
The Beneficial Effect of Mitochondrial Transfer Therapy in 5XFAD Mice via Liver–Serum–Brain Response
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mitochondria Isolation
2.2. Mice
2.3. Mitochondrial Transfer Treatment
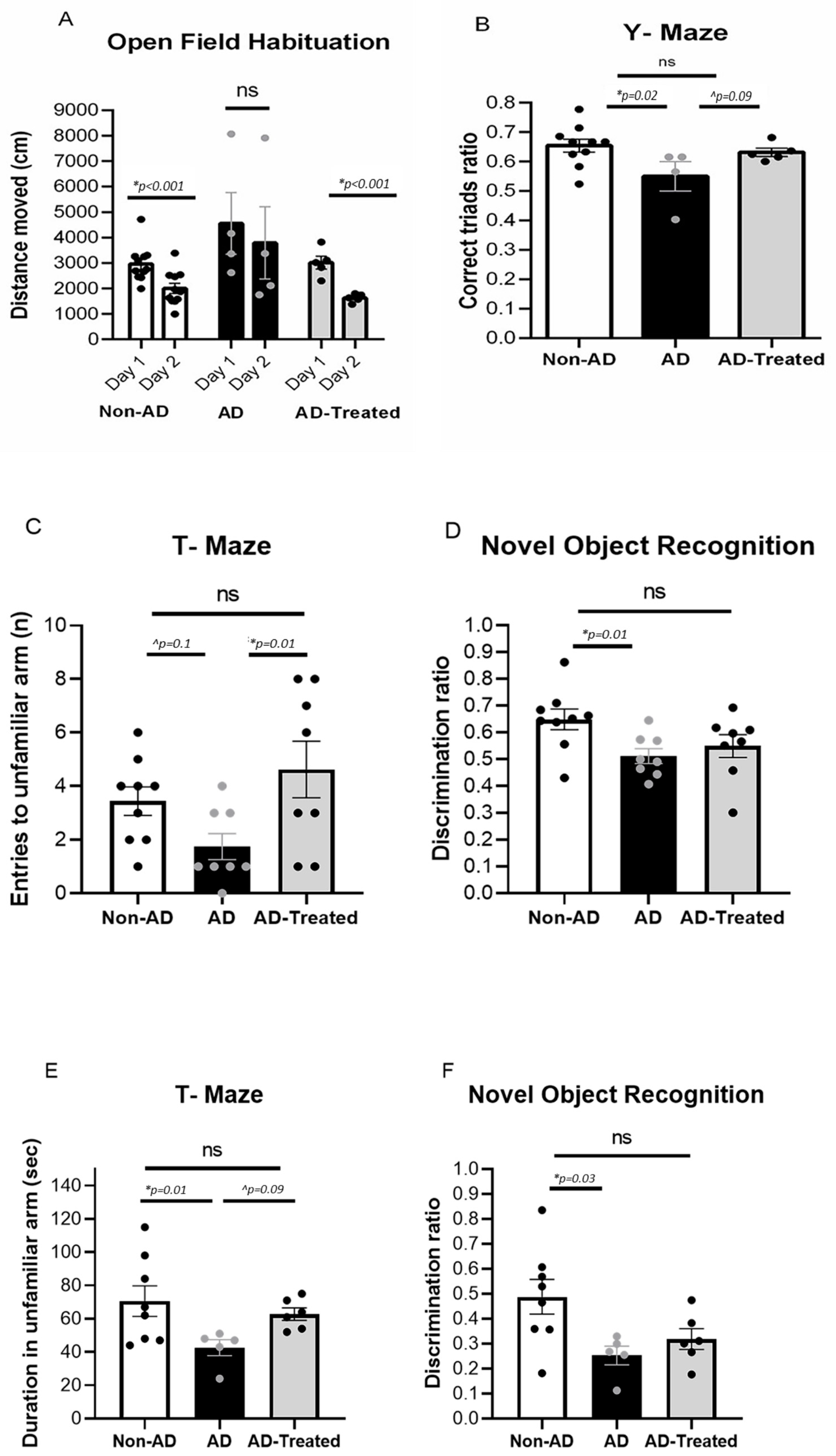
2.4. Behavioral Studies
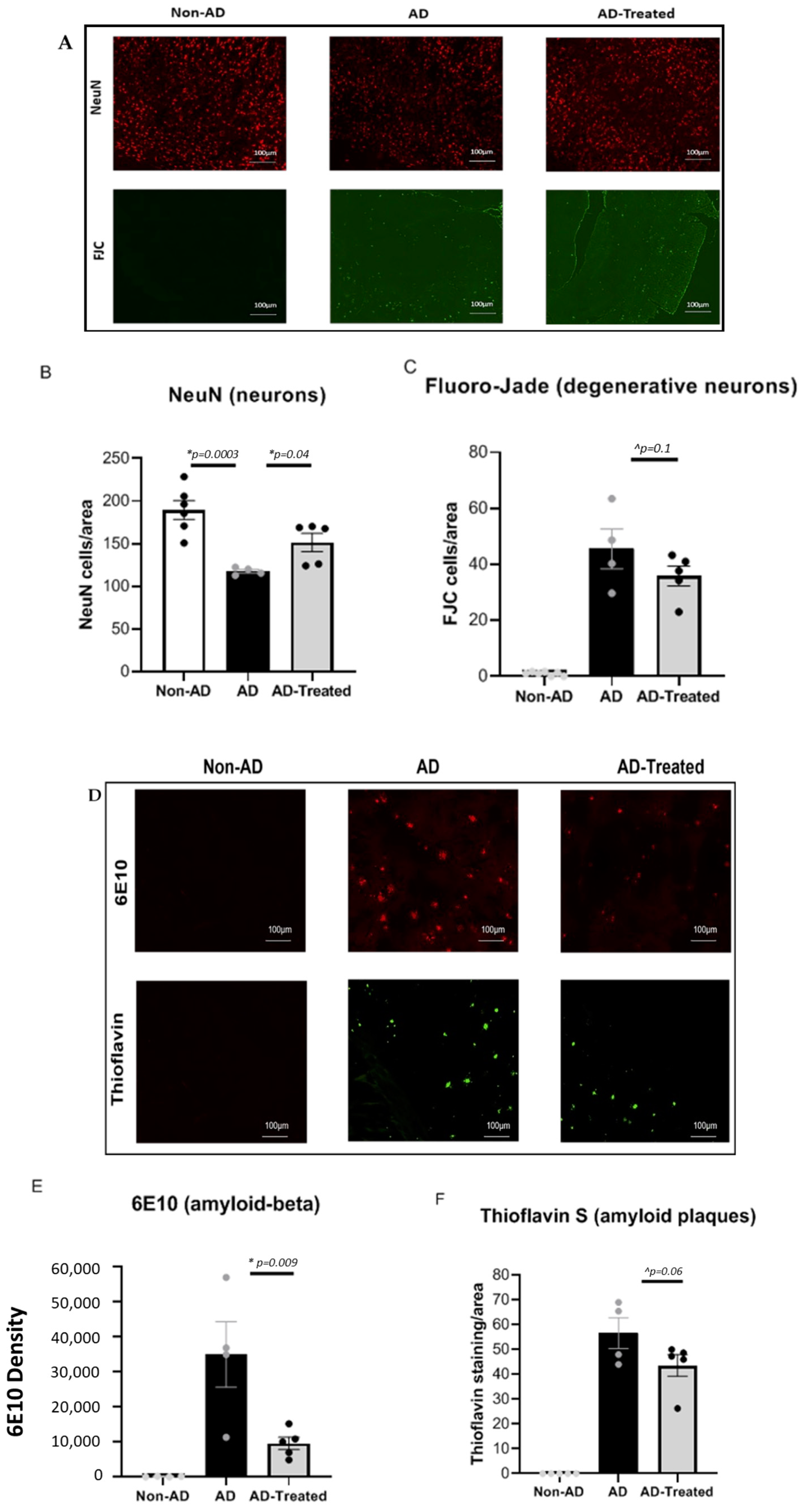
2.5. Histopathological Studies
2.6. Imaging Analysis
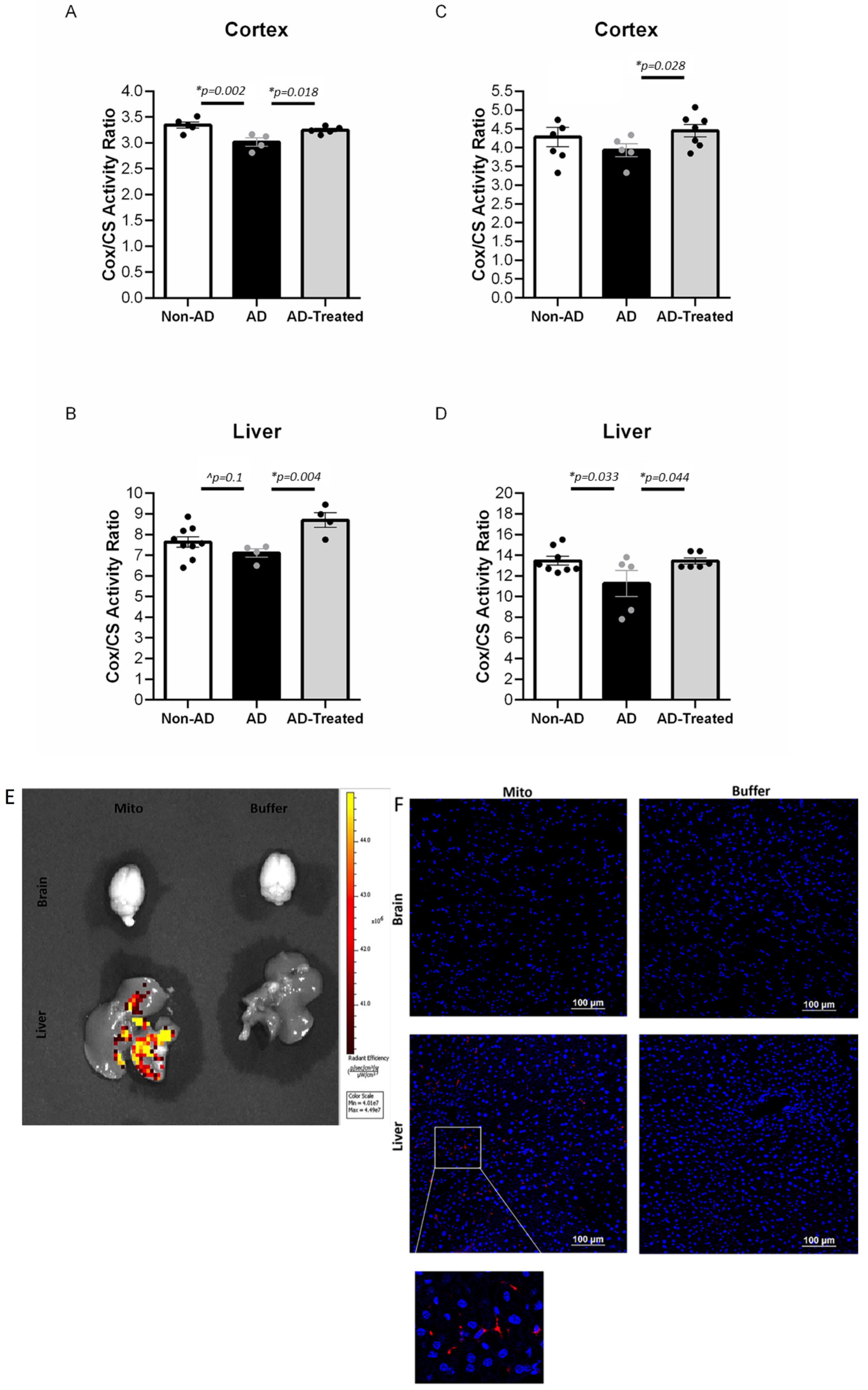
2.7. Mitochondrial Enzymatic Assays
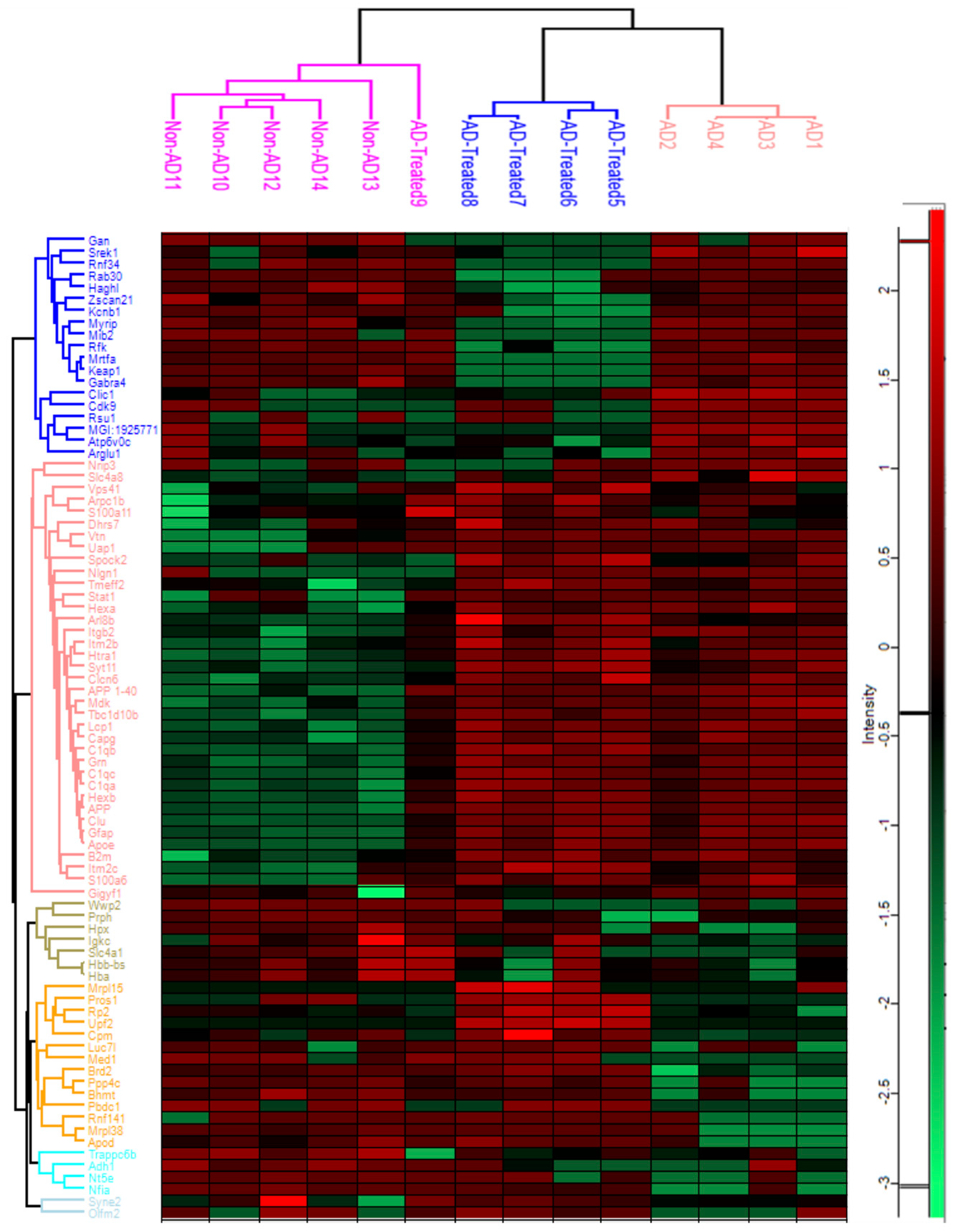
2.8. Proteomic Studies
2.9. Metabolomic Studies
2.10. Statistics
3. Results
3.1. Amelioration of Cognitive Deficits in AD Mice Treated with Mitochondrial Transfer
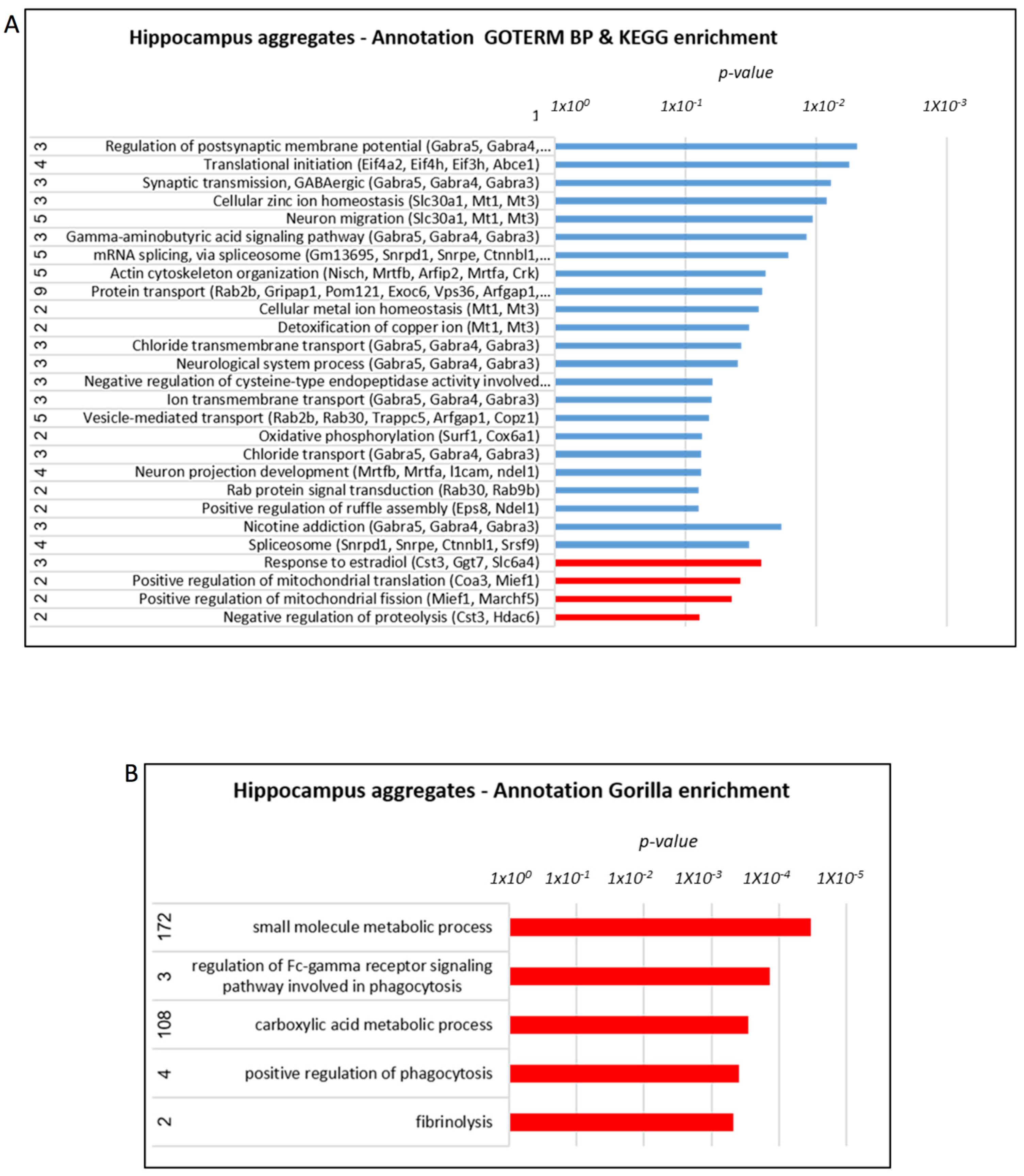
3.2. Reduced Neuronal Damage and Amyloid Burden in the Brain of AD Mice Treated with Mitochondrial Transfer
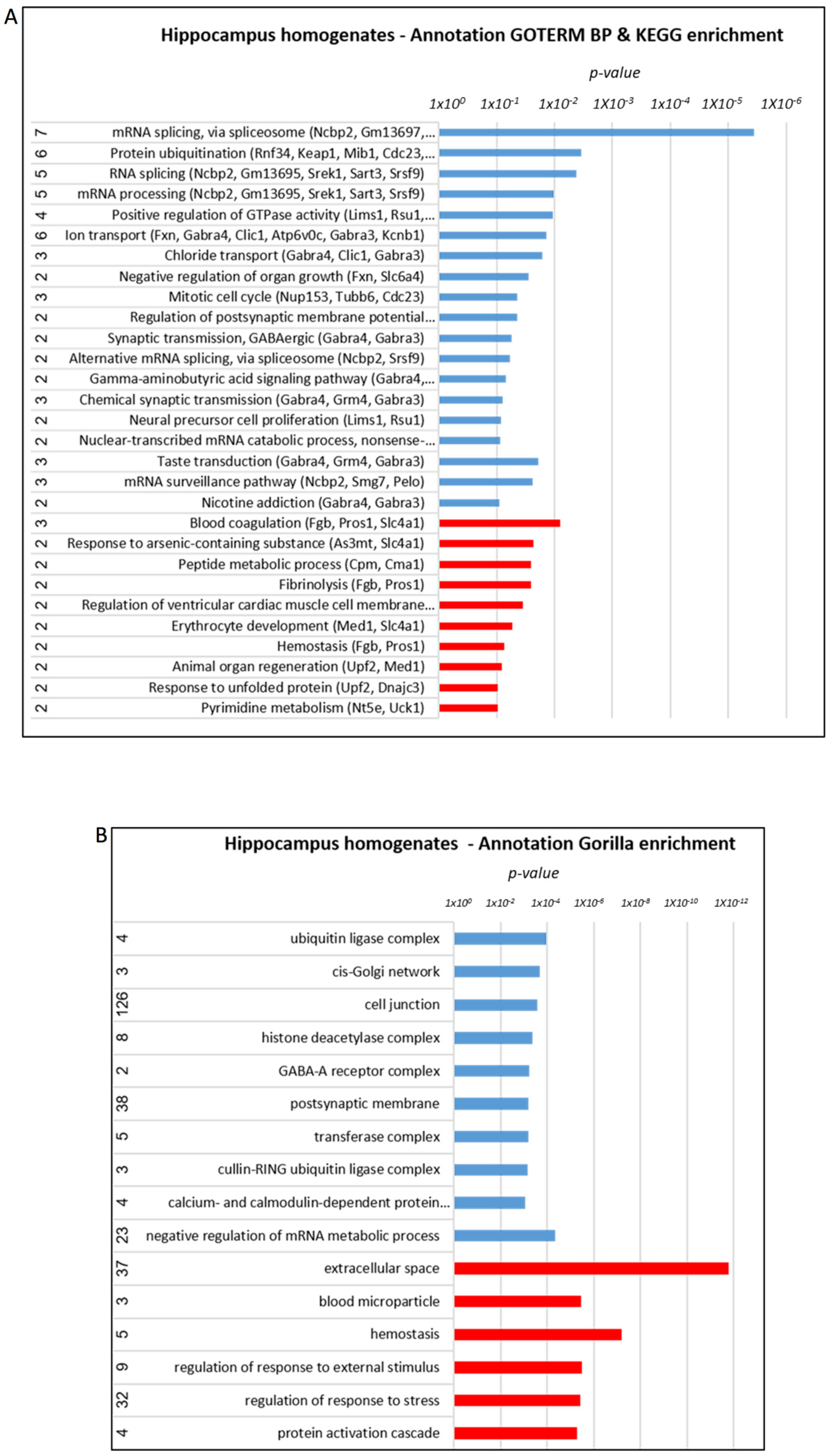
3.3. Increased Mitochondrial Enzymatic Activity in the Brain and Liver of AD Mice Treated with Mitochondrial Transfer
3.4. IV-Injected Mitochondria Are Detected in the Liver but Not in Brain
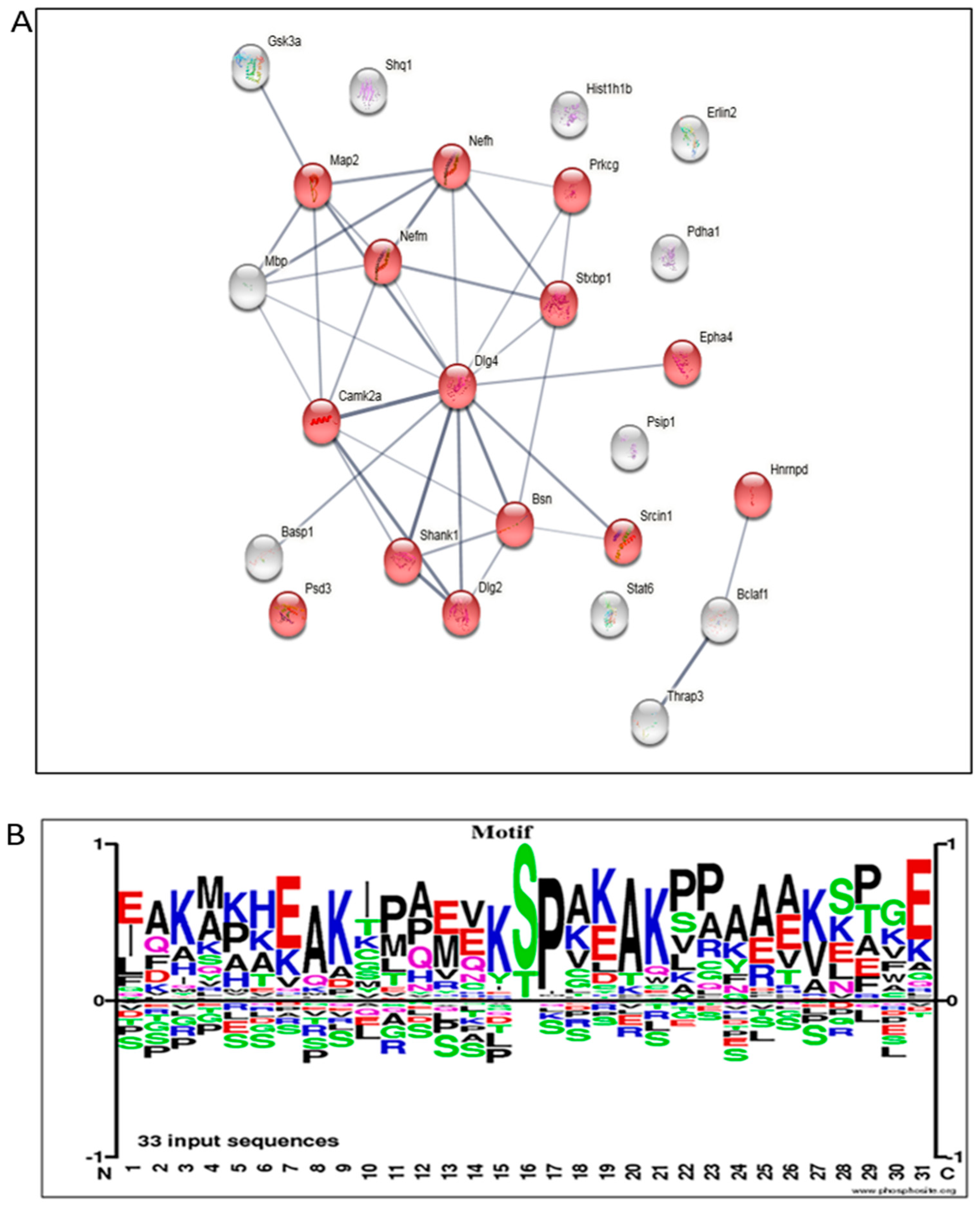
3.5. Proteome Alterations in the Brain of AD Mice Treated with Mitochondrial Transfer
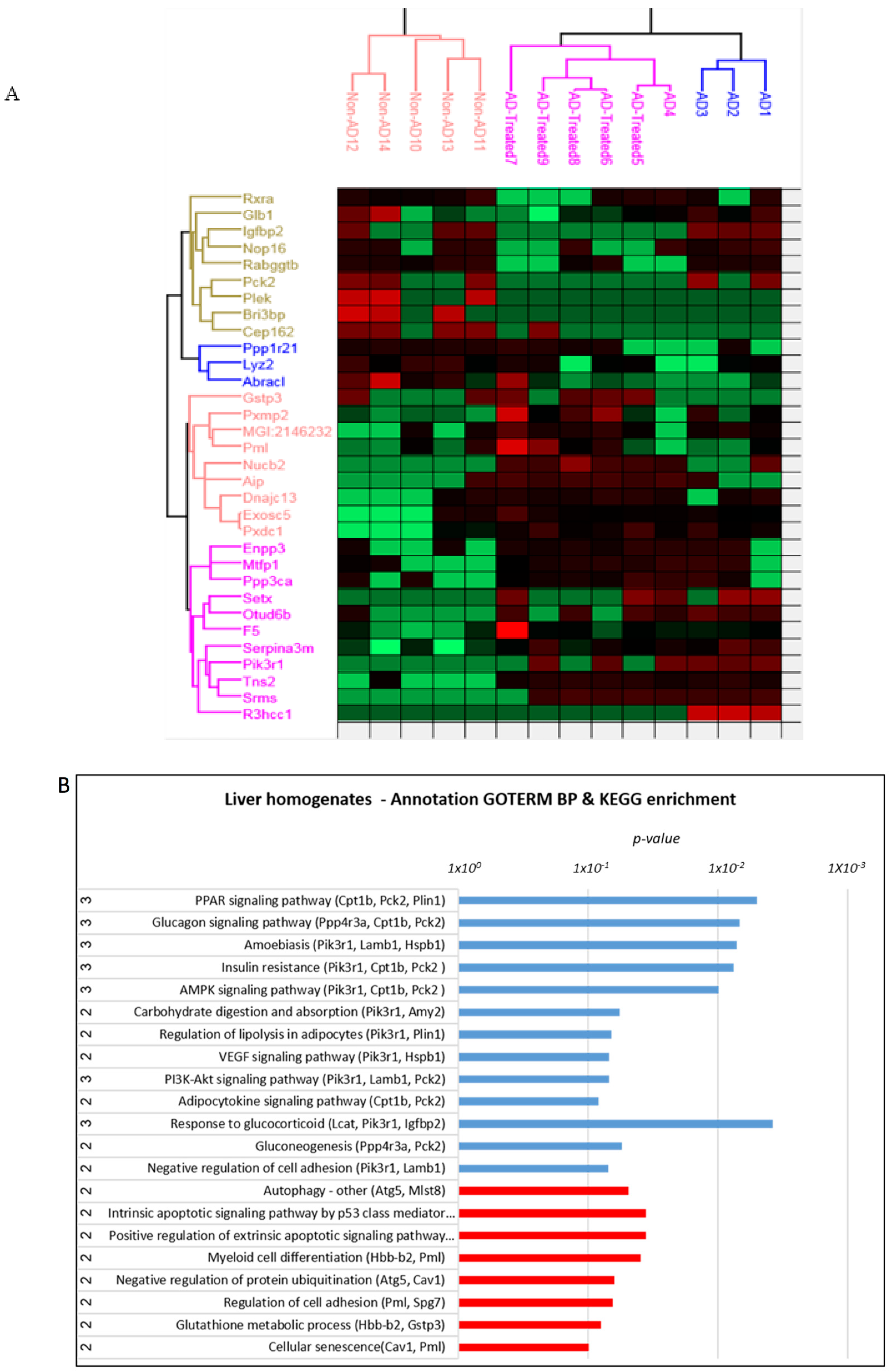
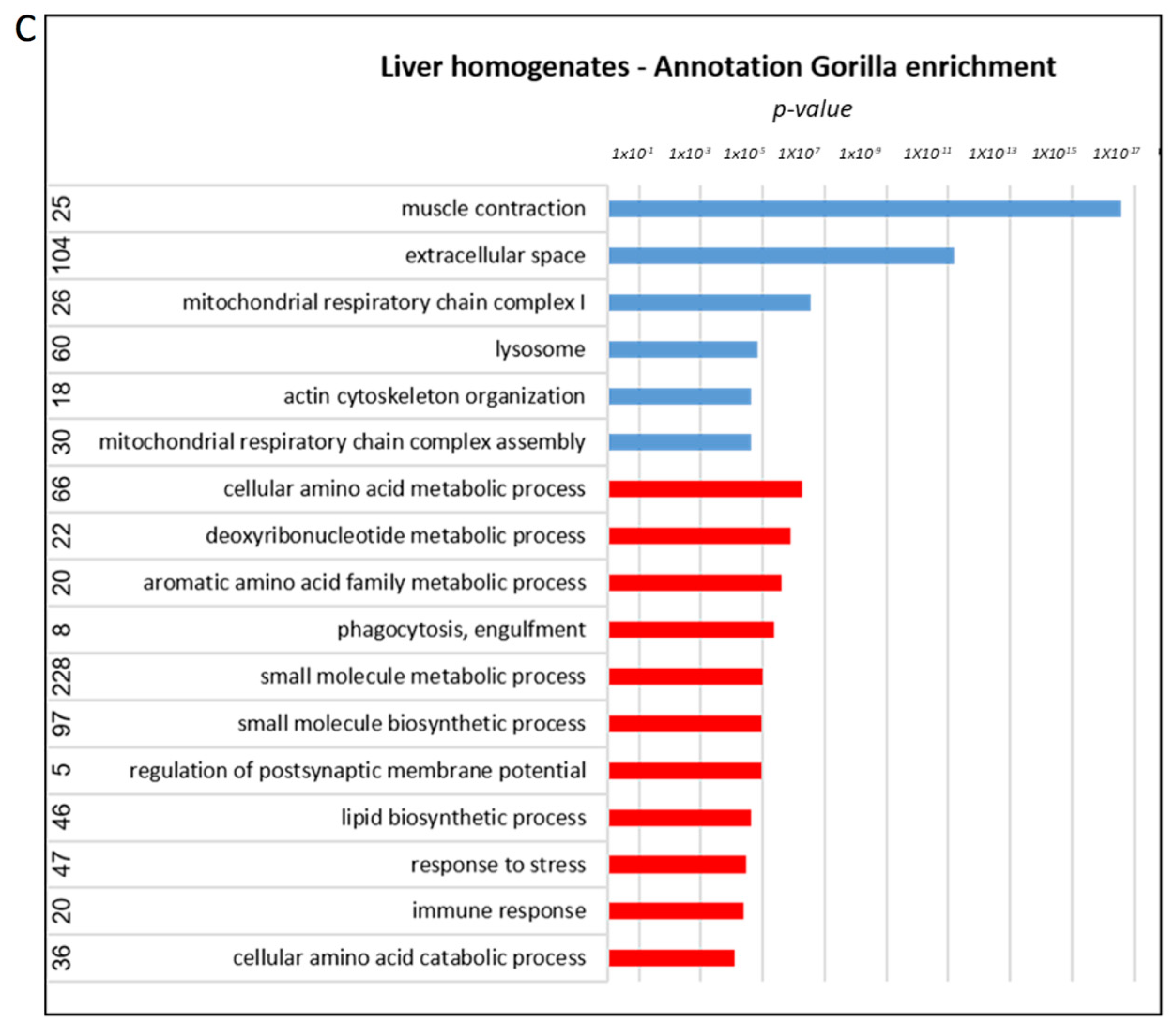
3.6. Proteome Alterations in the Liver of AD Mice Treated with Mitochondrial Transfer
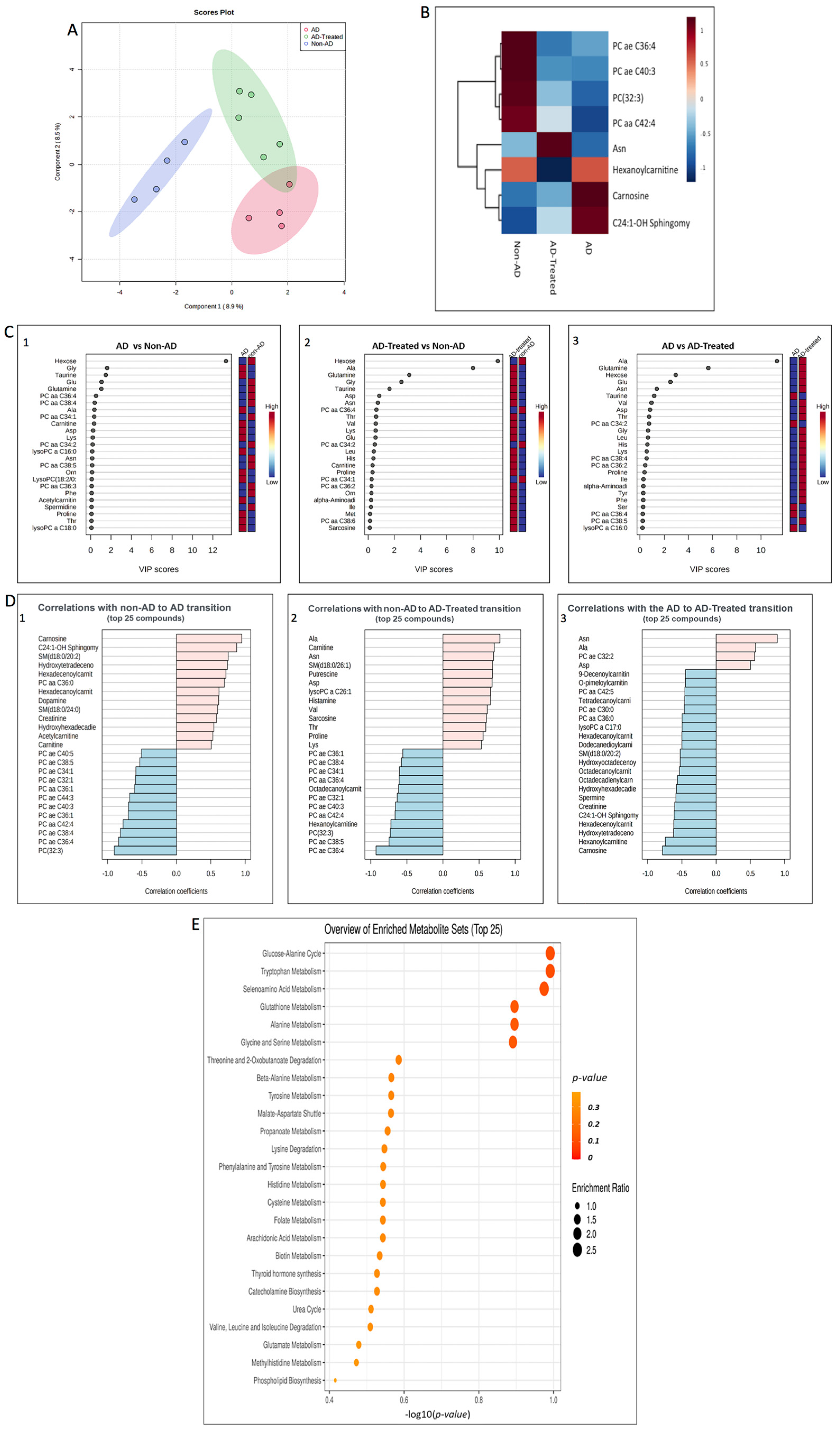
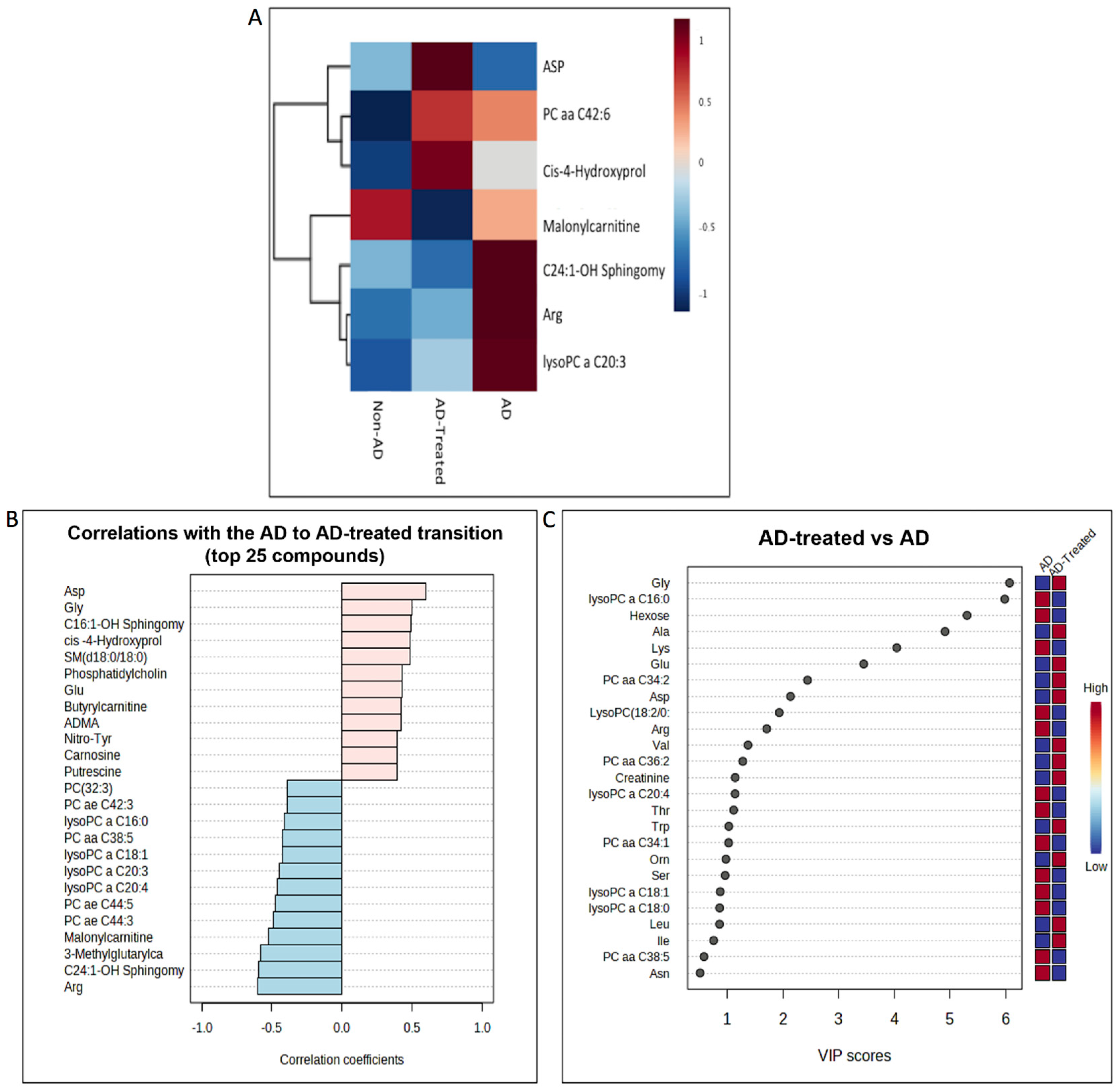
3.7. Metabolome Alterations in the Liver of AD Mice Treated with Mitochondrial Transfer
3.8. Integrative Analysis of Proteome and Metabolome Alterations in the Liver
3.9. Metabolome Alterations in the Serum of AD Mice Treated with Mitochondrial Transfer
4. Discussion
4.1. Beneficial Effects of the Mitochondrial Transfer Therapy in the 5XFAD Mice
4.2. A Possible Liver–Serum–Brain Route in the Mitochondrial Transfer Mechanism
4.3. Mitochondrial Transfer—Clinical Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Alzheimer’s disease (AD) |
| Intravenous (IV) |
| AMP-activated protein kinase (AMPK) |
| Amyloid precursor protein (APP) |
| Amyloid-beta (Aβ) |
| Analysis of Variance (ANOVA) |
| Citrate synthase (CS) |
| Cytochrome c oxidase (COX) |
| Discosoma sp. red fluorescent protein (DsRed) |
| Familial Alzheimer’s disease (FAD) |
| Five FAD mutations (5XFAD mice) |
| Fluoro-Jade C (FJC) |
| Gamma-Aminobutyric Acid (GABA) |
| Gene Ontology enRIchment anaLysis analysis and visuaLizAtion tool (GOrilla) |
