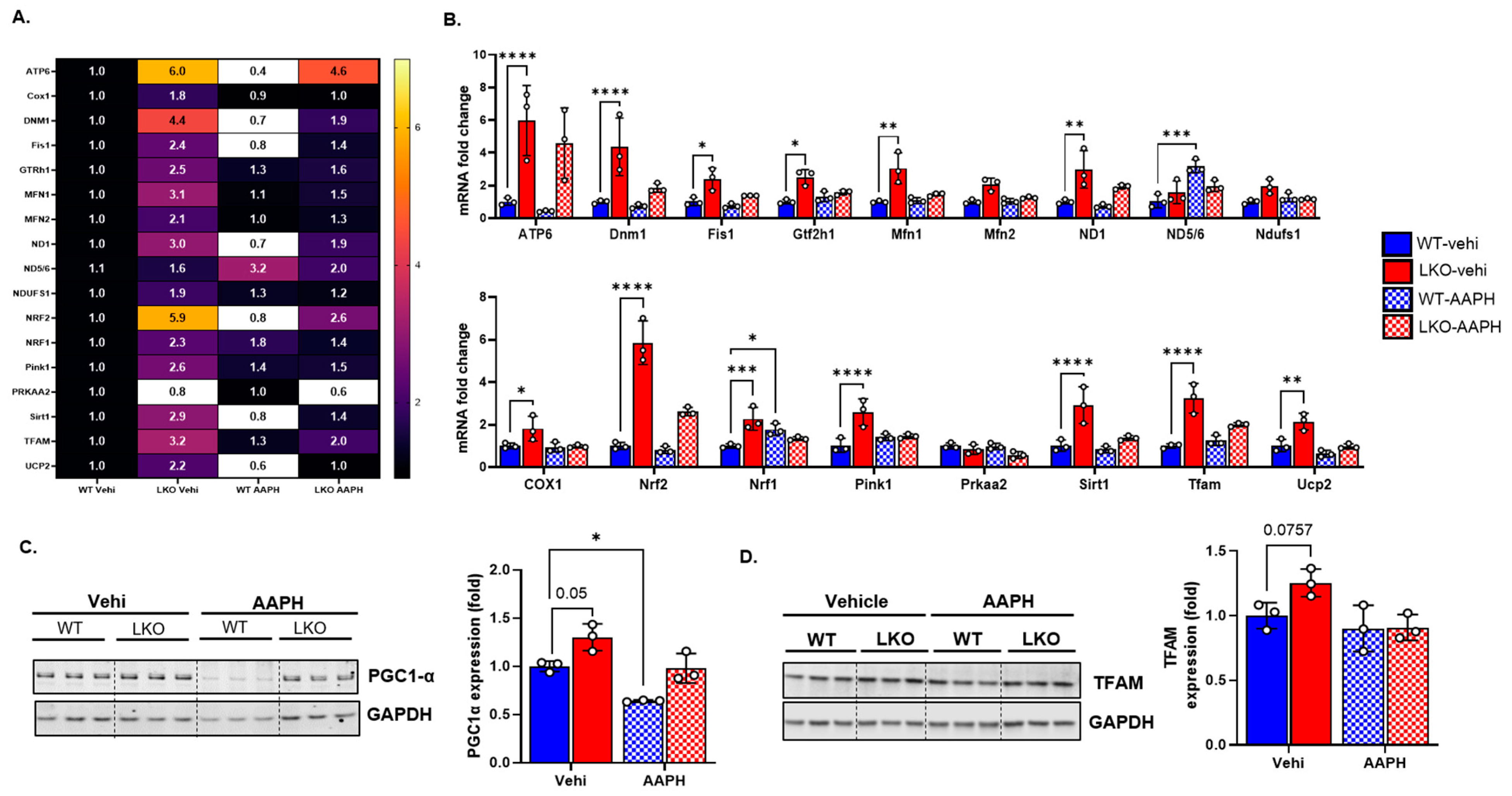
3.5. LRP1 Deficiency Upregulates Genes Related to Mitochondrial Biogenesis and Antioxidants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LoBue, C.; Munro Cullum, C.; Didehbani, N.; Yeatman, K.; Jones, B.; Kraut, M.A.; Hart, J. Neurodegenerative Dementias After Traumatic Brain Injury. J. Neuropsychiatry Clin. Neurosci. 2018, 30, 7. [Google Scholar] [CrossRef]
- Walker, K.R.; Tesco, G. Molecular Mechanisms of Cognitive Dysfunction Following Traumatic Brain Injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Center for Health Statistics: Mortality Data on CDC WONDER. 2023. Available online: https://wonder.cdc.gov/mcd.html (accessed on 1 April 2023).
