
Ischemic Tolerance—A Way to Reduce the Extent of Ischemia–Reperfusion Damage

{kind=link}
{kind=link}
Abstract
:1. Ischemia–Reperfusion (IR) Injury
2. Current Therapeutic Options for Influencing IR Damage
- A NO (Nitric Oxide) protective strategy [8];
- Adenosine [9];
- The influence of nuclear transcription factors [10];
- The inhibition of apoptosis [11];
- The inhibition of Ca 2+ excess in the cell [12];
- Inhibitors of Na+ H+ channels [15];
- Controlled reperfusion/reoxygenation [16];
- Intermittent ischemia [17];
- Aprotinin [20];
- Poly (ADP-ribose) polymerase (PARP inhibitors) [21];
- The blockade of the complement system [22];
- Hyperbaric oxygen therapy [27];
- Ischemic tolerance (conditioning).
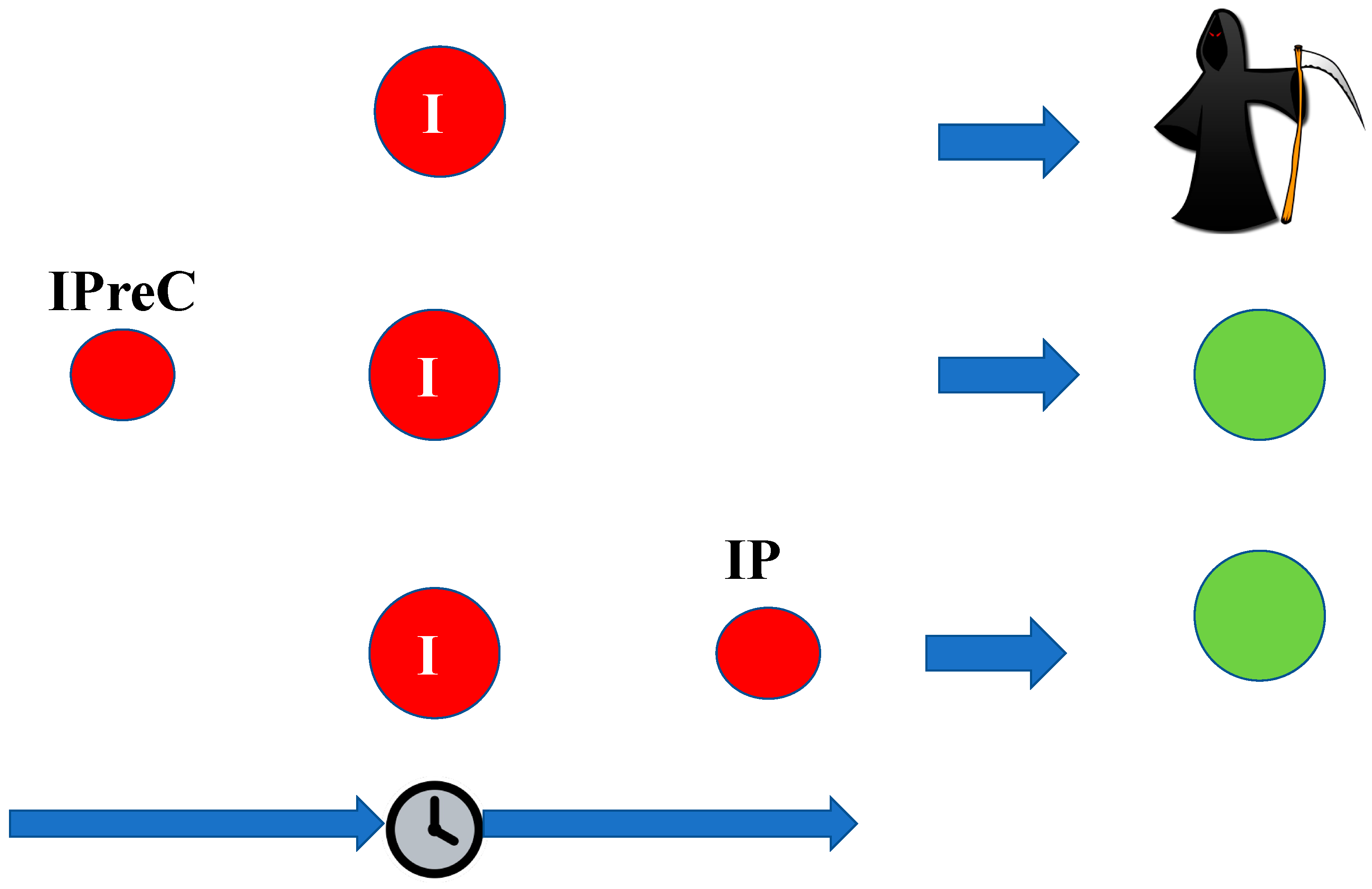
3. Ischemic Tolerance
- Ischemic preconditioning (IpreC) refers to the condition where the sublethal ischemia acts before the lethal ischemia itself;
- Ischemic perconditioning refers to the condition where the sublethal ischemia and the lethal ischemia act simultaneously;
- Ischemic postconditioning (IP) refers to the condition when the sublethal ischemia occurs after the lethal ischemia.