| Gene Ontology Term Enrichment (GOTERM) |
| Histone deacetylase (HDAC) |
| In vivo imaging system (IVIS) |
| Insulin-like growth factor-binding protein (Igfbp) |
| Intracerebroventricular (ICV) |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) |
| Label-free quantitation (LFQ) |
| Least significant difference (LSD) |
| Liquid chromatography-tandem mass spectrometry (LC-MSMS) |
| Neuronal nuclei (NeuN) |
| Partial least square discriminant analysis (PLS-DA) |
| Peroxisome proliferator-activated receptor (PPAR) |
| Phosphoinositide 3-kinase (PI3K) |
| Post-synaptic density (PSD) |
| Post-translational modification (PTM) |
| Presenilin1 (PS1) |
| Red fluorescent protein (RFP) |
| The Small Molecule Pathway Database (SMPDB) |
| Transgenic (Tg) |
| Vascular endothelial growth factor (VEGF) |
| VIP (measure of a variable’s importance) |
| Full names of proteins are presented in the Supplemental-proteomics-excels |
References
- Ramachandran, A.K.; Das, S.; Joseph, A.; Shenoy, G.G.; Alex, A.T.; Mudgal, J. Neurodegenerative Pathways in Alzheimer’s Disease: A Review. Curr. Neuropharmacol. 2021, 19, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Anil, D.A.; Aydin, B.O.; Yeliz, D.; Burcin, T. Design, synthesis, biological evaluation and molecular docking studies of novel 1 H-1,2,3-Triazole derivatives as potent inhibitors of carbonic anhydrase, acetylcholinesteraseand aldose reductase. J. Mol. Struct. 2022, 1257, 132613. [Google Scholar] [CrossRef]
- Scheffler, I.E. A century of mitochondrial research: Achievements and perspectives. Mitochondrion 2001, 1, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial dysfunction in aging and Alzheimer’s disease: Strategies to protect neurons. Antioxid Redox Signal 2007, 9, 1647–1658. [Google Scholar] [CrossRef]
- Desler, C.; Lillenes, M.S.; Tønjum, T.; Rasmussen, L.J. The role of mitochondrial dysfunction in the progression of Alzheimer’s disease. Curr. Med. Chem. 2017. [CrossRef] [Green Version]
- Demir, Y. Naphthoquinones, benzoquinones, and anthraquinones: Molecular docking, ADME and inhibition studies on human serum paraoxonase-1 associated with cardiovascular diseases. Drug Dev. Res. 2020, 81, 628–636. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef] [Green Version]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Núñez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; von Bernhardi, R.; Godoy, J.A.; Inestrosa, N.C.; Johnson, G.V. Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol. Dis. 2014, 71, 260–269. [Google Scholar] [CrossRef]