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta 2017, 1863, 1066. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial Control of Inflammation. Nat. Rev. Immunol. 2022, 23, 159–173. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619. [Google Scholar] [CrossRef]
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic Brain Injury and Mitochondrial Dysfunction. Am. J. Med. Sci. 2015, 350, 132–138. [Google Scholar] [CrossRef]
- Herz, J.; Hamann, U.; Rogne, S.; Myklebost, O.; Gausepohl, H.; Stanley, K.K. Surface Location and High Affinity for Calcium of a 500-Kd Liver Membrane Protein Closely Related to the LDL-Receptor Suggest a Physiological Role as Lipoprotein Receptor. EMBO J. 1988, 7, 4119. [Google Scholar] [CrossRef]
- Gaultier, A.; Arandjelovic, S.; Li, X.; Janes, J.; Dragojlovic, N.; Zhou, G.P.; Dolkas, J.; Myers, R.R.; Gonias, S.L.; Campana, W.M. A Shed Form of LDL Receptor-Related Protein-1 Regulates Peripheral Nerve Injury and Neuropathic Pain in Rodents. J. Clin. Investig. 2008, 118, 161–172. [Google Scholar] [CrossRef]
- Brifault, C.; Kwon, H.J.; Campana, W.M.; Gonias, S.L. LRP1 Deficiency in Microglia Blocks Neuro-Inflammation in the Spinal Dorsal Horn and Neuropathic Pain Processing. Glia 2019, 67, 1210. [Google Scholar] [CrossRef]
- Xian, X.; Ding, Y.; Dieckmann, M.; Zhou, L.; Plattner, F.; Liu, M.; Parks, J.S.; Hammer, R.E.; Boucher, P.; Tsai, S.; et al. LRP1 Integrates Murine Macrophage Cholesterol Homeostasis and Inflammatory Responses in Atherosclerosis. eLife 2017, 6, e29292. [Google Scholar] [CrossRef]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 Transports Amyloid-Β1–42 across the Blood-Brain Barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Kowal, R.C.; Herz, J.; Goldstein, J.L.; Esser, V.; Brown, M.S. Low Density Lipoprotein Receptor-Related Protein Mediates Uptake of Cholesteryl Esters Derived from Apoprotein E-Enriched Lipoproteins. Proc. Natl. Acad. Sci. USA 1989, 86, 5810. [Google Scholar] [CrossRef] [PubMed]
- Boucker, P.; Li, W.P.; Matz, R.L.; Takayama, Y.; Auwerx, J.; Anderson, R.G.W.; Herz, J. LRP1 Functions as an Atheroprotective Integrator of TGFβ and PDGF Signals in the Vascular Wall: Implications for Marfan Syndrome. PLoS ONE 2007, 2, e448. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Martens, Y.A.; Meneses, A.; Ryu, D.H.; Lu, W.; Raulin, A.C.; Li, F.; Zhao, J.; Chen, Y.; Jin, Y.; et al. LRP1 Is a Neuronal Receptor for α-Synuclein Uptake and Spread. Mol. Neurodegener. 2022, 17, 57. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 Is a Master Regulator of Tau Uptake and Spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Auderset, L.; Pitman, K.A.; Cullen, C.L.; Pepper, R.E.; Taylor, B.V.; Foa, L.; Young, K.M. Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Is a Negative Regulator of Oligodendrocyte Progenitor Cell Differentiation in the Adult Mouse Brain. Front. Cell Dev. Biol. 2020, 8, 1067. [Google Scholar] [CrossRef]
- Stiles, T.L.; Dickendesher, T.L.; Gaultier, A.; Fernandez-Castaneda, A.; Mantuano, E.; Giger, R.J.; Gonias, S.L. LDL Receptor-Related Protein-1 Is a Sialic-Acid-Independent Receptor for Myelin-Associatedglycoprotein That Functions in Neurite Outgrowth Inhibition by MAG and CNS Myelin. J. Cell Sci. 2013, 126, 209–220. [Google Scholar] [CrossRef]
- Mao, H.; Li, L.; Fan, Q.; Angelini, A.; Saha, P.K.; Coarfa, C.; Rajapakshe, K.; Perera, D.; Cheng, J.; Wu, H.; et al. Endothelium-Specific Depletion of LRP1 Improves Glucose Homeostasis through Inducing Osteocalcin. Nat. Commun. 2021, 12, 5296. [Google Scholar] [CrossRef]