4. Cross Tolerance
4.1. Hyperoxic and Hypoxic Preconditioning
4.2. Preconditioning Induced by Hyperthermia and Hypothermia
4.3. Chemical/Pharmacological Preconditioning
4.4. Electroacupuncture
4.5. Multipotent Adult Progenitor Cells and Tissue Engineering
5. Remote Conditioning
6. Mechanism of the Operation of Local Ischemic Preconditioning
6.1. Hypoxia-Inducible Factor (HIF-1α)
6.2. Glutamate Pathway
6.3. NO Oxide Synthase (NOS)
6.4. Pathway of the CD39-CD73-Adenosine Receptor
6.5. Immune Pathway
6.6. Enzymes and Receptors
6.7. Autophagy and Apoptosis
6.8. Energy Metabolism
6.9. Permeability of the Blood–Brain Barrier
6.10. Transcriptional Regulation
6.11. Genetic Reprogramming
6.12. Epigenetic Reprogramming
6.13. Activation of Ischemic Tolerance in the Brain
7. The Mechanism of Remote Ischemic Preconditioning
7.1. Humoral Pathway
7.2. Nerve Pathway
7.3. Inflammatory Pathway
7.4. Common Final Mechanisms
8. Cell Signaling Pathways in Ischemic Postconditioning
8.1. Akt Signaling Pathway Activation
8.2. The mTOR Signaling Pathway of Activation (Mammalian Target of Rapamycin = Thr/Ser Protein Kinase)
8.3. The Mitogen-Activated Protein Kinase (MAPK) Pathway
8.4. The Protein Kinase C (PKC) Pathway
8.5. The Toll-Like Receptor 4 (TLR4) Pathway
8.6. The Mitochondrial Role in Postconditioning
9. Pharmacological Postconditioning
10. Ischemic Tolerance (Closing Remarks)—Application Possibilities
- -
- Induction of ischemic tolerance;
- -
- Temporary elevation of blood pressure and blood flow.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krause, G.S.; Kumar, K.; White, B.C.; Aust, S.D.; Wiegenstein, J.G. Ischemia, resuscitation, and reperfusion: Mechanisms of tissue injury and prospects for protection. Am. Heart J. 1986, 111, 768–780. [Google Scholar] [CrossRef]
- Dorweiler, B.; Pruefer, D.; Andrasi, T.B.; Maksan, S.M.; Schmiedt, W.; Neufang, A.; Vahl, C.F. Ischemia-Reperfusion Injury: Pathophysiology and Clinical Implications. Eur. J. Trauma Emerg. Surg. 2007, 33, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Saat, T.C.; van den Akker, E.K.; IJzermans, J.N.; Dor, F.J.; de Bruin, R.W. Improving the outcome of kidney transplantation by ameliorating renal ischemia reperfusion injury: Lost in translation? J. Transl. Med. 2016, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuijs-Moeke, G.J.; Pischke, S.E.; Berger, S.P.; Sanders, J.S.F.; Pol, R.A.; Struys, M.; Ploeg, R.J.; Leuvenink, H.G.D. Ischemia and Reperfusion Injury in Kidney Transplantation: Relevant Mechanisms in Injury and Repair. J. Clin. Med. 2020, 9, 253. [Google Scholar] [CrossRef] [Green Version]
- Mico-Carnero, M.; Zaouali, M.A.; Rojano-Alfonso, C.; Maroto-Serrat, C.; Ben Abdennebi, H.; Peralta, C. A Potential Route to Reduce Ischemia/Reperfusion Injury in Organ Preservation. Cells 2022, 11, 2763. [Google Scholar] [CrossRef]
- Becker, L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004, 61, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Ahluwalia, A.; Foster, P.; Scotland, R.S.; McLean, P.G.; Mathur, A.; Perretti, M.; Moncada, S.; Hobbs, A.J. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2004, 101, 1386–1391. [Google Scholar] [CrossRef] [Green Version]
- De Marco, C.; Charron, T.; Rousseau, G. Adenosine in Acute Myocardial Infarction-Associated Reperfusion Injury: Does it Still Have a Role? Front. Pharmacol. 2022, 13, 856747. [Google Scholar] [CrossRef] [PubMed]
- Mata, A.; Cadenas, S. The Antioxidant Transcription Factor Nrf2 in Cardiac Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 11939. [Google Scholar] [CrossRef]
- Toldo, S.; Breckenridge, D.G.; Mezzaroma, E.; Van Tassell, B.W.; Shryock, J.; Kannan, H.; Phan, D.; Budas, G.; Farkas, D.; Lesnefsky, E.; et al. Inhibition of apoptosis signal-regulating kinase 1 reduces myocardial ischemia-reperfusion injury in the mouse. J. Am. Heart Assoc. 2012, 1, e002360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Wang, M.; He, S.; Sun, G.; Sun, X. Targeting Calcium Homeostasis in Myocardial Ischemia/Reperfusion Injury: An Overview of Regulatory Mechanisms and Therapeutic Reagents. Front. Pharmacol. 2020, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Retamal, C.; Schupper, D.; Vergara-Hernandez, D.; Saha, S.; Profumo, E.; Buttari, B.; Saso, L. Antioxidant Cardioprotection against Reperfusion Injury: Potential Therapeutic Roles of Resveratrol and Quercetin. Molecules 2022, 27, 2564. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Nagasaki, Y. Self-Assembling Antioxidants for Ischemia-Reperfusion Injuries. Antioxid. Redox Signal. 2022, 36, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Liu, Y.; Goto, M.; Tsuchida, A.; Miki, T.; Nakano, A.; Nishino, Y.; Ohnuma, Y.; Shimamoto, K. Mitochondrial ATP-sensitive K+ channels play a role in cardioprotection by Na+-H+ exchange inhibition against ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2001, 37, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Fischesser, D.M.; Bo, B.; Benton, R.P.; Su, H.; Jahanpanah, N.; Haworth, K.J. Controlling Reperfusion Injury with Controlled Reperfusion: Historical Perspectives and New Paradigms. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 504–523. [Google Scholar] [CrossRef]
- Sadeghi, M.; Horer, T.M.; Forsman, D.; Dogan, E.M.; Jansson, K.; Kindler, C.; Skoog, P.; Nilsson, K.F. Blood pressure targeting by partial REBOA is possible in severe hemorrhagic shock in pigs and produces less circulatory, metabolic and inflammatory sequelae than total REBOA. Injury 2018, 49, 2132–2141. [Google Scholar] [CrossRef]
