
Tau Transfer via Extracellular Vesicles Disturbs the Astrocytic Mitochondrial System
 , , , , , , , and
, , , , , , , and
Abstract
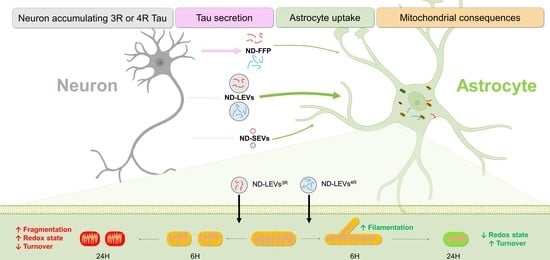
:
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Rat Primary Neuron and Astrocyte Cultures
2.3. Human iPSC-Derived Astrocyte Culture
2.4. Microfluidic System and Immunofluorescence
2.5. EVs Isolation from Primary Cultures
2.6. EV Isolation from Human Prefrontal Cortex Tissue
2.7. Nanoparticle Tracking Analysis (NTA)
2.8. Electron Microscopy [22,35]
2.9. Antibodies
2.10. ELISA
2.11. Quantification of Tau-V5 Levels in Primary Culture
2.12. Analysis of Mitochondrial Redox State, Morphology and Dynamics by MitoTimer
2.13. Sample Sizes, Calculations and Statistical Analysis
3. Results
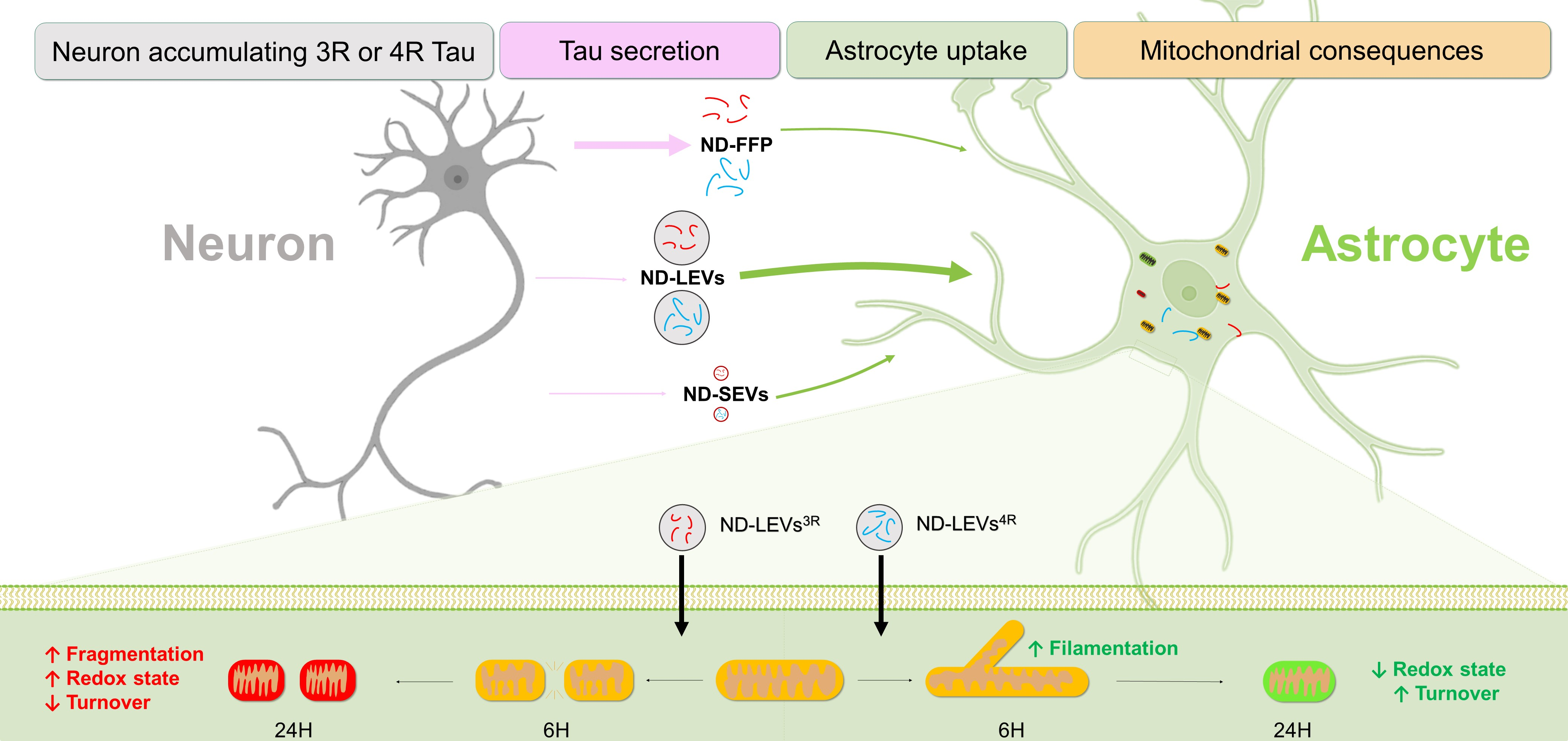
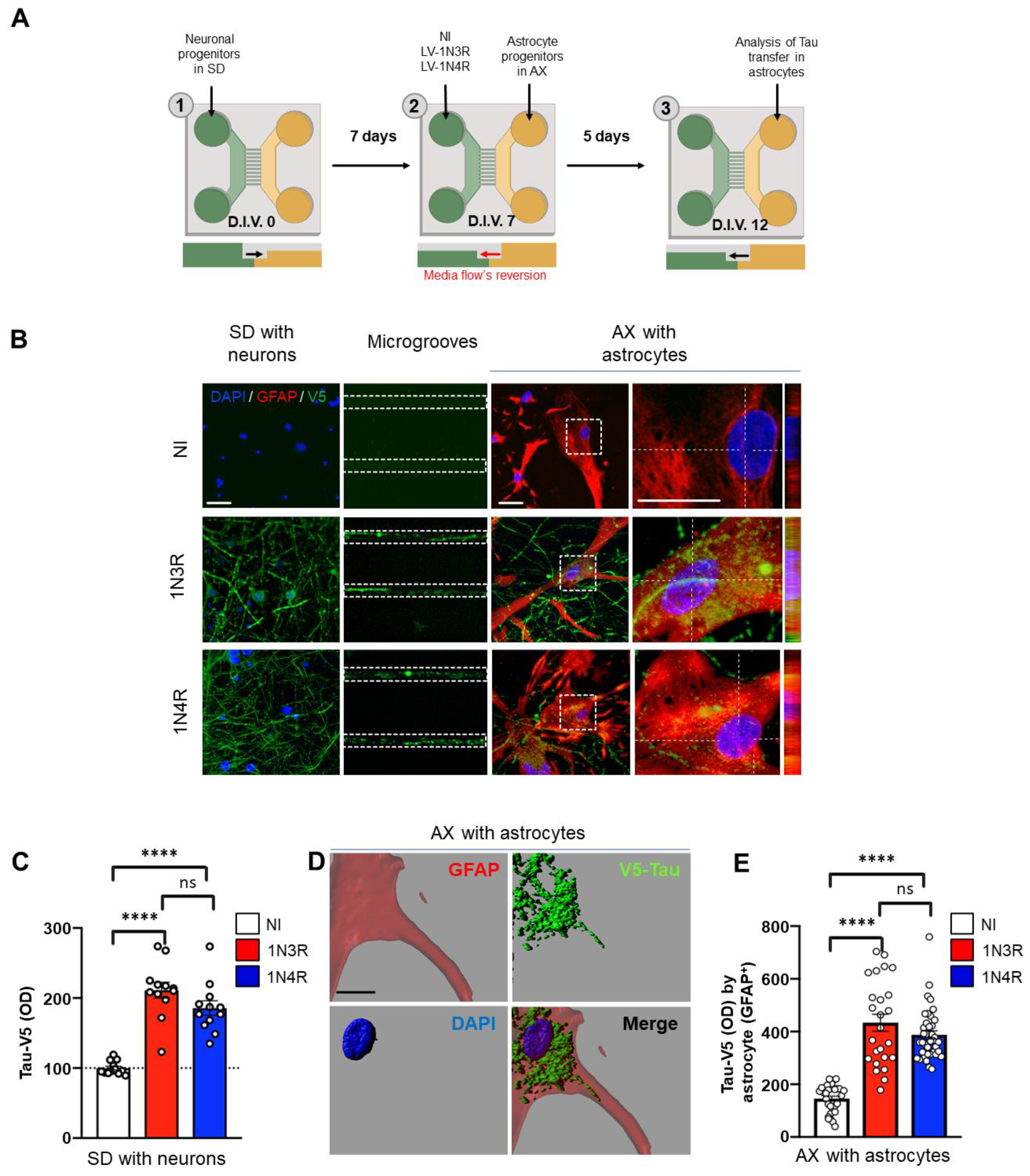
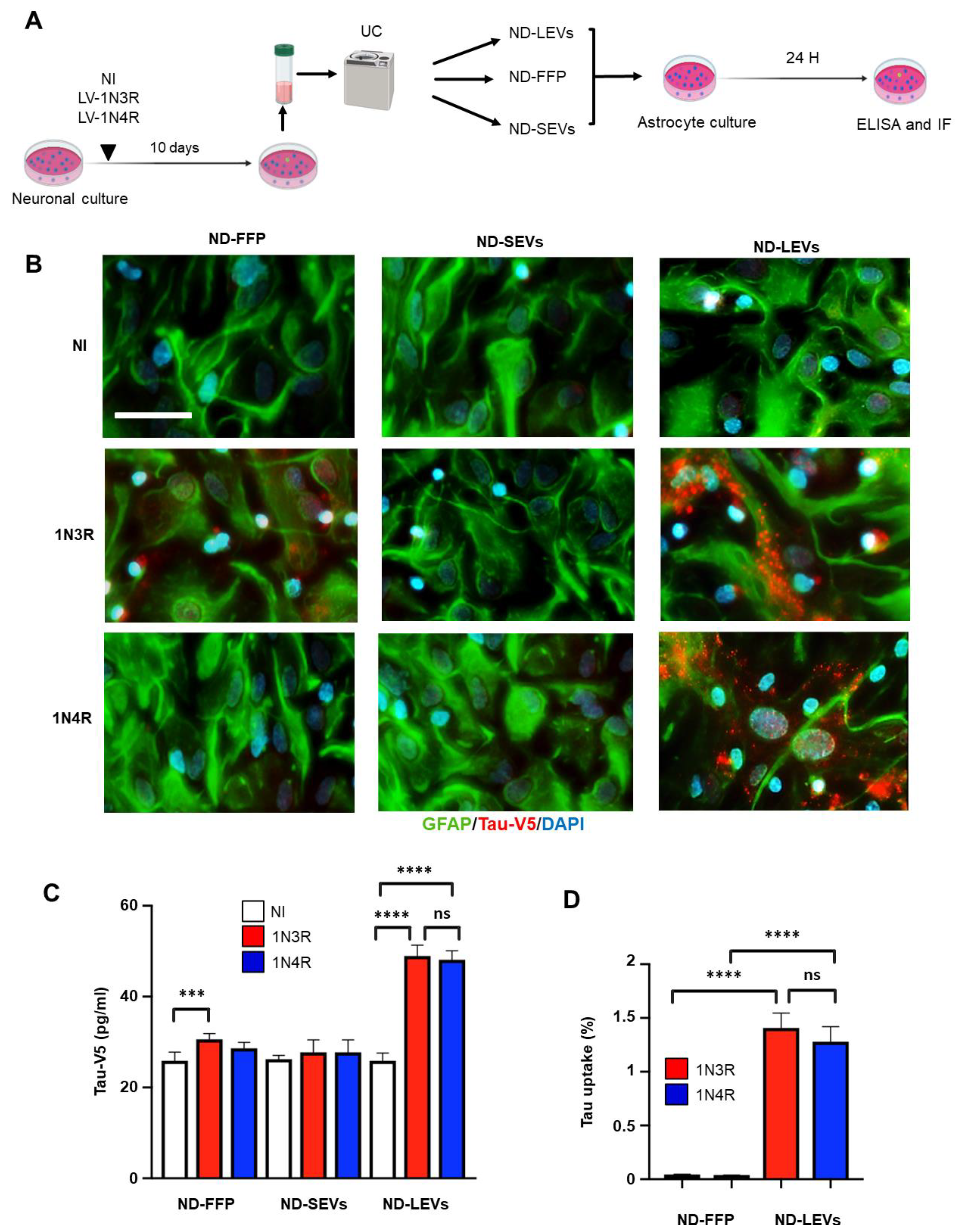
3.1. 3R and 4R Tau Transfer from Neurons to Astrocytes
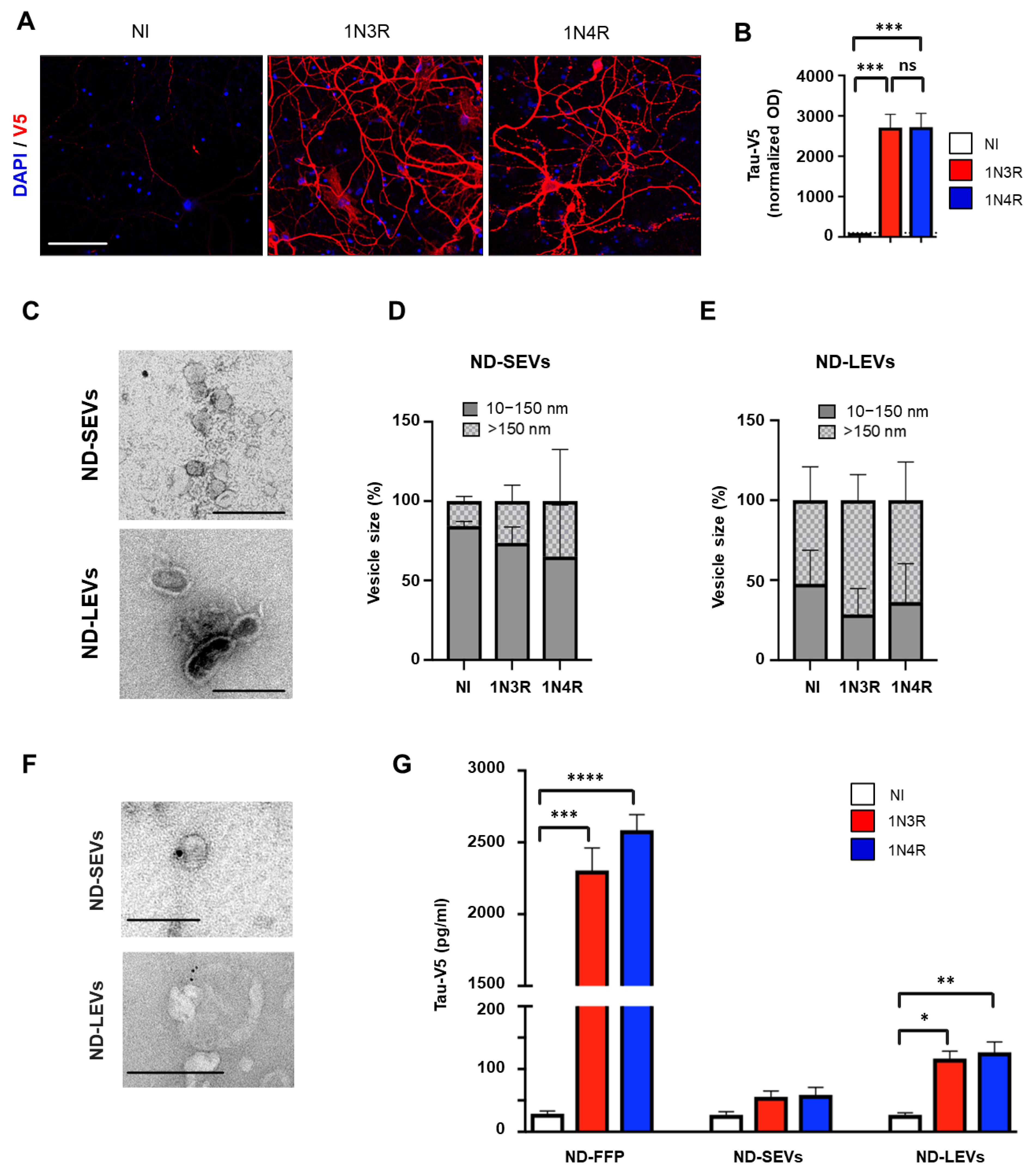
3.2. Neuronal Tau Is Mainly Secreted in the Free Form
3.3. Tau Isoforms Are Shuttled from Neurons to Astrocytes Mainly by EVs
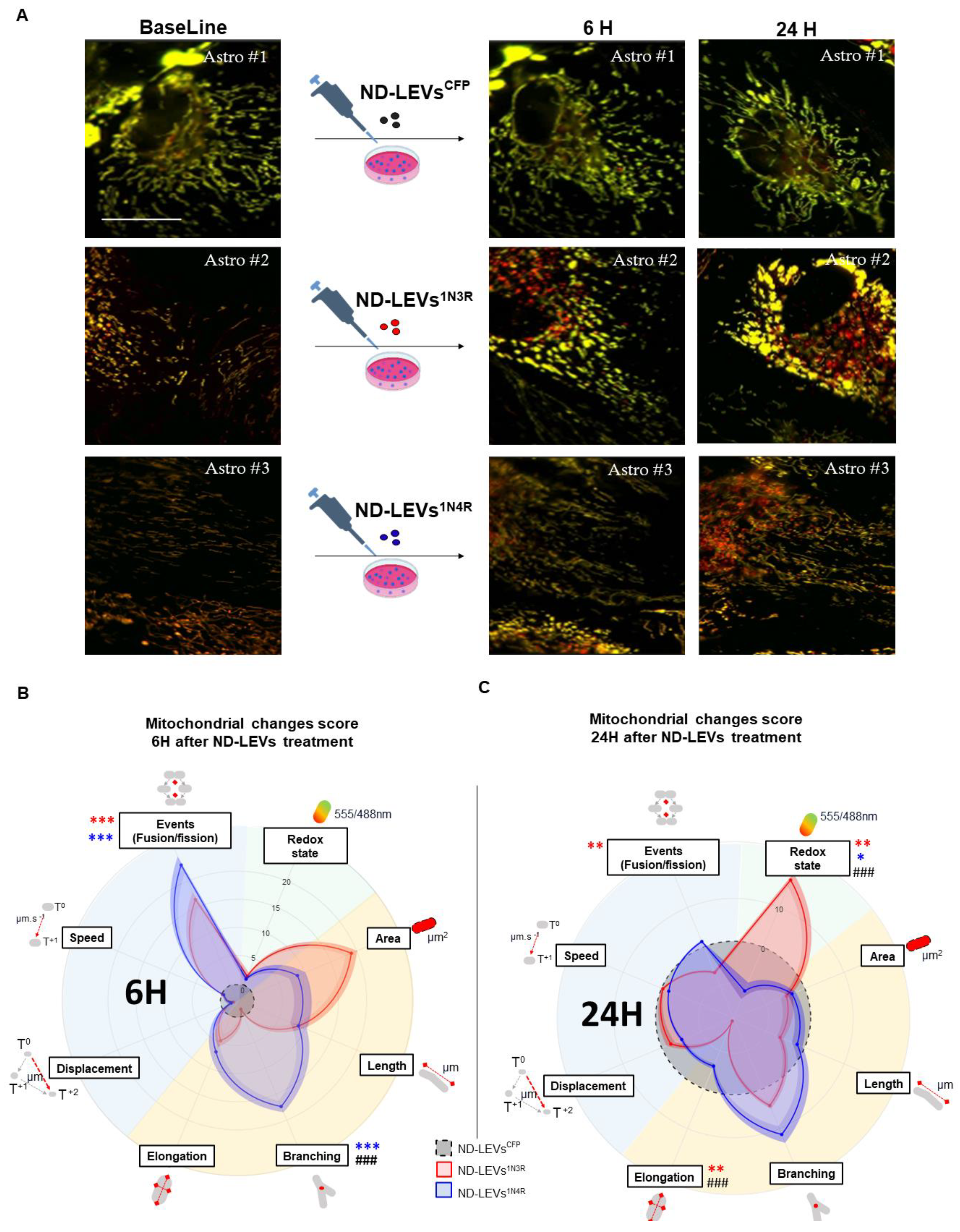
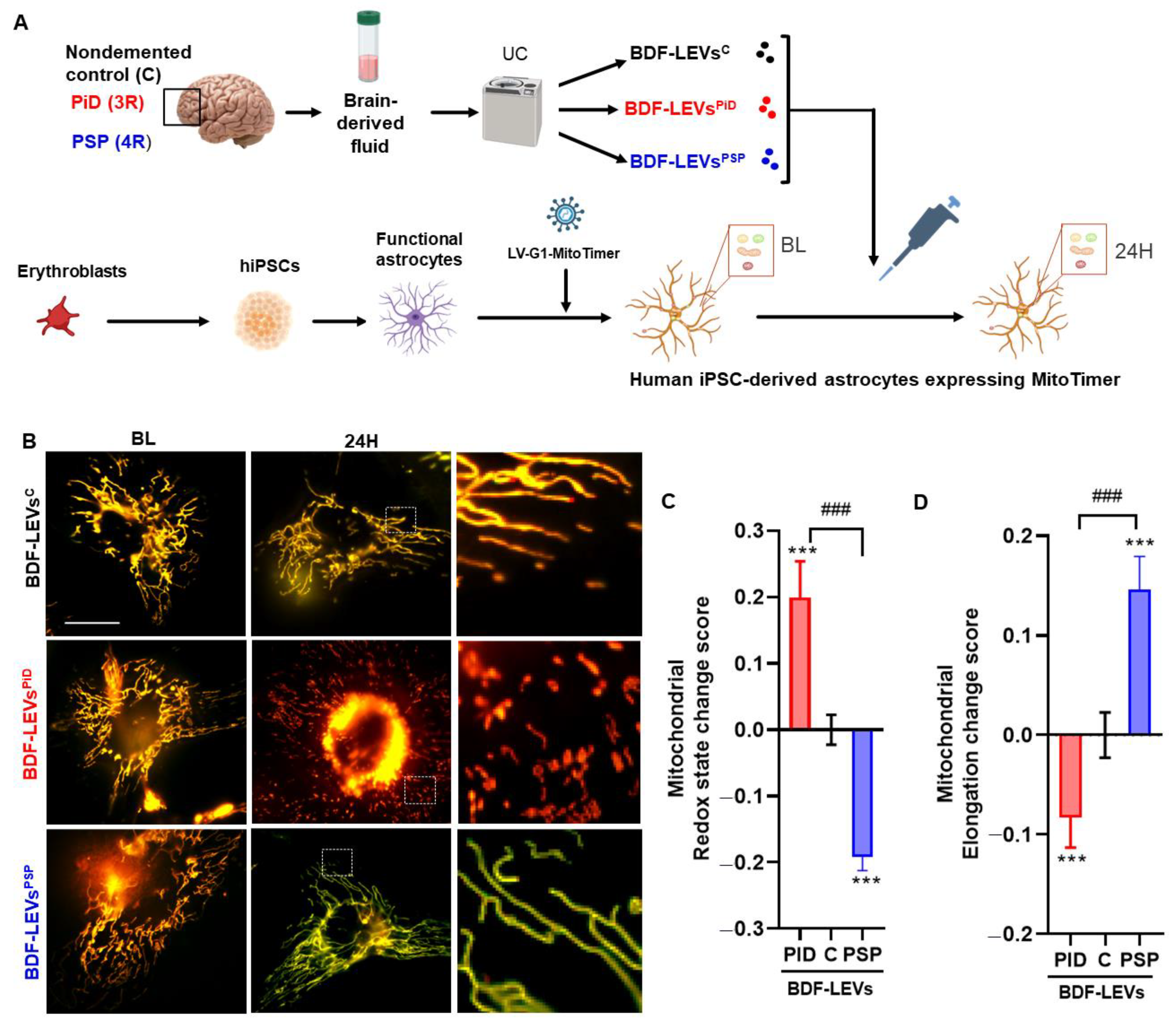
3.4. 3R and 4R Tau-Accumulating LEVs Treatments Induce Differential Mitochondrial Consequences in Astrocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W., Jr. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell Biol. 1989, 9, 1381–1388. [Google Scholar] [PubMed] [Green Version]
