
CRMP2 Participates in Regulating Mitochondrial Morphology and Motility in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Postmortem Brain Tissues
2.2. Animals
2.3. Genotyping
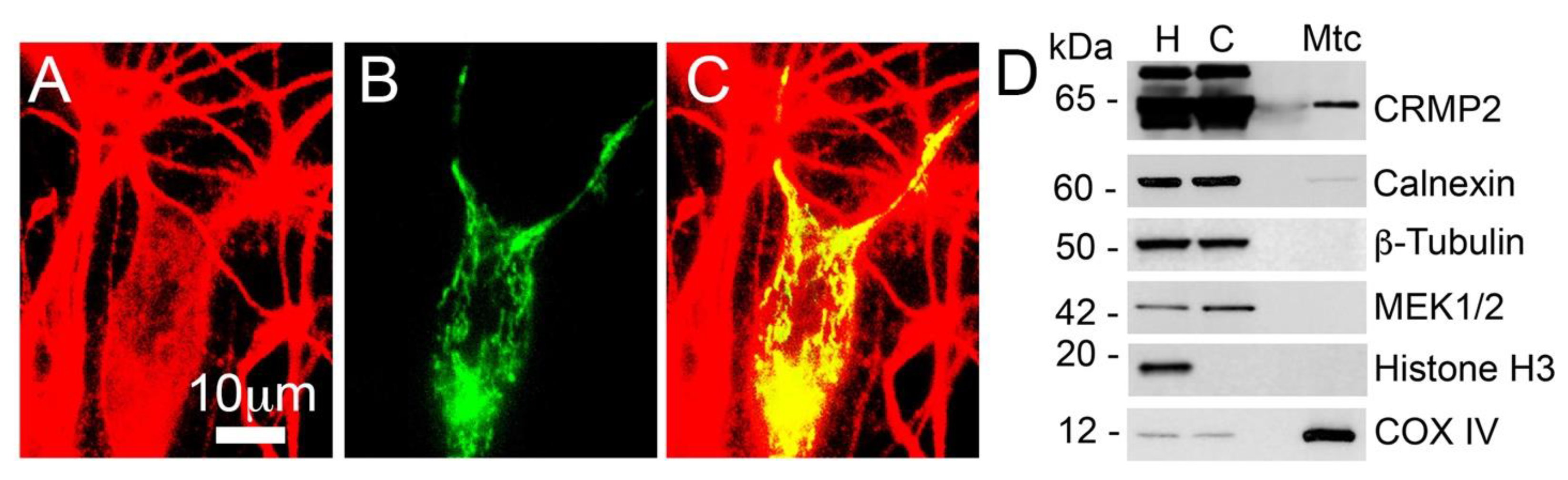
2.4. Isolation and Purification of Brain Synaptic Mitochondria
2.5. Isolation of Mitochondria from Cultured Neurons
2.6. Neuronal Cell Culture
2.7. Western Blotting
2.8. Co-Immunoprecipitation
2.9. Neuronal Transfection
2.10. Mitochondrial Morphology
2.11. Mitochondrial Motility
2.12. Neuronal Cell Death
2.13. Statistics
3. Results
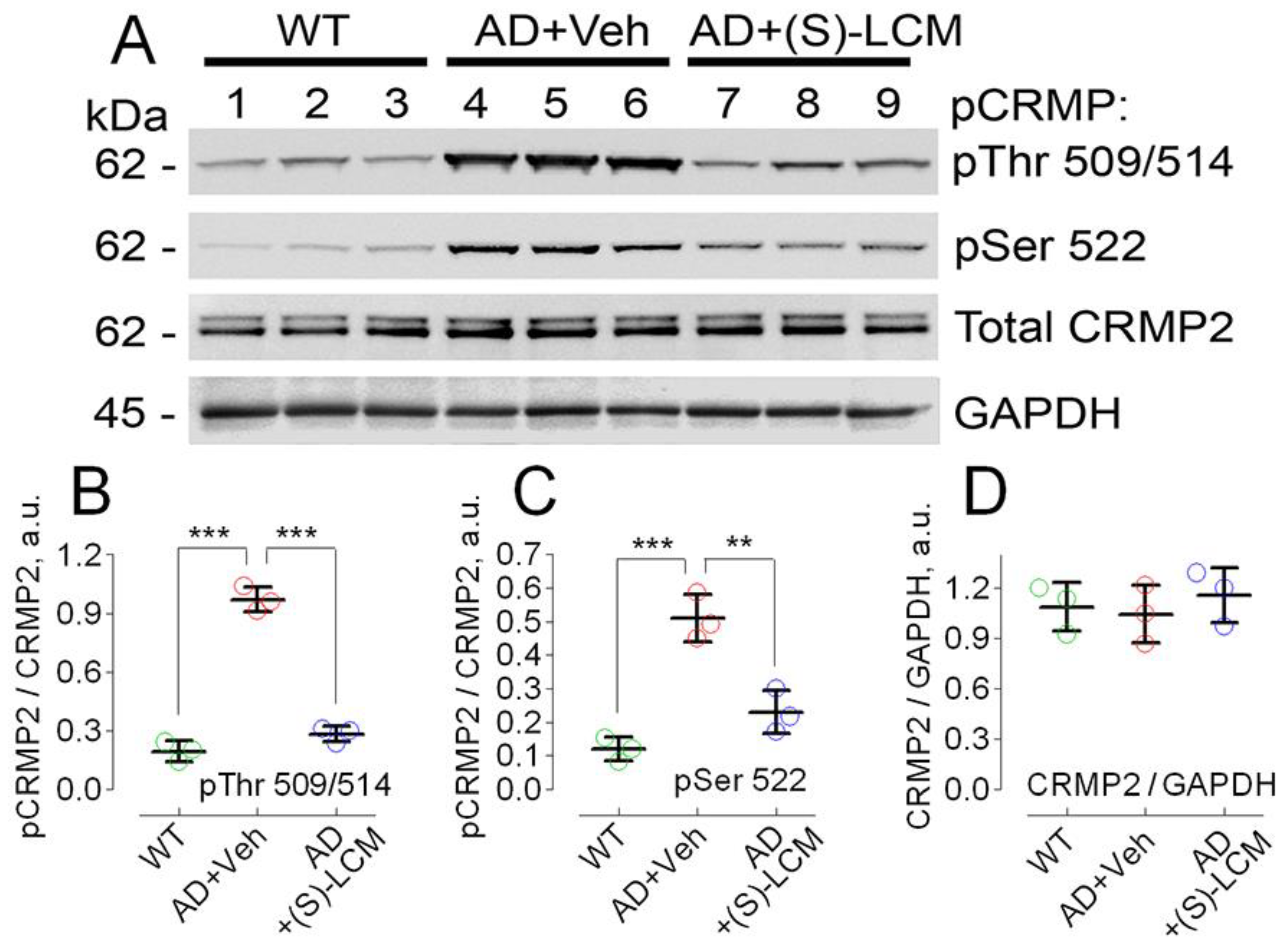
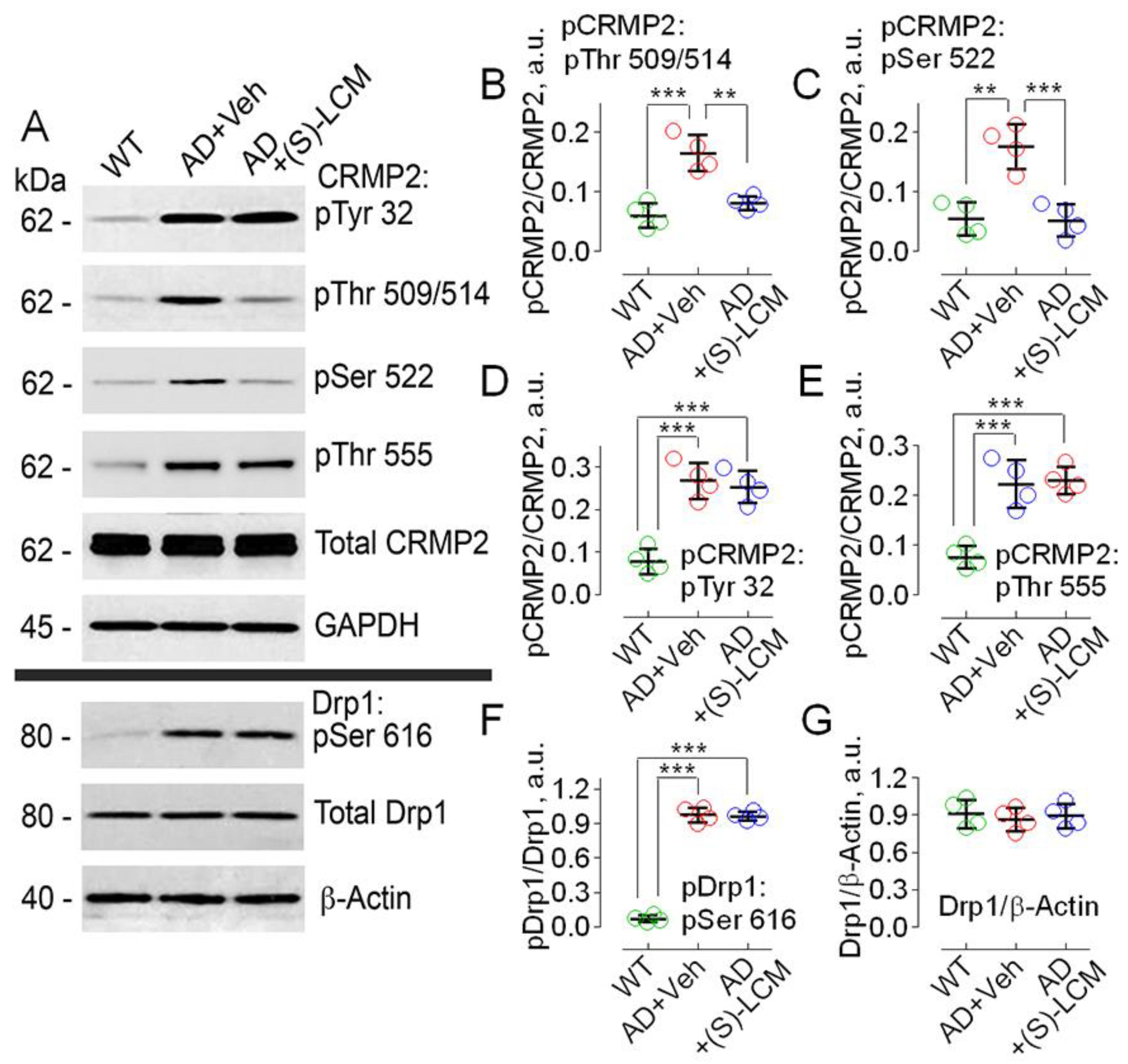
3.1. Hyperphosphorylation of CRMP2 in AD
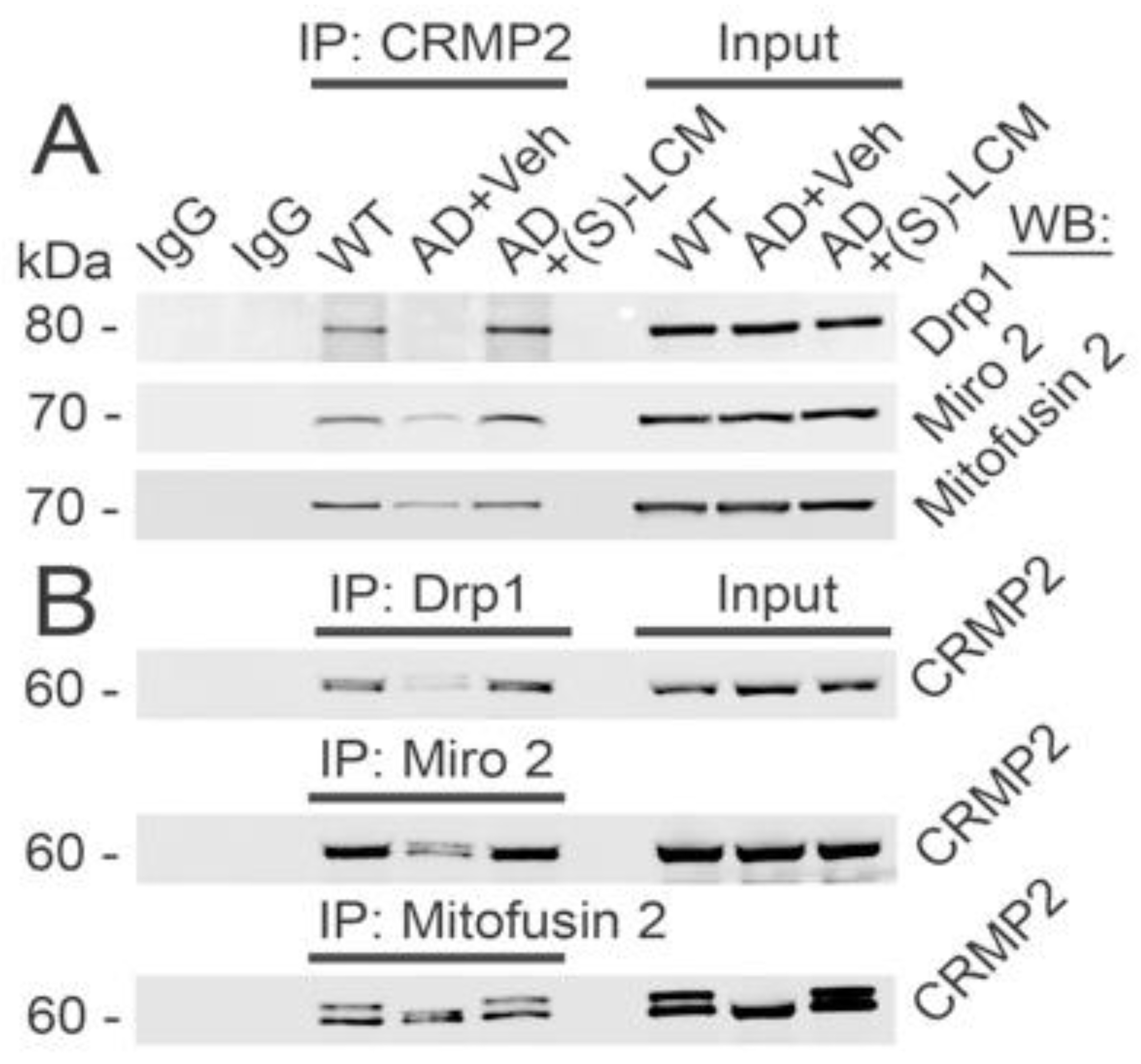
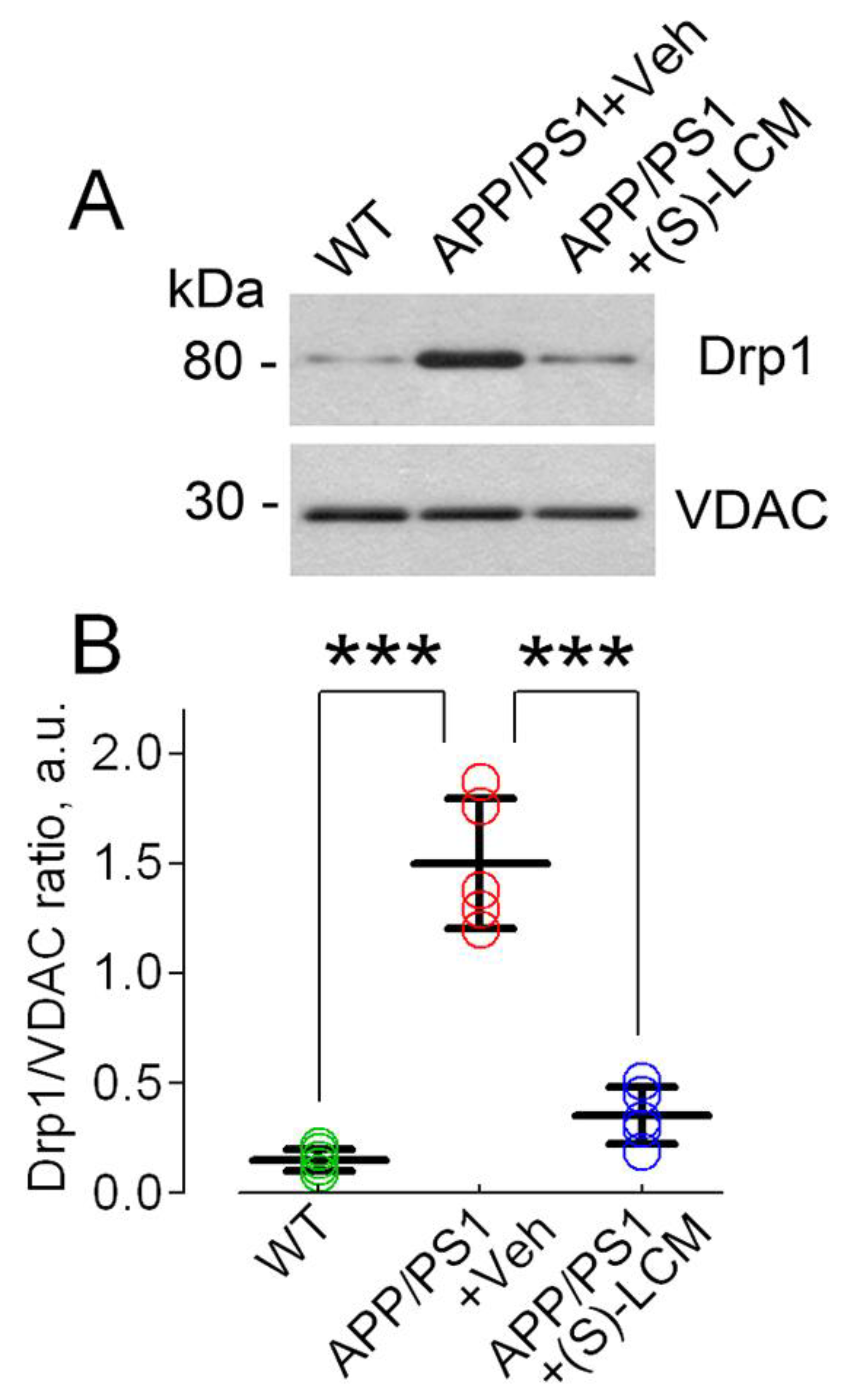
3.2. CRMP2 Interaction with Proteins Participating in Regulation of Mitochondrial Morphology and Motility in AD Neurons
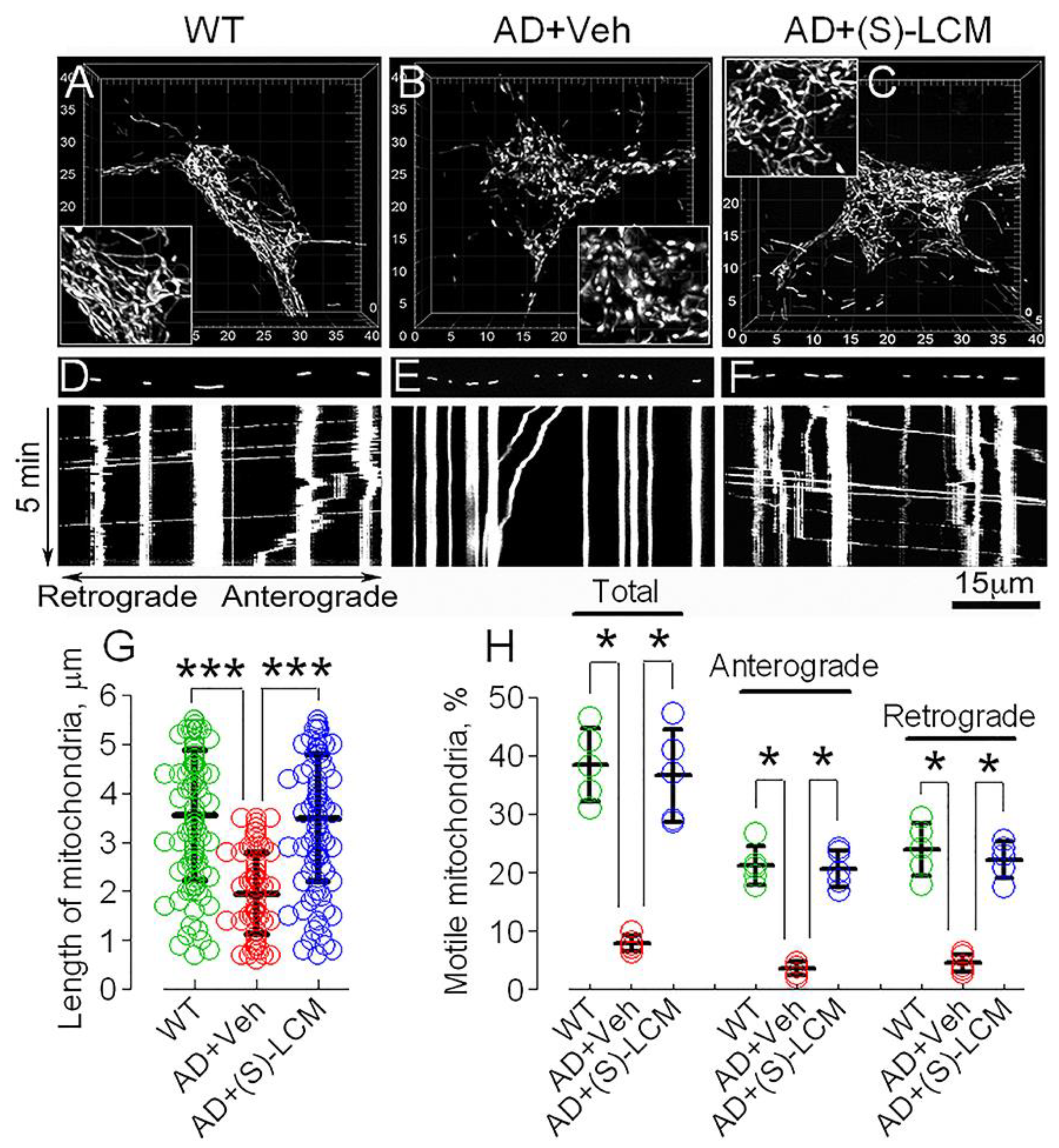
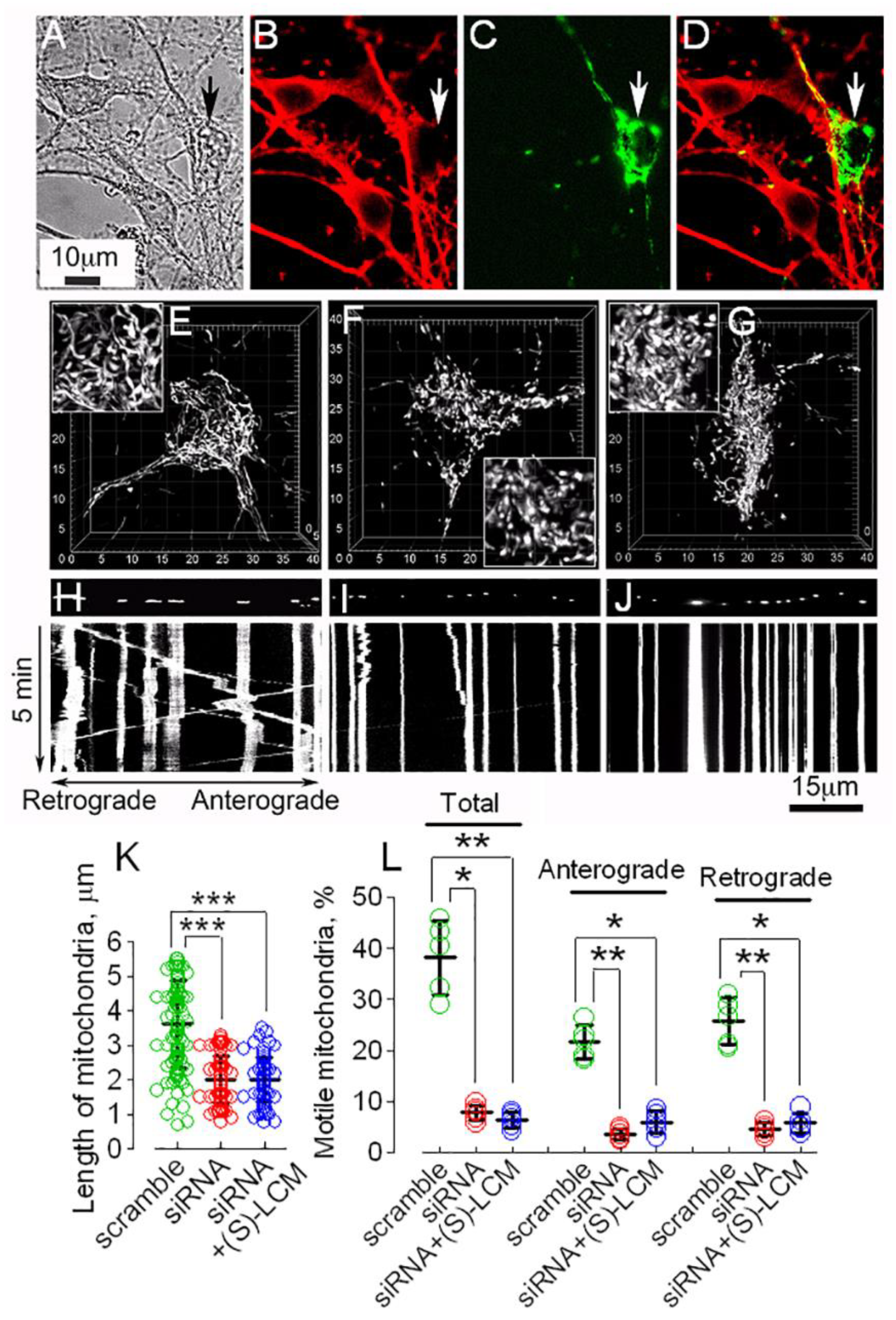
3.3. CRMP2 and Mitochondrial Motility and Morphology in AD Neurons
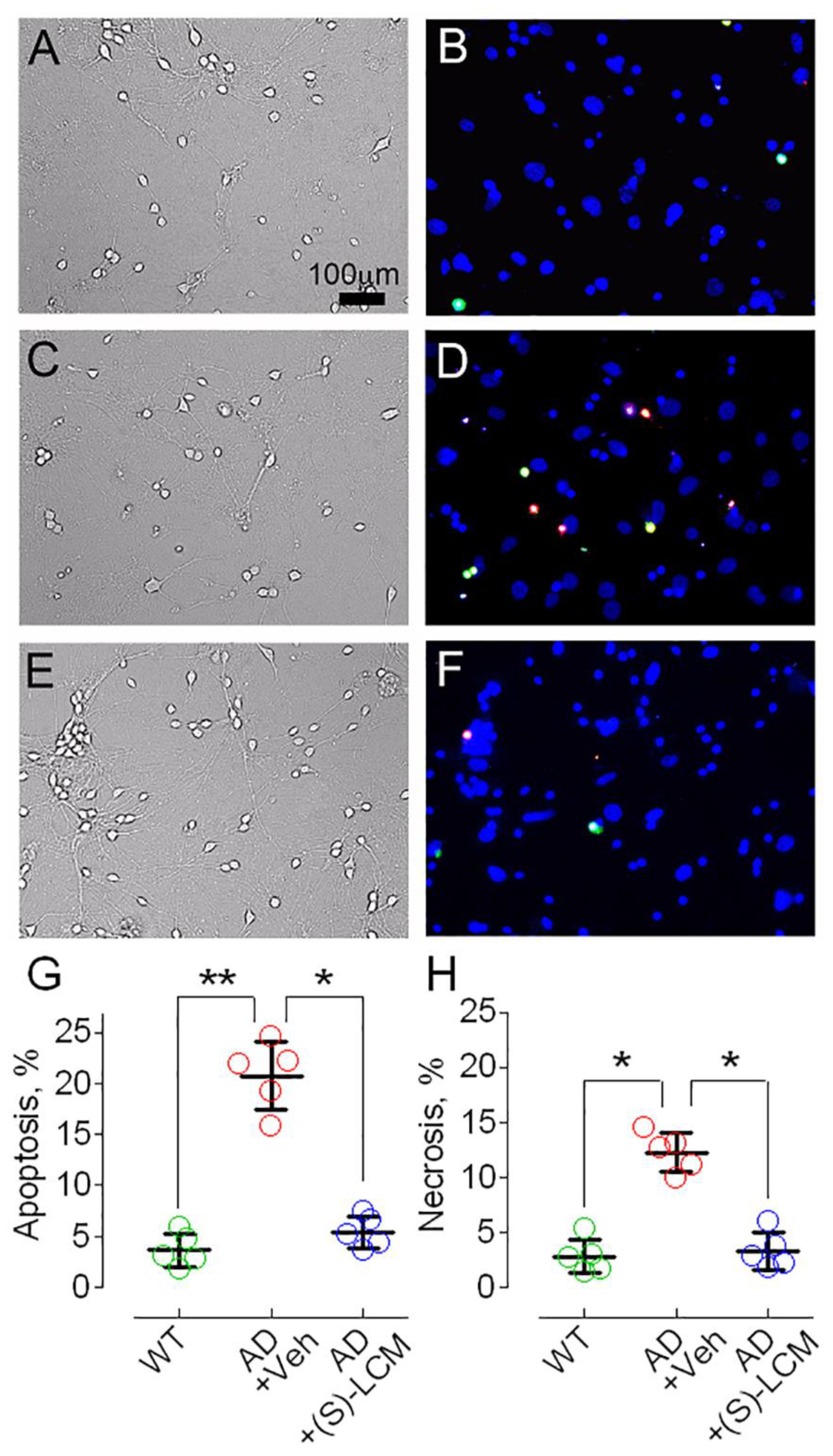
3.4. CRMP2 and Cell Viability of AD Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers. Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, J.E.; Lezi, E.; Lu, J.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.V.; Rivers-Auty, J.; Allan, S.M.; Lawrence, C.B. Mitochondrial Abnormalities and Synaptic Loss Underlie Memory Deficits Seen in Mouse Models of Obesity and Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 55, 915–932. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Pedros, I.; Petrov, D.; Allgaier, M.; Sureda, F.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallas, M.; Vazquez-Carrera, M.; Casadesus, G.; et al. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1556–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. Alzheimer’s disease: Diverse aspects of mitochondrial malfunctioning. Int. J. Clin. Exp. Pathol. 2010, 3, 570–581. [Google Scholar]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Khanna, R.; Wilson, S.M.; Brittain, J.M.; Weimer, J.; Sultana, R.; Butterfield, A.; Hensley, K. Opening Pandora’s jar: A primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012, 7, 749–771. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, T.; Pellman, J.J.; Yang, X.F.; Khanna, R.; Brustovetsky, N. Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J. Biol. Chem. 2014, 289, 7470–7482. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons. Cells 2021, 10, 2781. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. Involvement of CRMP2 in Regulation of Mitochondrial Morphology and Motility in Huntington’s Disease. Cells 2021, 10, 3172. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Watanabe, H.; Iwamatsu, A.; Kaibuchi, K. Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 2005, 93, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, S.H.; Kim, M.J.; Magee, K.A.; Aui, P.M.; Thomas, S.; Bakhuraysah, M.M.; Alrehaili, A.A.; Lee, J.Y.; Steer, D.L.; Kenny, R.; et al. Amyloid-beta-dependent phosphorylation of collapsin response mediator protein-2 dissociates kinesin in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1066–1080. [Google Scholar] [PubMed]
- Arimura, N.; Hattori, A.; Kimura, T.; Nakamuta, S.; Funahashi, Y.; Hirotsune, S.; Furuta, K.; Urano, T.; Toyoshima, Y.Y.; Kaibuchi, K. CRMP-2 directly binds to cytoplasmic dynein and interferes with its activity. J. Neurochem. 2009, 111, 380–390. [Google Scholar] [CrossRef]