- Lahiani-Cohen, I.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Rosenmann, H. Exposure to 3-Nitropropionic Acid Mitochondrial Toxin Induces Tau Pathology in Tangle-Mouse Model and in Wild Type-Mice. Front. Cell Dev. Biol. 2019, 7, 321. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.A.; Shay, J.W. Mitochondrial transformation of mammalian cells. Nature 1982, 295, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katrangi, E.; D’Souza, G.; Boddapati, S.V.; Kulawiec, M.; Singh, K.K.; Bigger, B.; Weissig, V. Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 2007, 10, 561–570. [Google Scholar] [CrossRef]
- Kitani, T.; Kami, D.; Matoba, S.; Gojo, S. Internalization of isolated functional mitochondria: Involvement of macropinocytosis. J. Cell Mol. Med. 2014, 18, 1694–1703. [Google Scholar] [CrossRef]
- Pacak, C.A.; Preble, J.M.; Kondo, H.; Seibel, P.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B.; McCully, J.D. Actin-dependent mitochondrial internalization in cardiomyocytes: Evidence for rescue of mitochondrial function. Biol. Open 2015, 4, 622–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesner, E.E.; Saada-Reich, A.; Lorberboum-Galski, H. Characteristics of Mitochondrial Transformation into Human Cells. Sci. Rep. 2016, 6, 26057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.C.; Lai, I.R. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats: Reply. Shock 2013, 39, 543. [Google Scholar] [CrossRef] [PubMed]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.J.; Kuo, C.C.; Lee, H.C.; Shen, C.I.; Cheng, F.C.; Wu, S.F.; Chang, J.C.; Pan, H.C.; Lin, S.Z.; Liu, C.S.; et al. Transferring Xenogenic Mitochondria Provides Neural Protection Against Ischemic Stress in Ischemic Rat Brains. Cell Transpl. 2016, 25, 913–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollihue, J.L.; Patel, S.P.; Eldahan, K.C.; Cox, D.H.; Donahue, R.R.; Taylor, B.K.; Sullivan, P.G.; Rabchevsky, A.G. Effects of Mitochondrial Transplantation on Bioenergetics, Cellular Incorporation, and Functional Recovery after Spinal Cord Injury. J. Neurotrauma 2018, 35, 1800–1818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, Z.; Yan, C.; Pu, K.; Wu, M.; Bai, J.; Li, Y.; Wang, Q. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav. Brain Res. 2019, 356, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Orfany, A.; Arriola, C.G.; Doulamis, I.P.; Guariento, A.; Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Rogers, C.; Del Nido, P.J.; et al. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 2020, 71, 1014–1026. [Google Scholar] [CrossRef]