- Muller, R.; Heinrich, M.; Heck, S.; Blohm, D.; Richter-Landsberg, C. Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell. Tissue Res. 1997, 288, 239–249. [Google Scholar] [CrossRef]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, J.R.; Lopez, E.; Diez-Ballesteros, J.C.; Avila, J.; Hernandez, F.; Bolos, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [Green Version]
- Rosler, T.W.; Tayaranian Marvian, A.; Brendel, M.; Nykanen, N.P.; Hollerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-repeat tauopathies. Prog. Neurobiol. 2019, 180, 101644. [Google Scholar] [CrossRef]
- Ikeda, M.; Shoji, M.; Kawarai, T.; Kawarabayashi, T.; Matsubara, E.; Murakami, T.; Sasaki, A.; Tomidokoro, Y.; Ikarashi, Y.; Kuribara, H.; et al. Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am. J. Pathol. 2005, 166, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [Green Version]
- Bolos, M.; Llorens-Martin, M.; Jurado-Arjona, J.; Hernandez, F.; Rabano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers Dis. 2016, 50, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buee, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amro, Z.; Yool, A.J.; Collins-Praino, L.E. The potential role of glial cells in driving the prion-like transcellular propagation of tau in tauopathies. Brain Behav. Immun. Health 2021, 14, 100242. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ying, J.; Wang, X.; Zheng, Q.; Zhao, T.; Yoon, S.; Yu, W.; Yang, D.; Fang, Y.; Hua, F. Astrocytes in Neural Circuits: Key Factors in Synaptic Regulation and Potential Targets for Neurodevelopmental Disorders. Front. Mol. Neurosci. 2021, 14, 729273. [Google Scholar] [CrossRef]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef] [PubMed]
- Mate de Gerando, A.; D’Orange, M.; Augustin, E.; Josephine, C.; Auregan, G.; Gaudin-Guerif, M.; Guillermier, M.; Herard, A.S.; Stimmer, L.; Petit, F.; et al. Neuronal tau species transfer to astrocytes and induce their loss according to tau aggregation state. Brain 2021, 144, 1167–1182. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, S.K.; Zhang, Y.; Rostami, A.; Kenkare, A.; Casella, G.; Yuan, Z.Q.; Li, X. Role of extracellular vesicles in neurodegenerative diseases. Prog. Neurobiol. 2021, 201, 102022. [Google Scholar] [CrossRef] [PubMed]
- Leroux, E.; Perbet, R.; Buee, L.; Colin, M. Extracellular vesicles in the central nervous system. Med. Sci. 2021, 37, 1133–1138. [Google Scholar] [CrossRef]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Ruan, Z.; Pathak, D.; Venkatesan Kalavai, S.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2020, 144, 288–309. [Google Scholar] [CrossRef] [PubMed]
