
Immune Cells Are Differentially Modulated in the Heart and the Kidney during the Development of Cardiorenal Syndrome 3
 , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Animal Experimental I/R Procedure
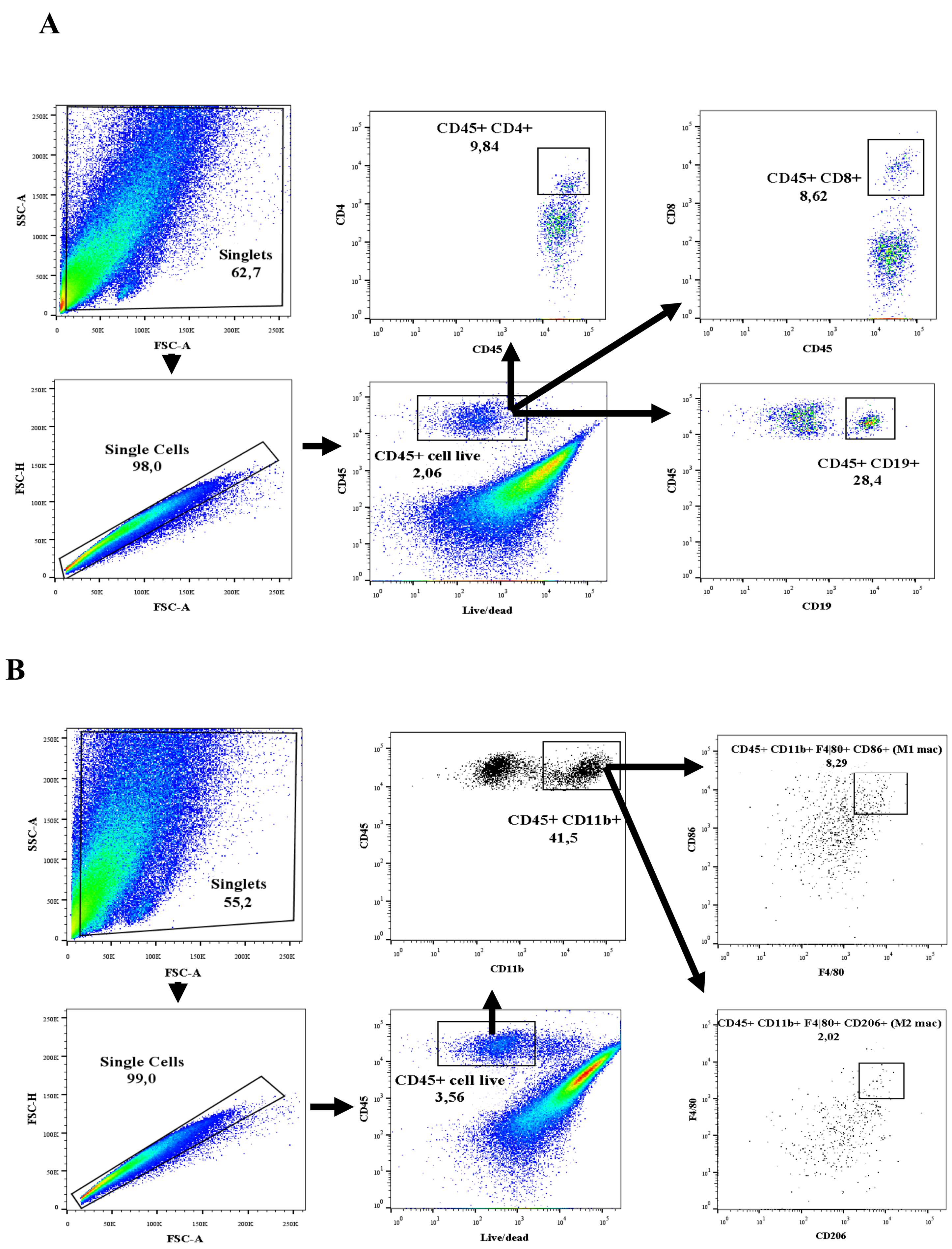
2.2. Isolation of Immune Cells and Flow Cytometry