- Blitzer, D.; Guariento, A.; Doulamis, I.P.; Shin, B.; Moskowitzova, K.; Barbieri, G.R.; Orfany, A.; Del Nido, P.J.; McCully, J.D. Delayed Transplantation of Autologous Mitochondria for Cardioprotection in a Porcine Model. Ann. Thorac. Surg. 2020, 109, 711–719. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Lu, Y.; Huang, S.; Xiao, R.; Zeng, X.; Zhang, X.; Li, J.; Wang, T.; Li, T.; et al. Mitochondrial transplantation attenuates hypoxic pulmonary vasoconstriction. Oncotarget 2016, 7, 31284–31298. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Wu, S.L.; Liu, K.H.; Chen, Y.H.; Chuang, C.S.; Cheng, F.C.; Su, H.L.; Wei, Y.H.; Kuo, S.J.; Liu, C.S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res. 2016, 170, 40–56. [Google Scholar] [CrossRef]
- Robicsek, O.; Ene, H.M.; Karry, R.; Ytzhaki, O.; Asor, E.; McPhie, D.; Cohen, B.M.; Ben-Yehuda, R.; Weiner, I.; Ben-Shachar, D. Isolated Mitochondria Transfer Improves Neuronal Differentiation of Schizophrenia-Derived Induced Pluripotent Stem Cells and Rescues Deficits in a Rat Model of the Disorder. Schizophr. Bull. 2017. [CrossRef] [Green Version]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Nascimento-Dos-Santos, G.; de-Souza-Ferreira, E.; Lani, R.; Faria, C.C.; Araújo, V.G.; Teixeira-Pinheiro, L.C.; Vasconcelos, T.; Gonçalo, T.; Santiago, M.F.; Linden, R.; et al. Neuroprotection from optic nerve injury and modulation of oxidative metabolism by transplantation of active mitochondria to the retina. Biochim. Biophys Acta Mol. Basis. Dis 2020, 1866, 165686. [Google Scholar] [CrossRef]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhamron, S.; Rozenstein-Tsalkovich, L.; Nitzan, K.; Abramsky, O.; Rosenmann, H. Phos-tau peptide immunization of amyloid-tg-mice reduced non-mutant phos-tau pathology, improved cognition and reduced amyloid plaques. Exp. Neurol. 2018, 303, 48–58. [Google Scholar] [CrossRef]
- Benhamron, S.; Nitzan, K.; Valitsky, M.; Lax, N.; Karussis, D.; Kassis, I.; Rosenmann, H. Cerebrospinal Fluid (CSF) Exchange Therapy with Artificial CSF Enriched with Mesenchymal Stem Cell Secretions Ameliorates Cognitive Deficits and Brain Pathology in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2020, 76, 369–385. [Google Scholar] [CrossRef]