- Leroux, E.; Perbet, R.; Caillierez, R.; Richetin, K.; Lieger, S.; Espourteille, J.; Bouillet, T.; Begard, S.; Danis, C.; Loyens, A.; et al. Extracellular vesicles: Major actors of heterogeneity in tau spreading among human tauopathies. Mol. Ther. 2022, 30, 782–797. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Pra, I. Amyloid beta-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [Green Version]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Begard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Lecolle, K.; Caillierez, R.; Begard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Auregan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef]
- Caillierez, R.; Begard, S.; Lecolle, K.; Deramecourt, V.; Zommer, N.; Dujardin, S.; Loyens, A.; Dufour, N.; Auregan, G.; Winderickx, J.; et al. Lentiviral Delivery of the Human Wild-type Tau Protein Mediates a Slow and Progressive Neurodegenerative Tau Pathology in the Rat Brain. Mol. Ther. 2013, 21, 1358–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dujardin, S.; Begard, S.; Caillierez, R.; Lachaud, C.; Carrier, S.; Lieger, S.; Gonzalez, J.A.; Deramecourt, V.; Deglon, N.; Maurage, C.A.; et al. Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol. Commun. 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Vega, C.; Pellerin, L.; Dantzer, R.; Magistretti, P.J. Long-term modulation of glucose utilization by IL-1 alpha and TNF-alpha in astrocytes: Na+ pump activity as a potential target via distinct signaling mechanisms. Glia 2002, 39, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Perriot, S.; Canales, M.; Mathias, A.; Du Pasquier, R. Differentiation of functional astrocytes from human-induced pluripotent stem cells in chemically defined media. STAR Protoc. 2021, 2, 100902. [Google Scholar] [CrossRef]
- Perriot, S.; Mathias, A.; Perriard, G.; Canales, M.; Jonkmans, N.; Merienne, N.; Meunier, C.; El Kassar, L.; Perrier, A.L.; Laplaud, D.A.; et al. Human Induced Pluripotent Stem Cell-Derived Astrocytes Are Differentially Activated by Multiple Sclerosis-Associated Cytokines. Stem Cell Rep. 2018, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Gotz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [Green Version]
- Dujardin, S.; Begard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Sablonniere, B.; Schraen-Maschke, S.; Ghestem, A.; Maurage, C.A.; Wattez, A.; Vermersch, P.; Delacourte, A. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum. Mol. Genet. 2001, 10, 2143–2155. [Google Scholar] [CrossRef] [Green Version]
- Troquier, L.; Caillierez, R.; Burnouf, S.; Fernandez-Gomez, F.J.; Grosjean, M.E.; Zommer, N.; Sergeant, N.; Schraen-Maschke, S.; Blum, D.; Buee, L. Targeting phospho-Ser422 by active Tau Immunotherapy in the THYTau22 mouse model: A suitable therapeutic approach. Curr. Alzheimer Res. 2012, 9, 397–405. [Google Scholar]
- Espourteille, J.; Zufferey, V.; Laurent, J.H.; Richetin, K. Live-imaging of Mitochondrial System in Cultured Astrocytes. J. Vis. Exp. 2021. [Google Scholar] [CrossRef]