- Wu, X.; You, D.; Cui, J.; Yang, L.; Lin, L.; Chen, Y.; Xu, C.; Lian, G.; Wan, J. Reduced Neutrophil Extracellular Trap Formation During Ischemia Reperfusion Injury in C3 KO Mice: C3 Requirement for NETs Release. Front. Immunol. 2022, 13, 781273. [Google Scholar] [CrossRef]
- Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.A.; Bianchi, C.; Voisine, P.; Feng, J.; Baker, J.; Hart, M.; Takahashi, M.; Stahl, G.; Sellke, F.W. Reduction of myocardial reperfusion injury by aprotinin after regional ischemia and cardioplegic arrest. J. Thorac. Cardiovasc. Surg. 2004, 128, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP-1 and PARthanatos in ischemic stroke. J. Neurochem. 2022, 160, 74–87. [Google Scholar] [CrossRef]
- Vogel, C.W. The Role of Complement in Myocardial Infarction Reperfusion Injury: An Underappreciated Therapeutic Target. Front. Cell Dev. Biol. 2020, 8, 606407. [Google Scholar] [CrossRef] [PubMed]
- Ergun, Y.; Darendeli, S.; Imrek, S.; Kilinc, M.; Oksuz, H. The comparison of the effects of anesthetic doses of ketamine, propofol, and etomidate on ischemia-reperfusion injury in skeletal muscle. Fundam. Clin. Pharmacol. 2010, 24, 215–222. [Google Scholar] [CrossRef]
- Lee, J.M.; Suh, J.K.; Jeong, J.S.; Cho, S.Y.; Kim, D.W. Antioxidant effect of lidocaine and procaine on reactive oxygen species-induced endothelial dysfunction in the rabbit abdominal aorta. Korean J. Anesthesiol. 2010, 59, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N. Engl. J. Med. 2002, 346, 549–556. [Google Scholar] [CrossRef]
- Lascarrou, J.B.; Merdji, H.; Le Gouge, A.; Colin, G.; Grillet, G.; Girardie, P.; Coupez, E.; Dequin, P.F.; Cariou, A.; Boulain, T.; et al. Targeted Temperature Management for Cardiac Arrest with Nonshockable Rhythm. N. Engl. J. Med. 2019, 381, 2327–2337. [Google Scholar] [CrossRef]
- Chen, W.; Lv, L.; Nong, Z.; Chen, X.; Pan, X.; Chen, C. Hyperbaric oxygen protects against myocardial ischemia-reperfusion injury through inhibiting mitochondria dysfunction and autophagy. Mol. Med. Rep. 2020, 22, 4254–4264. [Google Scholar] [CrossRef] [PubMed]
- Bonauer, A.; Carmona, G.; Iwasaki, M.; Mione, M.; Koyanagi, M.; Fischer, A.; Burchfield, J.; Fox, H.; Doebele, C.; Ohtani, K.; et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 2009, 324, 1710–1713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef]
- Rowart, P.; Erpicum, P.; Detry, O.; Weekers, L.; Gregoire, C.; Lechanteur, C.; Briquet, A.; Beguin, Y.; Krzesinski, J.M.; Jouret, F. Mesenchymal Stromal Cell Therapy in Ischemia/Reperfusion Injury. J. Immunol. Res. 2015, 2015, 602597. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhang, X.; Wan, Z.; Liu, X.; Chen, F.; Zhang, J.; Leng, Y. Mesenchymal stem cells against intestinal ischemia-reperfusion injury: A systematic review and meta-analysis of preclinical studies. Stem Cell Res. Ther. 2022, 13, 216. [Google Scholar] [CrossRef] [PubMed]
- Zemke, D.; Smith, J.L.; Reeves, M.J.; Majid, A. Ischemia and ischemic tolerance in the brain: An overview. Neurotoxicology 2004, 25, 895–904. [Google Scholar] [CrossRef]
- Gidday, J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 2006, 7, 437–448. [Google Scholar] [CrossRef]
- Burda, J.; Matiasova, M.; Gottlieb, M.; Danielisova, V.; Nemethova, M.; Garcia, L.; Salinas, M.; Burda, R. Evidence for a role of second pathophysiological stress in prevention of delayed neuronal death in the hippocampal CA1 region. Neurochem. Res. 2005, 30, 1397–1405. [Google Scholar] [CrossRef]
- Burda, J.; Danielisova, V.; Nemethova, M.; Gottlieb, M.; Matiasova, M.; Domorakova, I.; Mechirova, E.; Ferikova, M.; Salinas, M.; Burda, R. Delayed postconditionig initiates additive mechanism necessary for survival of selectively vulnerable neurons after transient ischemia in rat brain. Cell. Mol. Neurobiol. 2006, 26, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Cheng, O.; Ostrowski, R.P.; Wu, B.; Liu, W.; Chen, C.; Zhang, J.H. Cyclooxygenase-2 mediates hyperbaric oxygen preconditioning in the rat model of transient global cerebral ischemia. Stroke 2011, 42, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Bigdeli, M.R. Neuroprotection caused by hyperoxia preconditioning in animal stroke models. Sci. World J. 2011, 11, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Yunoki, M.; Nishio, S.; Ukita, N.; Anzivino, M.J.; Lee, K.S. Hypothermic preconditioning induces rapid tolerance to focal ischemic injury in the rat. Exp. Neurol. 2003, 181, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Xia, X.Y.; Xia, Y.X.; Ikenoue, T. Hyperthermic preconditioning prevents blood-brain barrier disruption produced by hypoxia-ischemia in newborn rat. Dev. Brain Res. 1999, 117, 53–58. [Google Scholar] [CrossRef]
- Schlegel, A.; Mueller, M.; Muller, X.; Eden, J.; Panconesi, R.; von Felten, S.; Steigmiller, K.; Da Silva, R.X.S.; de Rougemont, O.; Mabrut, J.Y.; et al. A multicenter randomized-controlled trial of hypothermic oxygenated perfusion (HOPE) for human liver grafts before transplantation. J. Hepatol. 2023. [Google Scholar] [CrossRef]
- Rampino, T.; Gregorini, M.; Germinario, G.; Pattonieri, E.F.; Erasmi, F.; Grignano, M.A.; Bruno, S.; Alomari, E.; Bettati, S.; Asti, A.; et al. Extracellular Vesicles Derived from Mesenchymal Stromal Cells Delivered during Hypothermic Oxygenated Machine Perfusion Repair Ischemic/Reperfusion Damage of Kidneys from Extended Criteria Donors. Biology 2022, 11, 350. [Google Scholar] [CrossRef]