- Koren, S.A.; Hamm, M.J.; Cloyd, R.; Fontaine, S.N.; Chishti, E.; Lanzillotta, C.; Rodriguez-Rivera, J.; Ingram, A.; Bell, M.; Galvis-Escobar, S.M.; et al. Broad Kinase Inhibition Mitigates Early Neuronal Dysfunction in Tauopathy. Int. J. Mol. Sci. 2021, 22, 1186. [Google Scholar] [CrossRef]
- Granit, A.M.; Barasch, D.; Peretz-Yablonski, T.E.; Kakhlon, O. Metabolomic profiling of triple negative breast cancer cells suggests that valproic acid can enhance the anticancer effect of cisplatin. Front. Cell Dev. Biol. 2022, 10, 1014798. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0, narrowing the gap between raw spectra and functional insights. Nucleic. Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPARgamma and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Recarte, D.; Palomer, X.; Wahli, W.; Vázquez-Carrera, M. The PPARβ/δ-AMPK Connection in the Treatment of Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 8555. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Guebre-Egziabher, F.; Zhao, X.; Feriod, C.; Dong, J.; Alves, T.C.; Ioja, S.; Pongratz, R.L.; Bhanot, S.; Roden, M.; et al. A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J. Biol. Chem. 2014, 289, 7257–7263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, A.; Baes, M. Role of PPARα in Hepatic Carbohydrate Metabolism. PPAR Res. 2010, 2010. [Google Scholar]
- Palabıyık, E.; Sulumer, A.N.; Uguz, H.; Avcı, B.; Askın, S.; Askın, H.; Demir, Y. Assessment of hypolipidemic and anti-inflammatory properties of walnut (Juglans regia) seed coat extract and modulates some metabolic enzymes activity in triton WR-1339-induced hyperlipidemia in rat kidney, liver, and heart. J. Mol. Recognit 2023, 36, e3004. [Google Scholar] [CrossRef]
- McCann, M.R.; George De la Rosa, M.V.; Rosania, G.R.; Stringer, K.A. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef]
- McGill, M.R.; Li, F.; Sharpe, M.R.; Williams, C.D.; Curry, S.C.; Ma, X.; Jaeschke, H. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch. Toxicol. 2014, 88, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Oskouian, B.; Saba, J.D. Cancer treatment strategies targeting sphingolipid metabolism. Adv. Exp. Med. Biol. 2010, 688, 185–205. [Google Scholar]