- Charrier, E.; Reibel, S.; Rogemond, V.; Aguera, M.; Thomasset, N.; Honnorat, J. Collapsin response mediator proteins (CRMPs): Involvement in nervous system development and adult neurodegenerative disorders. Mol. Neurobiol. 2003, 28, 51–64. [Google Scholar] [CrossRef]
- Cole, A.R.; Knebel, A.; Morrice, N.A.; Robertson, L.A.; Irving, A.J.; Connolly, C.N.; Sutherland, C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 2004, 279, 50176–50180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers. Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 864–879. [Google Scholar] [CrossRef]
- Tsai, L.H.; Lee, M.S.; Cruz, J. Cdk5, a therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta 2004, 1697, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Hamajima, N.; Ihara, Y. Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry 2000, 39, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.R.; Noble, W.; van, A.L.; Plattner, F.; Meimaridou, R.; Hogan, D.; Taylor, M.; LaFrancois, J.; Gunn-Moore, F.; Verkhratsky, A.; et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 2007, 103, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.; Thornhill, P.; Cole, A.R.; Sutherland, C. Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Curr. Alzheimer Res. 2009, 6, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Petratos, S.; Li, Q.X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.I.; Small, D.H. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2008, 131, 90–108. [Google Scholar] [CrossRef] [Green Version]
- Quach, T.T.; Moutal, A.; Khanna, R.; Deems, N.P.; Duchemin, A.M.; Barrientos, R.M. Collapsin Response Mediator Proteins: Novel Targets for Alzheimer’s Disease. J. Alzheimers. Dis. 2020, 77, 949–960. [Google Scholar] [CrossRef]