- Mao, H.; Lockyer, P.; Davin Townley-Tilson, W.H.; Xie, L.; Pi, X. LRP1 Regulates Retinal Angiogenesis by Inhibiting PARP-1 Activity and Endothelial Cell Proliferation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 350. [Google Scholar] [CrossRef]
- Wu, T.; Wang, X.J.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-Ribose) Polymerase-1 Modulates Nrf2-Dependent Transcription. Free. Radic. Biol. Med. 2014, 67, 69–80. [Google Scholar] [CrossRef]
- Liu, C.-C.; Hu, J.; Tsai, C.-W.; Yue, M.; Melrose, H.L.; Kanekiyo, T.; Bu, G. Neuronal LRP1 Regulates Glucose Metabolism and Insulin Signaling in the Brain. J. Neurosci. 2015, 35, 5851–5859. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.; Aledo, R.; Sendra, J.; Costales, P.; Juan-Babot, O.; Badimon, L.; Llorente-Cortés, V. Hypoxia Stimulates Low-Density Lipoprotein Receptor-Related Protein-1 Expression through Hypoxia-Inducible Factor-1α in Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Brifault, C.; Gilder, A.S.; Laudati, E.; Banki, M.; Gonias, S.L. LRP1 Shedding from Microglia Is Pro-Inflammatory Shedding of Membrane-Associated LDL Receptor-Related Protein-1 from Microglia Amplifies and Sustains Neuro-Inflammation. J. Biol. Chem. 2017, 292, 18699–18712. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Pang, J.; Huang, L.; Enkhjargal, B.; Zhang, T.; Mo, J.; Wu, P.; Xu, W.; Zuo, Y.; Peng, J.; et al. LRP1 Activation Attenuates White Matter Injury by Modulating Microglial Polarization through Shc1/PI3K/Akt Pathway after Subarachnoid Hemorrhage in Rats. Redox Biol. 2019, 21, 101121. [Google Scholar] [CrossRef]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in Traumatic Brain Injury and Mitochondrial-Targeted Multipotential Therapeutic Strategies. Br. J. Pharmacol. 2012, 167, 699. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651. [Google Scholar] [CrossRef]
- Vekaria, H.J.; Hubbard, W.B.; Scholpa, N.E.; Spry, M.L.; Gooch, J.L.; Prince, S.J.; Schnellmann, R.G.; Sullivan, P.G. Formoterol, a Β2-Adrenoreceptor Agonist, Induces Mitochondrial Biogenesis and Promotes Cognitive Recovery after Traumatic Brain Injury. Neurobiol. Dis. 2020, 140, 104866. [Google Scholar] [CrossRef]
- Kanno, T.; Utsumi, T.; Ide, A.; Takehara, Y.; Saibara, T.; Akiyama, J.; Yoshioka, T.; Utsumi, K. Dysfunction of Mouse Liver Mitochondria Induced by 2,2′-Azobis-(2-Amidinopropane) Dihydrochloride, A Radical Initiator, In Vitro And In Vivo. Free. Radic. Res. 2009, 21, 223–234. [Google Scholar] [CrossRef]
- Gautier, M.K.; Ginsberg, S.D. A Method for Quantification of Vesicular Compartments within Cells Using 3D Reconstructed Confocal Z-Stacks: Comparison of ImageJ and Imaris to Count Early Endosomes within Basal Forebrain Cholinergic Neurons. J. Neurosci. Methods 2021, 350, 109038. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 2012, 7, 376. [Google Scholar] [CrossRef]
- Fischer, T.D.; Hylin, M.J.; Zhao, J.; Moore, A.N.; Waxham, M.N.; Dash, P.K. Altered Mitochondrial Dynamics and TBI Pathophysiology. Front. Syst. Neurosci. 2016, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Impairing the Mitochondrial Fission and Fusion Balance: A New Mechanism of Neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 283. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1. [Google Scholar] [CrossRef]