- Eikelberg, D.; Lehmbecker, A.; Brogden, G.; Tongtako, W.; Hahn, K.; Habierski, A.; Hennermann, J.B.; Naim, H.Y.; Felmy, F.; Baumgärtner, W.; et al. Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human G. J. Clin. Med. 2020, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [Green Version]
- Donegan, J.J.; Boley, A.M.; Yamaguchi, J.; Toney, G.M.; Lodge, D.J. Modulation of extrasynaptic GABA. Nat. Commun. 2019, 10, 2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annamneedi, A.; Del Angel, M.; Gundelfinger, E.D.; Stork, O.; Çalışkan, G. The Presynaptic Scaffold Protein Bassoon in Forebrain Excitatory Neurons Mediates Hippocampal Circuit Maturation: Potential Involvement of TrkB Signalling. Int. J. Mol. Sci. 2021, 22, 7944. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.H.; Rasmussen, H.B.; Silahtaroglu, A. The DLGAP family: Neuronal expression, function and role in brain disorders. Mol. Brain 2017, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Penney, J.; Tsai, L.H. Histone deacetylases in memory and cognition. Sci. Signal 2014, 7, re12. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Nalivaeva, N.N.; Makova, N.Z.; Turner, A.J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: Implications for Alzheimer disease. EMBO Rep. 2009, 10, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Das, D.; Patra, S.; Bridwell-Rabb, J.; Barondeau, D.P. Mechanism of frataxin “bypass” in human iron-sulfur cluster biosynthesis with implications for Friedreich’s ataxia. J. Biol. Chem. 2019, 294, 9276–9284. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Zamolodchikov, D.; Ahn, H.J.; Strickland, S.; Norris, E.H. Fibrinogen and altered hemostasis in Alzheimer’s disease. J. Alzheimers. Dis. 2012, 32, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S. IGFBP-2 Signaling in the Brain: From Brain Development to Higher Order Brain Functions. Front. Endocrinol. 2019, 10, 822. [Google Scholar] [CrossRef] [Green Version]
- Torres-Aleman, I. Serum growth factors and neuroprotective surveillance: Focus on IGF-1. Mol. Neurobiol. 2000, 21, 153–160. [Google Scholar] [CrossRef]
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 437. [Google Scholar] [CrossRef]