- Allison, P.D. Change Scores as Dependent Variables in Regression Analysis. Sociol. Methodol. 1990, 20, 93–114. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q.; et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217, e20190783. [Google Scholar] [CrossRef] [PubMed]
- Gollihue, J.L.; Norris, C.M. Astrocyte mitochondria: Central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res. Rev. 2020, 59, 101039. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Robinson, M.B. Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef] [PubMed]
- Stoothoff, W.; Jones, P.B.; Spires-Jones, T.L.; Joyner, D.; Chhabra, E.; Bercury, K.; Fan, Z.; Xie, H.; Bacskai, B.; Edd, J.; et al. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 2009, 111, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef] [PubMed]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Gotz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Gotz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localizes within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca(2+) handling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential Effects of the Six Human TAU Isoforms: Somatic Retention of 2N-TAU and Increased Microtubule Number Induced by 4R-TAU. Front. Neurosci. 2021, 15, 643115. [Google Scholar] [CrossRef] [PubMed]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Schweers, O.; Schonbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar] [CrossRef] [PubMed]
- Amari, L.; Germain, M. Mitochondrial Extracellular Vesicles—Origins and Roles. Front. Mol. Neurosci. 2021, 14, 767219. [Google Scholar] [CrossRef]
- Davis, R.L.; Robertson, D.M. (Eds.) Textbook of Neuropathology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 918–921, 934–936. [Google Scholar]
- You, Y.; Muraoka, S.; Jedrychowski, M.P.; Hu, J.; McQuade, A.K.; Young-Pearse, T.; Aslebagh, R.; Shaffer, S.A.; Gygi, S.P.; Blurton-Jones, M.; et al. Human neural cell type-specific extracellular vesicle proteome defines disease-related molecules associated with activated astrocytes in Alzheimer’s disease brain. J. Extracell. Vesicles 2022, 11, e12183. [Google Scholar] [CrossRef]
- Muraoka, S.; DeLeo, A.M.; Sethi, M.K.; Yukawa-Takamatsu, K.; Yang, Z.; Ko, J.; Hogan, J.D.; Ruan, Z.; You, Y.; Wang, Y.; et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimers Dement. 2020, 16, 896–907. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Gender | Death (y) | PMD (h) | Tau Lesions | Braak | Thal | Cause of Death |
|---|---|---|---|---|---|---|---|
| Control | M | 78 | 19 | None | 0 | 0 | Invasive aspergillosis |
| F | 82 | NA | None | I | 1 | Pericarditis | |
| M | 23 | 24 | None | 0 | 0 | Myocarditis | |
| M | 59 | 13 | None | 0 | 0 | Septic shock | |
| M | 41 | 11 | None | 0 | 0 | Suffocation | |