- Rembutsu, M.; Soutar, M.P.; van, A.L.; Gourlay, R.; Hastie, C.J.; McLauchlan, H.; Morrice, N.A.; Cole, A.R.; Sutherland, C. Novel procedure to investigate the effect of phosphorylation on protein complex formation in vitro and in cells. Biochemistry 2008, 47, 2153–2161. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Z.-H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutal, A.; Francois-Moutal, L.; Perez-Miller, S.; Cottier, K.; Chew, L.A.; Yeon, S.K.; Dai, J.; Park, K.D.; Khanna, M.; Khanna, R. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Mol. Neurobiol. 2015, 53, 1959–1976. [Google Scholar] [CrossRef]
- Wilson, S.M.; Moutal, A.; Melemedjian, O.K.; Wang, Y.; Ju, W.; Francois-Moutal, L.; Khanna, M.; Khanna, R. The functionalized amino acid (S)-Lacosamide subverts CRMP2-mediated tubulin polymerization to prevent constitutive and activity-dependent increase in neurite outgrowth. Front Cell Neurosci. 2014, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noterman, M.F.; Chaubey, K.; Lin-Rahardja, K.; Rajadhyaksha, A.M.; Pieper, A.A.; Taylor, E.B. Dual-process brain mitochondria isolation preserves function and clarifies protein composition. Proc. Natl. Acad. Sci. USA 2021, 118, e2019046118. [Google Scholar] [CrossRef] [PubMed]
- Kushnareva, Y.E.; Wiley, S.E.; Ward, M.W.; Andreyev, A.Y.; Murphy, A.N. Excitotoxic injury to mitochondria isolated from cultured neurons. J. Biol. Chem. 2005, 280, 28894–28902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinsky, J.M. Intracellular calcium levels during the period of delayed excitotoxicity. J. Neurosci. 1993, 13, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calpain activation and Na(+)/Ca(2+) exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 2010, 88, 1317–1328. [Google Scholar] [PubMed] [Green Version]