- Wang, L.; Traystman, R.J.; Murphy, S.J. Inhalational anesthetics as preconditioning agents in ischemic brain. Curr. Opin. Pharmacol. 2008, 8, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, H.; Kirsch, J.R.; Hurn, P.D.; Murphy, S.J. Inhalational anesthetics as neuroprotectants or chemical preconditioning agents in ischemic brain. J. Cereb. Blood Flow Metab. 2007, 27, 1108–1128. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liang, Z.; Lei, S.; Wu, X.; Yuan, T.; Ma, K.; Chi, K. Sevoflurane Preconditioning Downregulates GRIA1 Expression to Attenuate Cerebral Ischemia-Reperfusion-Induced Neuronal Injury. Neurotox. Res. 2023, 41, 29–40. [Google Scholar] [CrossRef]
- Vartanian, K.B.; Stevens, S.L.; Marsh, B.J.; Williams-Karnesky, R.; Lessov, N.S.; Stenzel-Poore, M.P. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J. Neuroinflamm. 2011, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heurteaux, C.; Lauritzen, I.; Widmann, C.; Lazdunski, M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc. Natl. Acad. Sci. USA 1995, 92, 4666–4670. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Sun, S.; Li, H.; Xu, Y. Cerebral ischemic tolerance induced by 3-nitropropionic acid is associated with increased expression of erythropoietin in rats. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Danielisova, V.; Gottlieb, M.; Nemethova, M.; Kravcukova, P.; Domorakova, I.; Mechirova, E.; Burda, J. Bradykinin postconditioning protects pyramidal CA1 neurons against delayed neuronal death in rat hippocampus. Cell. Mol. Neurobiol. 2009, 29, 871–878. [Google Scholar] [CrossRef]
- Jackson, C.W.; Escobar, I.; Xu, J.; Perez-Pinzon, M.A. Effects of ischemic preconditioning on mitochondrial and metabolic neruoprotection: 5’ adenosine monophosphate-activated protein kinase and sirtuins. Brain Circ. 2018, 4, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, K.H.; Jang, Y.J.; Kim, H.N.; Bae, S.S.; Choi, B.T.; Shin, H.K. Electroacupuncture preconditioning reduces cerebral ischemic injury via BDNF and SDF-1alpha in mice. BMC Complement. Altern. Med. 2013, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Liebelt, B.; Papapetrou, P.; Ali, A.; Guo, M.; Ji, X.; Peng, C.; Rogers, R.; Curry, A.; Jimenez, D.; Ding, Y. Exercise preconditioning reduces neuronal apoptosis in stroke by up-regulating heat shock protein-70 (heat shock protein-72) and extracellular-signal-regulated-kinase 1/2. Neuroscience 2010, 166, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.I.; Park, J.H.; Park, M.Y.; Kim, N.G.; Park, K.J.; Choi, B.T.; Shin, Y.I.; Shin, H.K. Pre-conditioning with transcranial low-level light therapy reduces neuroinflammation and protects blood-brain barrier after focal cerebral ischemia in mice. Restor. Neurol. Neurosci. 2016, 34, 201–214. [Google Scholar] [CrossRef]
- Thompson, E.R.; Bates, L.; Ibrahim, I.K.; Sewpaul, A.; Stenberg, B.; McNeill, A.; Figueiredo, R.; Girdlestone, T.; Wilkins, G.C.; Wang, L.; et al. Novel delivery of cellular therapy to reduce ischemia reperfusion injury in kidney transplantation. Am. J. Transplant. 2021, 21, 1402–1414. [Google Scholar] [CrossRef]
- Grignano, M.A.; Bruno, S.; Viglio, S.; Avanzini, M.A.; Tapparo, M.; Ramus, M.; Croce, S.; Valsecchi, C.; Pattonieri, E.F.; Ceccarelli, G.; et al. CD73-Adenosinergic Axis Mediates the Protective Effect of Extracellular Vesicles Derived from Mesenchymal Stromal Cells on Ischemic Renal Damage in a Rat Model of Donation after Circulatory Death. Int. J. Mol. Sci. 2022, 23, 10681. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Her, A.Y.; Rha, S.W.; Choi, B.G.; Choi, S.Y.; Byun, J.K.; Mashaly, A.; Park, Y.; Jang, W.Y.; Kim, W.; et al. Impact of Trimetazidine Treatment on 5-year Clinical Outcomes in Patients with Significant Coronary Artery Spasm: A Propensity Score Matching Study. Am. J. Cardiovasc. Drugs 2018, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- St Peter, S.D.; Post, D.J.; Rodriguez-Davalos, M.I.; Douglas, D.D.; Moss, A.A.; Mulligan, D.C. Tacrolimus as a liver flush solution to ameliorate the effects of ischemia/reperfusion injury following liver transplantation. Liver Transpl. 2003, 9, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, M.; Castillo, R.L.; Beltran, J.F.; Herrera, L.; Farias, J.A.; Antileo, C.; Aguilar-Gallardo, C.; Pessoa, A.; Calle, Y.; Farias, J.G. Tackling Ischemic Reperfusion Injury With the Aid of Stem Cells and Tissue Engineering. Front. Physiol. 2021, 12, 705256. [Google Scholar] [CrossRef] [PubMed]
- Kerendi, F.; Kin, H.; Halkos, M.E.; Jiang, R.; Zatta, A.J.; Zhao, Z.Q.; Guyton, R.A.; Vinten-Johansen, J. Remote postconditioning. Brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res. Cardiol. 2005, 100, 404–412. [Google Scholar] [CrossRef]
- Wang, C.; Weihrauch, D.; Schwabe, D.A.; Bienengraeber, M.; Warltier, D.C.; Kersten, J.R.; Pratt, P.F., Jr.; Pagel, P.S. Extracellular signal-regulated kinases trigger isoflurane preconditioning concomitant with upregulation of hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in rats. Anesth. Analg. 2006, 103, 281–288. [Google Scholar] [CrossRef]
- Hao, Y.; Xin, M.; Feng, L.; Wang, X.; Wang, X.; Ma, D.; Feng, J. Review Cerebral Ischemic Tolerance and Preconditioning: Methods, Mechanisms, Clinical Applications, and Challenges. Front. Neurol. 2020, 11, 812. [Google Scholar] [CrossRef]
- Fox, C.; Walsh, P.; Mulhall, K.J. Molecular Mechanism of Ischaemic Preconditioning of Skeletal Muscle In Vitro. Cureus 2018, 10, e3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Tae, H.J.; Kim, I.H.; Cho, J.H.; Lee, T.K.; Park, J.H.; Ahn, J.H.; Choi, S.Y.; Bai, H.C.; Shin, B.N.; et al. Roles of HIF-1alpha, VEGF, and NF-kappaB in Ischemic Preconditioning-Mediated Neuroprotection of Hippocampal CA1 Pyramidal Neurons Against a Subsequent Transient Cerebral Ischemia. Mol. Neurobiol. 2017, 54, 6984–6998. [Google Scholar] [CrossRef]
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. AJNR Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar] [PubMed]