- Klein, J.A.; Ackerman, S.L. Oxidative Stress, Cell Cycle, and Neurodegeneration. J. Clin. Investig. 2003, 111, 785. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, D.; Zhou, B.; Luan, Y.; Yao, Q.; Liu, Y.; Yang, S.; Jia, J.; Xu, Y.; Bie, X.; et al. Decrease of MtDNA Copy Number Affects Mitochondrial Function and Involves in the Pathological Consequences of Ischaemic Stroke. J. Cell. Mol. Med. 2022, 26, 4157. [Google Scholar] [CrossRef]
- Pandya, J.D.; Readnower, R.D.; Patel, S.P.; Yonutas, H.M.; Pauly, J.R.; Goldstein, G.A.; Rabchevsky, A.G.; Sullivan, P.G. N-Acetylcysteine Amide Confers Neuroprotection, Improves Bioenergetics and Behavioral Outcome Following TBI. Exp. Neurol. 2014, 257, 106–113. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Harwood, C.L.; Geisler, J.G.; Vekaria, H.J.; Sullivan, P.G. Mitochondrial Uncoupling Prodrug Improves Tissue Sparing, Cognitive Outcome, and Mitochondrial Bioenergetics after Traumatic Brain Injury in Male Mice. J. Neurosci. Res. 2018, 96, 1677–1688. [Google Scholar] [CrossRef]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed Mitochondrial Dynamics and Neurodegenerative Disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505. [Google Scholar] [CrossRef]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chen, M.; Chen, C.; Cui, J.; Johnson, C.E.; Cheng, J.; Wang, X.; Swerdlow, R.H.; DePalma, R.G.; Xia, W.; et al. Proteomic Analysis and Biochemical Correlates of Mitochondrial Dysfunction after Low-Intensity Primary Blast Exposure. J. Neurotrauma 2019, 36, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Dong, J.; Xu, X.; Zhang, B.; Liu, B. Mitochondrial Division Inhibitor 1 Prevents Early-Stage Induction of Mitophagy and Accelerated Cell Death in a Rat Model of Moderate Controlled Cortical Impact Brain Injury. World Neurosurg. 2019, 122, e1090–e1101. [Google Scholar] [CrossRef]
- Auderset, L.; Cullen, C.L.; Young, K.M. Low Density Lipoprotein-Receptor Related Protein 1 Is Differentially Expressed by Neuronal and Glial Populations in the Developing and Mature Mouse Central Nervous System. PLoS ONE 2016, 11, e0155878. [Google Scholar] [CrossRef]
- May, P.; Rohlmann, A.; Bock, H.H.; Zurhove, K.; Marth, J.D.; Schomburg, E.D.; Noebels, J.L.; Beffert, U.; Sweatt, J.D.; Weeber, E.J.; et al. Neuronal LRP1 Functionally Associates with Postsynaptic Proteins and Is Required for Normal Motor Function in Mice. Mol. Cell. Biol. 2004, 24, 8872. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Trotter, J.; Zhang, J.; Peters, M.M.; Cheng, H.; Bao, J.; Han, X.; Weeber, E.J.; Bu, G. Neuronal LRP1 Knockout in Adult Mice Leads to Impaired Brain Lipid Metabolism and Progressive, Age-Dependent Synapse Loss and Neurodegeneration. J. Neurosci. 2010, 30, 17068. [Google Scholar] [CrossRef]
- Zhu, Y.; Romero, M.I.; Ghosh, P.; Ye, Z.; Charnay, P.; Rushing, E.J.; Marth, J.D.; Parada, L.F. Ablation of NF1 Function in Neurons Induces Abnormal Development of Cerebral Cortex and Reactive Gliosis in the Brain. Genes. Dev. 2001, 15, 859. [Google Scholar] [CrossRef]
- Van Gool, B.; Storck, S.E.; Reekmans, S.M.; Lechat, B.; Gordts, P.L.S.M.; Pradier, L.; Pietrzik, C.U.; Roebroek, A.J.M. LRP1 Has a Predominant Role in Production over Clearance of Aβ in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7234. [Google Scholar] [CrossRef]