- van Kruining, D.; Luo, Q.; van Echten-Deckert, G.; Mielke, M.M.; Bowman, A.; Ellis, S.; Oliveira, T.G.; Martinez-Martinez, P. Sphingolipids as prognostic biomarkers of neurodegeneration, neuroinflammation, and psychiatric diseases and their emerging role in lipidomic investigation methods. Adv. Drug Deliv. Rev. 2020, 159, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Caraci, F.; Jolivet, R.B. Pivotal role of carnosine in the modulation of brain cells activity: Multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog. Neurobiol. 2019, 175, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS ONE 2011, 6, e17971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Lu, X.Y.; Zhu, W.L.; Liu, X.Q.; Li, B.Y.; Song, L.; Liu, H.F.; Cai, W.W.; Deng, Y.X.; Xu, T.T.; et al. Carnosine ameliorates age-related dementia via improving mitochondrial dysfunction in SAMP8 mice. Food Funct. 2020, 11, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Zhang, H.; Liu, P. Behavioral and neurochemical effects of acute putrescine depletion by difluoromethylornithine in rats. Neuroscience 2009, 161, 691–706. [Google Scholar] [CrossRef]
- Morrison, L.D.; Kish, S.J. Brain polyamine levels are altered in Alzheimer’s disease. Neurosci. Lett. 1995, 197, 5–8. [Google Scholar] [CrossRef]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Hayakawa, K. Extracellular Mitochondria Signals in CNS Disorders. Front. Cell Dev. Biol. 2021, 9, 642853. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C.; O’Rourke, B. Mitochondrial transplantation in humans: “magical” cure or cause for concern? J. Clin. Investig. 2018, 128, 5191–5194. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V. Mitochondrial Transplantation: A Critical Analysis. Biochemistry 2020, 85, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Filipović, B.; Marković, O.; Đurić, V. Cognitive Changes and Brain Volume Reduction in Patients with Nonalcoholic Fatty Liver Disease. Can. J. Gastroenterol. Hepatol. 2018, 2018, 9638797. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.; Xu, A. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolomics | Proteomics | |
|---|---|---|
| Amino acid metabolism | Altered metabolites: | Enriched terms: Cellular amino acids metabolic process, small molecule metabolic/biosynthetic process |
| Asn, Asp, Ala, Gly, Glu, glutamine, carnosine, putrascine. | ||
| Enriched pathways: | ||
| Tryptophan metabolism, alanine metabolism, glutathione metabolism, glycine and serine metabolism | ||
| Glutathione metabolism | Enriched pathways: Glutathione metabolism | Altered proteins: |
| Gstp3 | ||
| Enriched pathways/terms: | ||