| PSP | M | 74 | 9 | NFT and GFT | 0 | 1 | |
| M | 90 | 36 | NFT and GFT | 0 | 2 | ||
| M | 88 | 3 | NFT and GFT | 0 | 4 | ||
| M | 69 | 17 | NFT and GFT | 0 | 0 | ||
| F | 79 | 4 | NFT and GFT | 0 | 0 | ||
| PiD | M | 57 | 22 | Pick bodies | NA | 0 | |
| M | 71 | 21 | Pick bodies | NA | 3 | ||
| F | 78 | 11 | Pick bodies and NFT | NA | 0 | ||
| M | 68 | 15 | Pick bodies | NA | 0 | ||
| M | 68 | 8 | Pick bodies | NA | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perbet, R.; Zufferey, V.; Leroux, E.; Parietti, E.; Espourteille, J.; Culebras, L.; Perriot, S.; Du Pasquier, R.; Bégard, S.; Deramecourt, V.; et al. Tau Transfer via Extracellular Vesicles Disturbs the Astrocytic Mitochondrial System. Cells 2023, 12, 985. https://doi.org/10.3390/cells12070985
Perbet R, Zufferey V, Leroux E, Parietti E, Espourteille J, Culebras L, Perriot S, Du Pasquier R, Bégard S, Deramecourt V, et al. Tau Transfer via Extracellular Vesicles Disturbs the Astrocytic Mitochondrial System. Cells. 2023; 12(7):985. https://doi.org/10.3390/cells12070985
Chicago/Turabian StylePerbet, Romain, Valentin Zufferey, Elodie Leroux, Enea Parietti, Jeanne Espourteille, Lucas Culebras, Sylvain Perriot, Renaud Du Pasquier, Séverine Bégard, Vincent Deramecourt, and et al. 2023. "Tau Transfer via Extracellular Vesicles Disturbs the Astrocytic Mitochondrial System" Cells 12, no. 7: 985. https://doi.org/10.3390/cells12070985