- Watters, O.; O’Connor, J.J. A role for tumor necrosis factor-alpha in ischemia and ischemic preconditioning. J. Neuroinflammation 2011, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Feng, L.; Cheng, Y.; Xin, M.; You, J.; Yin, X.; Hao, Y.; Cui, L.; Feng, J. Astrocytic gap junction inhibition by carbenoxolone enhances the protective effects of ischemic preconditioning following cerebral ischemia. J. Neuroinflammation 2018, 15, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centeno, J.M.; Orti, M.; Salom, J.B.; Sick, T.J.; Perez-Pinzon, M.A. Nitric oxide is involved in anoxic preconditioning neuroprotection in rat hippocampal slices. Brain Res. 1999, 836, 62–69. [Google Scholar] [CrossRef]
- Lee, N.T.; Ong, L.K.; Gyawali, P.; Nassir, C.; Mustapha, M.; Nandurkar, H.H.; Sashindranath, M. Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease. Biomolecules 2021, 11, 994. [Google Scholar] [CrossRef]
- Ulker, P.; Ozkan, O.; Amoroso, M.; Aslan, M.; Bassorgun, I.; Ubur, M.C.; Unal, K.; Ozcan, F.; Ozkan, O. Does ischemic preconditioning increase flap survival by ADORA2B receptor activation? Clin. Hemorheol. Microcirc. 2020, 75, 151–162. [Google Scholar] [CrossRef]
- Hart, M.L.; Gorzolla, I.C.; Schittenhelm, J.; Robson, S.C.; Eltzschig, H.K. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 2010, 184, 4017–4024. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.L.; Much, C.; Gorzolla, I.C.; Schittenhelm, J.; Kloor, D.; Stahl, G.L.; Eltzschig, H.K. Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 2008, 135, 1739–1750.e3. [Google Scholar] [CrossRef]
- Eckle, T.; Krahn, T.; Grenz, A.; Kohler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M.A.; Osswald, H.; Thompson, L.F.; Unertl, K.; et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.B.; Xu, Z.; Novitskaya, T.; Zhang, B.; Chepurko, E.; Pu, X.A.; Wheeler, D.G.; Ziolo, M.; Gumina, R.J. Impact of cardiac-specific expression of CD39 on myocardial infarct size in mice. Life Sci. 2017, 179, 54–59. [Google Scholar] [CrossRef]
- Dwyer, K.M. Burnstock oration—purinergic signalling in kidney transplantation. Purinergic Signal. 2022, 18, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Gong, X.; Kuang, G.; Jiang, R.; Wan, J.; Wang, B. Sesamin protects against renal ischemia reperfusion injury by promoting CD39-adenosine-A2AR signal pathway in mice. Am. J. Transl. Res. 2016, 8, 2245–2254. [Google Scholar] [PubMed]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinzon, M.A.; Vitro, T.M.; Dietrich, W.D.; Sick, T.J. The effect of rapid preconditioning on the microglial, astrocytic and neuronal consequences of global cerebral ischemia. Acta Neuropathol. 1999, 97, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, V.; Gordon, W.C.; Mukherjee, P.K.; Trivedi, P.; Ottino, P. Downregulation of COX-2 and JNK expression after induction of ischemic tolerance in the gerbil brain. Brain Res. 2004, 1016, 195–200. [Google Scholar] [CrossRef]
- Lee, J.C.; Tae, H.J.; Cho, G.S.; Kim, I.H.; Ahn, J.H.; Park, J.H.; Chen, B.H.; Cho, J.H.; Shin, B.N.; Cho, J.H.; et al. Ischemic preconditioning protects neurons from damage and maintains the immunoreactivity of kynurenic acid in the gerbil hippocampal CA1 region following transient cerebral ischemia. Int. J. Mol. Med. 2015, 35, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.Y.; Lusardi, T.A.; Williams-Karnesky, R.L.; Lan, J.Q.; Poulsen, D.J.; Boison, D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J. Cereb. Blood Flow Metab. 2011, 31, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Saito, A.; Okuno, S.; Ferrand-Drake, M.; Dodd, R.L.; Chan, P.H. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J. Cereb. Blood Flow Metab. 2005, 25, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Khor, S.; Hackett, S.R.; Rabinowitz, J.D.; Perlman, D.H.; White, E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol. Cell 2014, 55, 916–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.Y.; Li, W.; Qian, H.R.; Yao, S.; Liu, J.G.; Qi, X.K. Ischemia preconditioning is neuroprotective in a rat cerebral ischemic injury model through autophagy activation and apoptosis inhibition. Braz. J. Med. Biol. Res. 2013, 46, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.B.; Toledo-Pereyra, L.H. Caspase-independent programmed cell death following ischemic stroke. J. Investig. Surg. 2008, 21, 141–147. [Google Scholar] [CrossRef]
- Tohyama, Y.; Sako, K.; Yonemasu, Y. Hypothermia attenuates hyperglycolysis in the periphery of ischemic core in rat brain. Exp. Brain Res. 1998, 122, 333–338. [Google Scholar] [CrossRef]
- Wacker, B.K.; Freie, A.B.; Perfater, J.L.; Gidday, J.M. Junctional protein regulation by sphingosine kinase 2 contributes to blood-brain barrier protection in hypoxic preconditioning-induced cerebral ischemic tolerance. J. Cereb. Blood Flow Metab. 2012, 32, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef]
- Jones, N.M.; Bergeron, M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J. Cereb. Blood Flow Metab. 2001, 21, 1105–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burda, J.; Hrehorovska, M.; Bonilla, L.G.; Danielisova, V.; Cizkova, D.; Burda, R.; Nemethova, M.; Fando, J.L.; Salinas, M. Role of protein synthesis in the ischemic tolerance acquisition induced by transient forebrain ischemia in the rat. Neurochem. Res. 2003, 28, 1213–1219. [Google Scholar] [CrossRef]
- Dhodda, V.K.; Sailor, K.A.; Bowen, K.K.; Vemuganti, R. Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis. J. Neurochem. 2004, 89, 73–89. [Google Scholar] [CrossRef]