- Fuentealba, R.A.; Liu, Q.; Zhang, J.; Kanekiyo, T.; Hu, X.; Lee, J.M.; Ladu, M.J.; Bu, G. Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Mediates Neuronal Aβ42 Uptake and Lysosomal Trafficking. PLoS ONE 2010, 5, e11884. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.C.; Zheng, H.; Kanekiyo, T.; Atagi, Y.; Jia, L.; Wang, D.; N’songo, A.; Can, D.; Xu, H.; et al. LRP1 Modulates the Microglial Immune Response via Regulation of JNK and NF-ΚB Signaling Pathways. J. Neuroinflamm. 2016, 13, 304. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J.; Tran, H.; Verbeek, M.M.; Reiss, K.; Estus, S.; Bu, G. LRP1 Shedding in Human Brain: Roles of ADAM10 and ADAM17. Mol. Neurodegener. 2009, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Gorovoy, M.; Gaultier, A.; Campana, W.M.; Firestein, G.S.; Gonias, S.L. Inflammatory Mediators Promote Production of Shed LRP1/CD91, Which Regulates Cell Signaling and Cytokine Expression by Macrophages. J. Leukoc. Biol. 2010, 88, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Klingener, M.; Aguirre, A. TGFβ Signaling Regulates the Timing of CNS Myelination by Modulating Oligodendrocyte Progenitor Cell Cycle Exit through SMAD3/4/FoxO1/Sp1. J. Neurosci. 2014, 34, 7917. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Fang, L.; Tang, X.; Lu, S.; Tamm, M.; Stolz, D.; Roth, M. TGF-β Upregulated Mitochondria Mass through the SMAD2/3→C/EBPβ→PRMT1 Signal Pathway in Primary Human Lung Fibroblasts. J. Immunol. 2019, 202, 37–47. [Google Scholar] [CrossRef]
- Abe, Y.; Sakairi, T.; Beeson, C.; Kopp, J.B. TGF-Β1 Stimulates Mitochondrial Oxidative Phosphorylation and Generation of Reactive Oxygen Species in Cultured Mouse Podocytes, Mediated in Part by the MTOR Pathway. Am. J. Physiol. Renal Physiol. 2013, 305, F1477. [Google Scholar] [CrossRef]
- Scholpa, N.E.; Williams, H.; Wang, W.; Corum, D.; Narang, A.; Tomlinson, S.; Sullivan, P.G.; Rabchevsky, A.G.; Schnellmann, R.G. Pharmacological Stimulation of Mitochondrial Biogenesis Using the Food and Drug Administration-Approved Β2-Adrenoreceptor Agonist Formoterol for the Treatment of Spinal Cord Injury. J. Neurotrauma 2019, 36, 962. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yoshida, M.; Yamato, M.; Ide, T.; Wu, Z.; Ochi-Shindou, M.; Kanki, T.; Kang, D.; Sunagawa, K.; Tsutsui, H.; et al. Reverse of Age-Dependent Memory Impairment and Mitochondrial DNA Damage in Microglia by an Overexpression of Human Mitochondrial Transcription Factor A in Mice. J. Neurosci. 2008, 28, 8624. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Matsusaka, H.; Kang, D.; Matsushima, S.; Ide, T.; Kubota, T.; Fujiwara, T.; Hamasaki, N.; Takeshita, A.; Sunagawa, K.; et al. Overexpression of Mitochondrial Transcription Factor a Ameliorates Mitochondrial Deficiencies and Cardiac Failure after Myocardial Infarction. Circulation 2005, 112, 683–690. [Google Scholar] [CrossRef]
- Borcherding, N.; Jia, W.; Giwa, R.; Field, R.L.; Moley, J.R.; Kopecky, B.J.; Chan, M.M.; Yang, B.Q.; Sabio, J.M.; Walker, E.C.; et al. Dietary Lipids Inhibit Mitochondria Transfer to Macrophages to Divert Adipocyte-Derived Mitochondria into the Blood. Cell Metab. 2022, 34, 1499–1513.e8. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Joseph, B.; Spry, M.; Vekaria, H.J.; Saatman, K.E.; Sullivan, P.G. Acute Mitochondrial Impairment Underlies Prolonged Cellular Dysfunction after Repeated Mild Traumatic Brain Injuries. J. Neurotrauma 2019, 36, 1252. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Spry, M.L.; Gooch, J.L.; Cloud, A.L.; Vekaria, H.J.; Burden, S.; Powell, D.K.; Berkowitz, B.A.; Geldenhuys, W.J.; Harris, N.G.; et al. Clinically Relevant Mitochondrial-Targeted Therapy Improves Chronic Outcomes after Traumatic Brain Injury. Brain 2021, 144, 3788. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Clouthier, D.E.; Hammer, R.E. LDL Receptor-Related Protein Internalizes and Degrades UPA-PAI-1 Complexes and Is Essential for Embryo Implantation. Cell 1992, 71, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Leung, L.Y.; Yang, X.; Flerlage, W.J.; Gilsdorf, J.S.; Deng-Bryant, Y.; Shear, D.A. Comprehensive Profile of Acute Mitochondrial Dysfunction in a Preclinical Model of Severe Penetrating TBI. Front. Neurol. 2019, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A. Brain Ischemia/Reperfusion Injury and Mitochondrial Complex I Damage. Biochemistry 2019, 84, 1411–1423. [Google Scholar] [CrossRef]
- Pope, S.; Land, J.M.; Heales, S.J.R. Oxidative Stress and Mitochondrial Dysfunction in Neurodegeneration; Cardiolipin a Critical Target? Biochim. Biophys. Acta (BBA) 2008, 1777, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Leslie, P.L.; Franklin, D.A.; Liu, Y.; Zhang, Y. P53 Regulates the Expression of LRP1 and Apoptosis through a Stress Intensity-Dependent MicroRNA Feedback Loop. Cell Rep. 2018, 24, 1484–1495. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Gene Symbol | Assay ID | |
|---|---|---|---|
| 1 | TFAM | Transcription Factor A, Mitochondrial | Mm00447485_m1 |
| 2 | Nrf1 | Nuclear respiratory factor 1 | Mm01135606_m1 |
| 3 | Prkaa2 | Protein Kinase AMP-Activated Catalytic Subunit Alpha 2 | Mm01264789_m1 |
| 4 | ND1 | NADH-ubiquinone oxidoreductase chain 1 | Mm04225274_s1 |
| 5 | Ucp2 | Uncoupling protein 2 | Mm00627599_m1 |
| 6 | Pink1 | PTEN-induced putative kinase 1 | Mm00550827 |
| 7 | Gtf2h1 | General transcription factor IIH subunit 1 | Mm00500417_m1 |
| 8 | Mfn1 | Mitofusin 1 | Mm00612599_m1 |
| 9 | Fis1 | Fission protein 1 | Mm00481580 |
| 10 | ATP6 | ATP Synthase Membrane Subunit 6 | Mm03649417_g1 |
| 11 | Nfe212 (Nrf2) | Nuclear factor erythroid 2-related factor 2 | Mm00477784_m1 |
| 12 | Sirt1 | Sirtuin 1 | Mm01168521_m1 |
| 13 | Mfn2 | Mitofusin 2 | Mm00500120 |
| 14 | Dnm1 | Dynamin 1 | Mm01342903_m1 |
| 15 | COX1 | Cytochrome c oxidase I | Mm04225243_g1 |
| 16 | Ndufs1 | NADH:Ubiquinone Oxidoreductase Core Subunit S1 | Mm005236040_m1 |
| 17 | ND6/ND5 | NADH-ubiquinone oxidoreductase chain 6 protein/5 | Mm04225325_g1 |
| 18 | 18sRNA | Mm03928990_g1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velmurugan, G.V.; Hubbard, W.B.; Prajapati, P.; Vekaria, H.J.; Patel, S.P.; Rabchevsky, A.G.; Sullivan, P.G. LRP1 Deficiency Promotes Mitostasis in Response to Oxidative Stress: Implications for Mitochondrial Targeting after Traumatic Brain Injury. Cells 2023, 12, 1445. https://doi.org/10.3390/cells12101445
Velmurugan GV, Hubbard WB, Prajapati P, Vekaria HJ, Patel SP, Rabchevsky AG, Sullivan PG. LRP1 Deficiency Promotes Mitostasis in Response to Oxidative Stress: Implications for Mitochondrial Targeting after Traumatic Brain Injury. Cells. 2023; 12(10):1445. https://doi.org/10.3390/cells12101445
Chicago/Turabian StyleVelmurugan, Gopal V., W. Brad Hubbard, Paresh Prajapati, Hemendra J. Vekaria, Samir P. Patel, Alexander G. Rabchevsky, and Patrick G. Sullivan. 2023. "LRP1 Deficiency Promotes Mitostasis in Response to Oxidative Stress: Implications for Mitochondrial Targeting after Traumatic Brain Injury" Cells 12, no. 10: 1445. https://doi.org/10.3390/cells12101445