- Brittain, J.M.; Chen, L.; Wilson, S.M.; Brustovetsky, T.; Gao, X.; Ashpole, N.M.; Molosh, A.I.; You, H.; Hudmon, A.; Shekhar, A.; et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 2011, 286, 37778–37792. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.X.; Schmutzler, B.S.; Brittain, J.M.; Wang, Y.; Hingtgen, C.M.; Nicol, G.D.; Khanna, R. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 2009, 122, 4351–4362. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, T.; Li, V.; Brustovetsky, N. Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium 2009, 46, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Moutal, A.; Chew, L.A.; Yang, X.; Wang, Y.; Yeon, S.K.; Telemi, E.; Meroueh, S.; Park, K.D.; Shrinivasan, R.; Gilbraith, K.B.; et al. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain 2016, 157, 1448–1463. [Google Scholar] [CrossRef] [Green Version]
- Trinchese, F.; Liu, S.; Ninan, I.; Puzzo, D.; Jacob, J.P.; Arancio, O. Cell cultures from animal models of Alzheimer’s disease as a tool for faster screening and testing of drug efficacy. J. Mol. Neurosci. 2004, 24, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Shalbuyeva, N.; Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calcium-dependent spontaneously reversible remodeling of brain mitochondria. J. Biol. Chem. 2006, 281, 37547–37558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.H.; Nakamura, T.; Lipton, S.A. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol. Life Sci. 2010, 67, 3435–3447. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Bossy-Wetzel, E.; Barsoum, M.J.; Godzik, A.; Schwarzenbacher, R.; Lipton, S.A. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 2003, 15, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Obrenovich, M.E.; Reddy, V.P.; Shenk, J.C.; Moreira, P.I.; Nunomura, A.; Zhu, X.; Smith, M.A.; Perry, G. Antioxidant therapy in Alzheimer’s disease: Theory and practice. Mini. Rev. Med. Chem. 2008, 8, 1395–1406. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 1483–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Manczak, M.; Pradeepkiran, J.A.; Morton, H.; Reddy, P.H. A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic tau mouse model of Alzheimer disease. Hum. Mol. Genet. 2021, 31, 1788–1805. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Goshima, Y.; Nakamura, F.; Strittmatter, P.; Strittmatter, S.M. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 1995, 376, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 2011, 43, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Inagaki, N.; Chihara, K.; Menager, C.; Nakamura, N.; Amano, M.; Iwamatsu, A.; Goshima, Y.; Kaibuchi, K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 2000, 275, 23973–23980. [Google Scholar] [CrossRef] [Green Version]
- Arimura, N.; Menager, C.; Kawano, Y.; Yoshimura, T.; Kawabata, S.; Hattori, A.; Fukata, Y.; Amano, M.; Goshima, Y.; Inagaki, M.; et al. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell Biol. 2005, 25, 9973–9984. [Google Scholar] [CrossRef] [Green Version]
- Uchida, Y.; Ohshima, T.; Yamashita, N.; Ogawara, M.; Sasaki, Y.; Nakamura, F.; Goshima, Y. Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. J. Biol. Chem. 2009, 284, 27393–27401. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.A.; Pimplikar, S.W. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J. Cell Biol. 2005, 171, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Khanna, R.; Moutal, A.; Perez-Miller, S.; Chefdeville, A.; Boinon, L.; Patek, M. Druggability of CRMP2 for Neurodegenerative Diseases. ACS Chem. Neurosci. 2020, 11, 2492–2505. [Google Scholar] [CrossRef]
- Yang, Z.; Kuboyama, T.; Tohda, C. A Systematic Strategy for Discovering a Therapeutic Drug for Alzheimer’s Disease and Its Target Molecule. Front Pharmacol. 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, R.; van, A.L.; Mann, D.M.; Platt, B.; Plattner, F.; Bedford, L.; Mayer, J.; Howlett, D.; Usardi, A.; Sutherland, C.; et al. CRMP2 hyperphosphorylation is characteristic of Alzheimer’s disease and not a feature common to other neurodegenerative diseases. J. Alzheimers. Dis. 2011, 27, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, C.; Rutten, B.P.; Pielen, A.; Schafer, S.; Wirths, O.; Tremp, G.; Czech, C.; Blanchard, V.; Multhaup, G.; Rezaie, P.; et al. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 2004, 164, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Bayer, T.A. Neuron loss in transgenic mouse models of Alzheimer’s disease. Int. J. Alzheimers. Dis. 2010, 2010, 723782. [Google Scholar] [CrossRef] [Green Version]
- Comes, G.; Manso, Y.; Escrig, A.; Fernandez-Gayol, O.; Sanchis, P.; Molinero, A.; Giralt, M.; Carrasco, J.; Hidalgo, J. Influence of Transgenic Metallothionein-1 on Gliosis, CA1 Neuronal Loss, and Brain Metal Levels of the Tg2576 Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 251. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, M.E.; Wiederhold, K.H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron loss in APP transgenic mice. Nature 1998, 395, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calza, L. Vulnerability of primary neurons derived from Tg2576 Alzheimer mice to oxygen and glucose deprivation: Role of intraneuronal amyloid-beta accumulation and astrocytes. Dis. Model. Mech. 2017, 10, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Stowers, R.S.; Megeath, L.J.; Gorska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isono, T.; Yamashita, N.; Obara, M.; Araki, T.; Nakamura, F.; Kamiya, Y.; Alkam, T.; Nitta, A.; Nabeshima, T.; Mikoshiba, K.; et al. Amyloid-β25–35 induces impairment of cognitive function and long-term potentiation through phosphorylation of collapsin response mediator protein 2. Neurosci. Res. 2013, 77, 180–185. [Google Scholar] [CrossRef]
- Yamashita, N.; Ohshima, T.; Nakamura, F.; Kolattukudy, P.; Honnorat, J.; Mikoshiba, K.; Goshima, Y. Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J. Neurosci. 2012, 32, 1360–1365. [Google Scholar] [CrossRef] [Green Version]
- Mileusnic, R.; Rose, S.P. The memory enhancing effect of the APP-derived tripeptide Ac-rER is mediated through CRMP2. J. Neurochem. 2011, 118, 616–625. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 Participates in Regulating Mitochondrial Morphology and Motility in Alzheimer’s Disease. Cells 2023, 12, 1287. https://doi.org/10.3390/cells12091287
Brustovetsky T, Khanna R, Brustovetsky N. CRMP2 Participates in Regulating Mitochondrial Morphology and Motility in Alzheimer’s Disease. Cells. 2023; 12(9):1287. https://doi.org/10.3390/cells12091287
Chicago/Turabian StyleBrustovetsky, Tatiana, Rajesh Khanna, and Nickolay Brustovetsky. 2023. "CRMP2 Participates in Regulating Mitochondrial Morphology and Motility in Alzheimer’s Disease" Cells 12, no. 9: 1287. https://doi.org/10.3390/cells12091287