- Atochin, D.N.; Clark, J.; Demchenko, I.T.; Moskowitz, M.A.; Huang, P.L. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke 2003, 34, 1299–1303. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Yamamoto, T.; Tsuboi, A. Molecular mechanisms underlying activity-dependent ischemic tolerance in the brain. Neurosci. Res. 2023, 186, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, Y.; Anzai, N.; Kinouchi, H.; Koizumi, S. P2X7 Receptors in Astrocytes: A Switch for Ischemic Tolerance. Molecules 2022, 27, 3655. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Hirayama, Y. Ischemic Tolerance Induced by Glial Cells. Neurochem. Res. 2022, 47, 2522–2528. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Tropak, M.; Diaz, R.J.; Suto, F.; Surendra, H.; Kuzmin, E.; Li, J.; Gross, G.; Wilson, G.J.; Callahan, J.; et al. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: Evidence suggesting cross-species protection. Clin. Sci. 2009, 117, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Kanoria, S.; Jalan, R.; Seifalian, A.M.; Williams, R.; Davidson, B.R. Protocols and mechanisms for remote ischemic preconditioning: A novel method for reducing ischemia reperfusion injury. Transplantation 2007, 84, 445–458. [Google Scholar] [CrossRef]
- Burda, R.; Danielisova, V.; Burda, J. The End Effector of Ischemic Tolerance Present in Blood Plasma from Double Conditioned Donors Ameliorates Trimethyltin Provoked Damage in Brain. OBM Neurobiol. 2019, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Burda, R.; Morochovič, R.; Némethová, M.; Burda, J. Remote ischemic postconditioning as well as blood plasma from double-conditioned donor ameliorate reperfusion syndrome in skeletal muscle. J. Plast. Surg. Hand Surg. 2020, 54, 59–65. [Google Scholar] [CrossRef]
- Wei, D.; Ren, C.; Chen, X.; Zhao, H. The chronic protective effects of limb remote preconditioning and the underlying mechanisms involved in inflammatory factors in rat stroke. PLoS ONE 2012, 7, e30892. [Google Scholar] [CrossRef]
- Mastitskaya, S.; Marina, N.; Gourine, A.; Gilbey, M.P.; Spyer, K.M.; Teschemacher, A.G.; Kasparov, S.; Trapp, S.; Ackland, G.L.; Gourine, A.V. Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre-ganglionic neurones. Cardiovasc. Res. 2012, 95, 487–494. [Google Scholar] [CrossRef]
- Sun, Z.; Baker, W.; Hiraki, T.; Greenberg, J.H. The effect of right vagus nerve stimulation on focal cerebral ischemia: An experimental study in the rat. Brain Stimul. 2012, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.; Buchholz, B.; Rodriguez, M.; Perez, V.; Inserte, J.; Garcia-Dorado, D.; Gelpi, R.J. Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp. Physiol. 2013, 98, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Steensrud, T.; Li, J.; Dai, X.; Manlhiot, C.; Kharbanda, R.K.; Tropak, M.; Redington, A. Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1598–H1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Yellon, D.M.; Hausenloy, D.J. The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res. Cardiol. 2010, 105, 651–655. [Google Scholar] [CrossRef]
- Konstantinov, I.E.; Arab, S.; Kharbanda, R.K.; Li, J.; Cheung, M.M.; Cherepanov, V.; Downey, G.P.; Liu, P.P.; Cukerman, E.; Coles, J.G.; et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol. Genom. 2004, 19, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Saxena, P.; Konstantinov, I.E.; Cherepanov, V.; Cheung, M.M.; Wearden, P.; Zhangdong, H.; Schmidt, M.; Downey, G.P.; Redington, A.N. Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J. Surg. Res. 2010, 158, 155–161. [Google Scholar] [CrossRef]
- Weber, C. Far from the heart: Receptor cross-talk in remote conditioning. Nat. Med. 2010, 16, 760–762. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, Z.; Chao, X.; Gu, J.; Cai, W.; Zhou, W.; Yin, G.; Li, Q. Ischemic preconditioning enhances autophagy but suppresses autophagic cell death in rat spinal neurons following ischemia-reperfusion. Brain Res. 2014, 1562, 76–86. [Google Scholar] [CrossRef]
- Lv, S.; Ju, C.; Peng, J.; Liang, M.; Zhu, F.; Wang, C.; Huang, K.; Cheng, M.; Zhang, F. 25-Hydroxycholesterol protects against myocardial ischemia-reperfusion injury via inhibiting PARP activity. Int. J. Biol. Sci. 2020, 16, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.K.; Yu, L.N.; Zhang, F.J.; Yang, M.J.; Yu, J.; Yan, M.; Chen, G. Postconditioning with sevoflurane protects against focal cerebral ischemia and reperfusion injury via PI3K/Akt pathway. Brain Res. 2010, 1357, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Meller, R.; Inoue, K.; Ordonez, A.N.; Ashley, M.D.; Xiong, Z.; Gala, R.; Simon, R.P. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: Ischemic postconditioning. J. Cereb. Blood Flow Metab. 2008, 28, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, Z.; Zhang, X.; Zhang, X.; Dong, L.; Xing, Y.; Li, Y.; Liu, Z.; Chen, L.; Qiao, H.; et al. Protection by silibinin against experimental ischemic stroke: Up-regulated pAkt, pmTOR, HIF-1alpha and Bcl-2, down-regulated Bax, NF-kappaB expression. Neurosci. Lett. 2012, 529, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Wang, P.; Ji, X.; Zhao, H. Ischemic post-conditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J. Neurochem. 2013, 127, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.Z.; Shan, X.Y.; Li, X.S.; Tao, H.M. Remote ischemic postconditioning protects the brain from focal ischemia/reperfusion injury by inhibiting autophagy through the mTOR/p70S6K pathway. Neurol. Res. 2018, 40, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, H.; Takahashi, T.; Hsieh, J.; Liao, J.; Steinberg, G.K.; Zhao, H. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 2008, 105, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimohata, T.; Zhao, H.; Steinberg, G.K. Epsilon PKC may contribute to the protective effect of hypothermia in a rat focal cerebral ischemia model. Stroke 2007, 38, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Zhou, F.; Li, S.; Zong, Y.; Zhang, M.; Lin, Y.; Zhang, X.; Yang, H.; Zou, Y.; Qi, C.; et al. Remote ischemic postconditioning protects ischemic brain from injury in rats with focal cerebral ischemia/reperfusion associated with suppression of TLR4 and NF-small ka, CyrillicB expression. Neuroreport 2016, 27, 469–475. [Google Scholar] [CrossRef]