| Glutathione metabolic process | ||
| Glucose metabolism | Altered metabolites: Hexose Enriched pathways: Glucose alanine cycle | Altered proteins: |
| Pck2, Glb1, Igfbp2. | ||
| Enriched pathways: | ||
| PPAR signaling, glucagon signaling, insulin resistance, AMPK signaling, PI3K/AKT signaling, gluconeogenesis | ||
| Sphingolipid metabolism | Altered metabolites: C24:1-OH sphingomyelin SM(d18:0/20:2) | Altered proteins: |
| Pik3r1, Glb1 | ||
| Enriched terms: | ||
| Lipid biosynthetic pathway | ||
| Acylcarnitines (and mitochondria) | Altered metabolites: Hydroxytetradecenoylcarnitine, hexadecenoylcarnitine, hexanoylcarnitine | Altered proteins: |
| Pck2 | ||
| Enriched terms: | ||
| Mitochondrial respiratory chain complex assembly |
| Metabolite | Liver | Serum |
|---|---|---|
| Acylcarnitines | ||
| Hydroxytetradecenoylcarnitine | decrease | - |
| Hexadecenoylcarnitine | decrease | - |
| Hexanoylcarnitine | decrease | - |
| Sphingolipids | ||
| C24:1-OH Sphingomyelin | decrease | decrease |
| SM(d18:0/20:2) | decrease | - |
| Amino acids | ||
| Ala | increase | increase |
| Asp | increase | increase |
| Asn | increase | - |
| Gly | increase | increase |
| Glu | increase | increase |
| Glutamine | increase | - |
| Biogenic amines | ||
| Carnosine | decrease | increase |
| Putrescine | increase | increase |
| Taurine | decrease | - |
| Monosaccharides | ||
| Hexose | increase | decrease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sweetat, S.; Nitzan, K.; Suissa, N.; Haimovich, Y.; Lichtenstein, M.; Zabit, S.; Benhamron, S.; Akarieh, K.; Mishra, K.; Barasch, D.; et al. The Beneficial Effect of Mitochondrial Transfer Therapy in 5XFAD Mice via Liver–Serum–Brain Response. Cells 2023, 12, 1006. https://doi.org/10.3390/cells12071006
Sweetat S, Nitzan K, Suissa N, Haimovich Y, Lichtenstein M, Zabit S, Benhamron S, Akarieh K, Mishra K, Barasch D, et al. The Beneficial Effect of Mitochondrial Transfer Therapy in 5XFAD Mice via Liver–Serum–Brain Response. Cells. 2023; 12(7):1006. https://doi.org/10.3390/cells12071006
Chicago/Turabian StyleSweetat, Sahar, Keren Nitzan, Nir Suissa, Yael Haimovich, Michal Lichtenstein, Samar Zabit, Sandrine Benhamron, Karameh Akarieh, Kumudesh Mishra, Dinorah Barasch, and et al. 2023. "The Beneficial Effect of Mitochondrial Transfer Therapy in 5XFAD Mice via Liver–Serum–Brain Response" Cells 12, no. 7: 1006. https://doi.org/10.3390/cells12071006