- Feng, R.; Li, S.; Li, F. Toll-like receptor 4 is involved in ischemic tolerance of postconditioning in hippocampus of tree shrews to thrombotic cerebral ischemia. Brain Res. 2011, 1384, 118–127. [Google Scholar] [CrossRef]
- Burda, J.; Martin, M.E.; Garcia, A.; Alcazar, A.; Fando, J.L.; Salinas, M. Phosphorylation of the alpha subunit of initiation factor 2 correlates with the inhibition of translation following transient cerebral ischaemia in the rat. Biochem. J. 1994, 302 Pt 2, 335–338. [Google Scholar] [CrossRef]
- Garcia, L.; Burda, J.; Hrehorovska, M.; Burda, R.; Martin, M.E.; Salinas, M. Ischaemic preconditioning in the rat brain: Effect on the activity of several initiation factors, Akt and extracellular signal-regulated protein kinase phosphorylation, and GRP78 and GADD34 expression. J. Neurochem. 2004, 88, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, P.; Femmino, S.; Popara, J.; Penna, C. Mitochondria in Cardiac Postconditioning. Front. Physiol. 2018, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Argaud, L.; Gateau-Roesch, O.; Augeul, L.; Couture-Lepetit, E.; Loufouat, J.; Gomez, L.; Robert, D.; Ovize, M. Increased mitochondrial calcium coexists with decreased reperfusion injury in postconditioned (but not preconditioned) hearts. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H386–H391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaud, L.; Gateau-Roesch, O.; Raisky, O.; Loufouat, J.; Robert, D.; Ovize, M. Postconditioning inhibits mitochondrial permeability transition. Circulation 2005, 111, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Dongworth, R.K.; Hall, A.R.; Burke, N.; Hausenloy, D.J. Targeting mitochondria for cardioprotection: Examining the benefit for patients. Future Cardiol. 2014, 10, 255–272. [Google Scholar] [CrossRef]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Garlid, K.D.; Halestrap, A.P. The mitochondrial K(ATP) channel–fact or fiction? J. Mol. Cell Cardiol. 2012, 52, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Di Lisa, F.; Canton, M.; Carpi, A.; Kaludercic, N.; Menabo, R.; Menazza, S.; Semenzato, M. Mitochondrial injury and protection in ischemic pre- and postconditioning. Antioxid. Redox Signal. 2011, 14, 881–891. [Google Scholar] [CrossRef]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res. Cardiol. 2006, 101, 180–189. [Google Scholar] [CrossRef]
- Serviddio, G.; Di Venosa, N.; Federici, A.; D’Agostino, D.; Rollo, T.; Prigigallo, F.; Altomare, E.; Fiore, T.; Vendemiale, G. Brief hypoxia before normoxic reperfusion (postconditioning) protects the heart against ischemia-reperfusion injury by preventing mitochondria peroxyde production and glutathione depletion. FASEB J. 2005, 19, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Bopassa, J.C.; Ferrera, R.; Gateau-Roesch, O.; Couture-Lepetit, E.; Ovize, M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc. Res. 2006, 69, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Perrelli, M.G.; Pagliaro, P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid. Redox Signal. 2013, 18, 556–599. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Granata, R.; Tocchetti, C.G.; Gallo, M.P.; Alloatti, G.; Pagliaro, P. Endogenous Cardioprotective Agents: Role in Pre and Postconditioning. Curr. Drug Targets 2015, 16, 843–867. [Google Scholar] [CrossRef] [Green Version]
- Obal, D.; Dettwiler, S.; Favoccia, C.; Scharbatke, H.; Preckel, B.; Schlack, W. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth. Analg. 2005, 101, 1252–1260. [Google Scholar] [CrossRef]
- Pravdic, D.; Mio, Y.; Sedlic, F.; Pratt, P.F.; Warltier, D.C.; Bosnjak, Z.J.; Bienengraeber, M. Isoflurane protects cardiomyocytes and mitochondria by immediate and cytosol-independent action at reperfusion. Br. J. Pharmacol. 2010, 160, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Zhang, F.J.; Wang, S.P.; Chen, G.; Chen, C.C.; Yan, M. Postconditioning of sevoflurane and propofol is associated with mitochondrial permeability transition pore. J. Zhejiang Univ. Sci. B 2008, 9, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Steenbergen, C. What makes the mitochondria a killer? Can we condition them to be less destructive? Biochim. Biophys. Acta 2011, 1813, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Perrelli, M.G.; Raimondo, S.; Tullio, F.; Merlino, A.; Moro, F.; Geuna, S.; Mancardi, D.; Pagliaro, P. Postconditioning induces an anti-apoptotic effect and preserves mitochondrial integrity in isolated rat hearts. Biochim. Biophys. Acta 2009, 1787, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Seewald, M.; Coles, J.A., Jr.; Sigg, D.C.; Iaizzo, P.A. Featured Article: Pharmacological postconditioning with delta opioid attenuates myocardial reperfusion injury in isolated porcine hearts. Exp. Biol. Med. 2017, 242, 986–995. [Google Scholar] [CrossRef]
- Khan, H.; Kashyap, A.; Kaur, A.; Singh, T.G. Pharmacological postconditioning: A molecular aspect in ischemic injury. J. Pharm. Pharmacol. 2020, 72, 1513–1527. [Google Scholar] [CrossRef]
- Bahde, R.; Spiegel, H.U. Hepatic ischaemia-reperfusion injury from bench to bedside. Br. J. Surg. 2010, 97, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Meybohm, P.; Gruenewald, M.; Zacharowski, K.D.; Albrecht, M.; Lucius, R.; Fosel, N.; Hensler, J.; Zitta, K.; Bein, B. Mild hypothermia alone or in combination with anesthetic post-conditioning reduces expression of inflammatory cytokines in the cerebral cortex of pigs after cardiopulmonary resuscitation. Crit. Care 2010, 14, R21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pateliya, B.B.; Singh, N.; Jaggi, A.S. Possible role of opioids and KATP channels in neuroprotective effect of postconditioning in mice. Biol. Pharm. Bull. 2008, 31, 1755–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burda, J.; Danielisova, V.; Nemethova, M.; Gottlieb, M.; Kravcukova, P.; Domorakova, I.; Mechirova, E.; Burda, R. Postconditioning and anticonditioning: Possibilities to interfere to evoked apoptosis. Cell. Mol. Neurobiol. 2009, 29, 821–825. [Google Scholar] [CrossRef]
- Korthals, J.K.; Maki, T.; Gieron, M.A. Nerve and muscle vulnerability to ischemia. J. Neurol. Sci. 1985, 71, 283–290. [Google Scholar] [CrossRef]
- Jenkins, D.P.; Pugsley, W.B.; Alkhulaifi, A.M.; Kemp, M.; Hooper, J.; Yellon, D.M. Ischaemic preconditioning reduces troponin T release in patients undergoing coronary artery bypass surgery. Heart 1997, 77, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Burda, R. How to Influence the Extent of Reperfusion Syndromme in Replantations and Revascularisations, Faculty of Medicine, Pavol Jozef Šafárik University, Košice, Slovakia. 2014. Available online: https://opac.crzp.sk/?fn=detailBiblioForm&sid=CB068CE69013CFB6D36EE4794CD3 (accessed on 20 February 2023).
- Tsubota, H.; Marui, A.; Esaki, J.; Bir, S.C.; Ikeda, T.; Sakata, R. Remote postconditioning may attenuate ischaemia-reperfusion injury in the murine hindlimb through adenosine receptor activation. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 804–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignataro, G.; Esposito, E.; Sirabella, R.; Vinciguerra, A.; Cuomo, O.; Di Renzo, G.; Annunziato, L. nNOS and p-ERK involvement in the neuroprotection exerted by remote postconditioning in rats subjected to transient middle cerebral artery occlusion. Neurobiol. Dis. 2013, 54, 105–114. [Google Scholar] [CrossRef]
- Kolbenschlag, J.; Sogorski, A.; Kapalschinski, N.; Harati, K.; Lehnhardt, M.; Daigeler, A.; Hirsch, T.; Goertz, O. Remote Ischemic Conditioning Improves Blood Flow and Oxygen Saturation in Pedicled and Free Surgical Flaps. Plast. Reconstr. Surg. 2016, 138, 1089–1097. [Google Scholar] [CrossRef]
- Heusch, G.; Gersh, B.J. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: A continual challenge. Eur. Heart J. 2017, 38, 774–784. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Candilio, L.; Laing, C.; Kunst, G.; Pepper, J.; Kolvekar, S.; Evans, R.; Robertson, S.; Knight, R.; Ariti, C.; et al. Effect of remote ischemic preconditioning on clinical outcomes in patients undergoing coronary artery bypass graft surgery (ERICCA): Rationale and study design of a multi-centre randomized double-blinded controlled clinical trial. Clin. Res. Cardiol. 2012, 101, 339–348. [Google Scholar] [CrossRef]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Boning, A.; Niemann, B.; et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Heusch, G. Extracellular signalling molecules in the ischaemic/reperfused heart—druggable and translatable for cardioprotection? Br. J. Pharmacol. 2015, 172, 2010–2025. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Singh, N.; Virdi, J.K.; Jaggi, A.S. Diabetes abolish cardioprotective effects of remote ischemic conditioning: Evidences and possible mechanisms. J. Physiol. Biochem. 2019, 75, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Schwartz Longacre, L.; Kloner, R.A.; Arai, A.E.; Baines, C.P.; Bolli, R.; Braunwald, E.; Downey, J.; Gibbons, R.J.; Gottlieb, R.A.; Heusch, G.; et al. New horizons in cardioprotection: Recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation 2011, 124, 1172–1179. [Google Scholar] [CrossRef]
- Burda, R.; Danielisova, V.; Gottlieb, M.; Nemethova, M.; Bonova, P.; Matiasova, M.; Morochovic, R.; Burda, J. Delayed remote ischemic postconditioning protects against transient cerebral ischemia/reperfusion as well as kainate-induced injury in rats. Acta Histochem. 2014, 116, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Z.N.; Liu, X.; Dai, G.; Jin, S.Q. Effects of preconditioned plasma collected during the late phase of remote ischaemic preconditioning on ventricular arrhythmias caused by myocardial ischaemia reperfusion in rats. J. Int. Med. Res. 2018, 46, 1370–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, N.C.; Riedemann, I.; Smit, K.F.; Zitta, K.; van de Vondervoort, D.; Zuurbier, C.J.; Hollmann, M.W.; Preckel, B.; Albrecht, M. Plasma from human volunteers subjected to remote ischemic preconditioning protects human endothelial cells from hypoxia-induced cell damage. Basic Res. Cardiol. 2015, 110, 17. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burda, R.; Burda, J.; Morochovič, R. Ischemic Tolerance—A Way to Reduce the Extent of Ischemia–Reperfusion Damage. Cells 2023, 12, 884. https://doi.org/10.3390/cells12060884
Burda R, Burda J, Morochovič R. Ischemic Tolerance—A Way to Reduce the Extent of Ischemia–Reperfusion Damage. Cells. 2023; 12(6):884. https://doi.org/10.3390/cells12060884
Chicago/Turabian StyleBurda, Rastislav, Jozef Burda, and Radoslav Morochovič. 2023. "Ischemic Tolerance—A Way to Reduce the Extent of Ischemia–Reperfusion Damage" Cells 12, no. 6: 884. https://doi.org/10.3390/cells12060884