2.2.1. Tissue Digestion and Cell Preparation
2.2.2. Flow Cytometric Analysis
2.3. Gene Expression Analysis Using Quantitative Real-Time PCR (qRT-PCR)
2.4. Statistical Analysis
3. Results
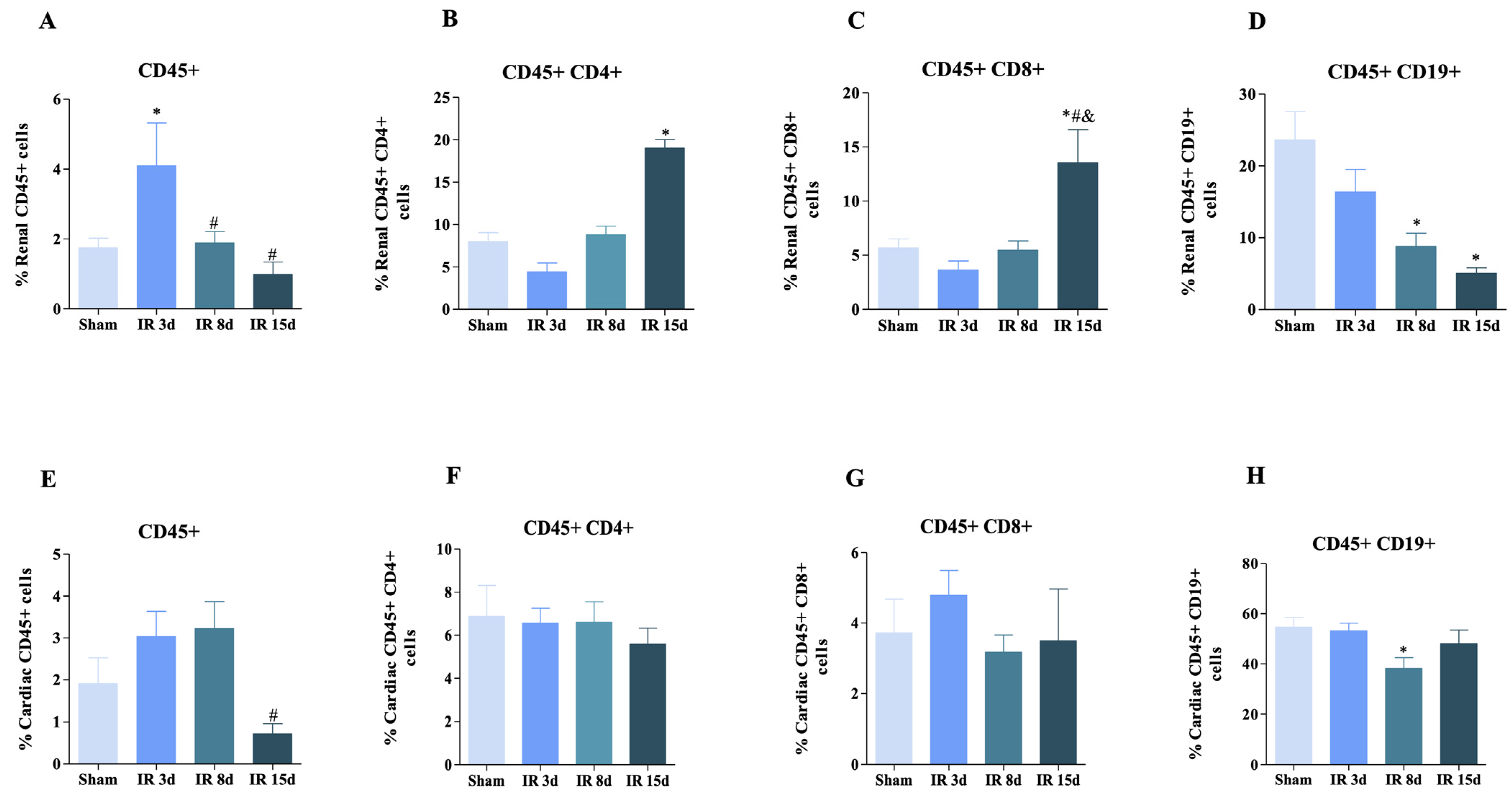
3.1. T and B Lymphocyte Populations Are Activated in Renal and Cardiac Tissue after Renal I/R
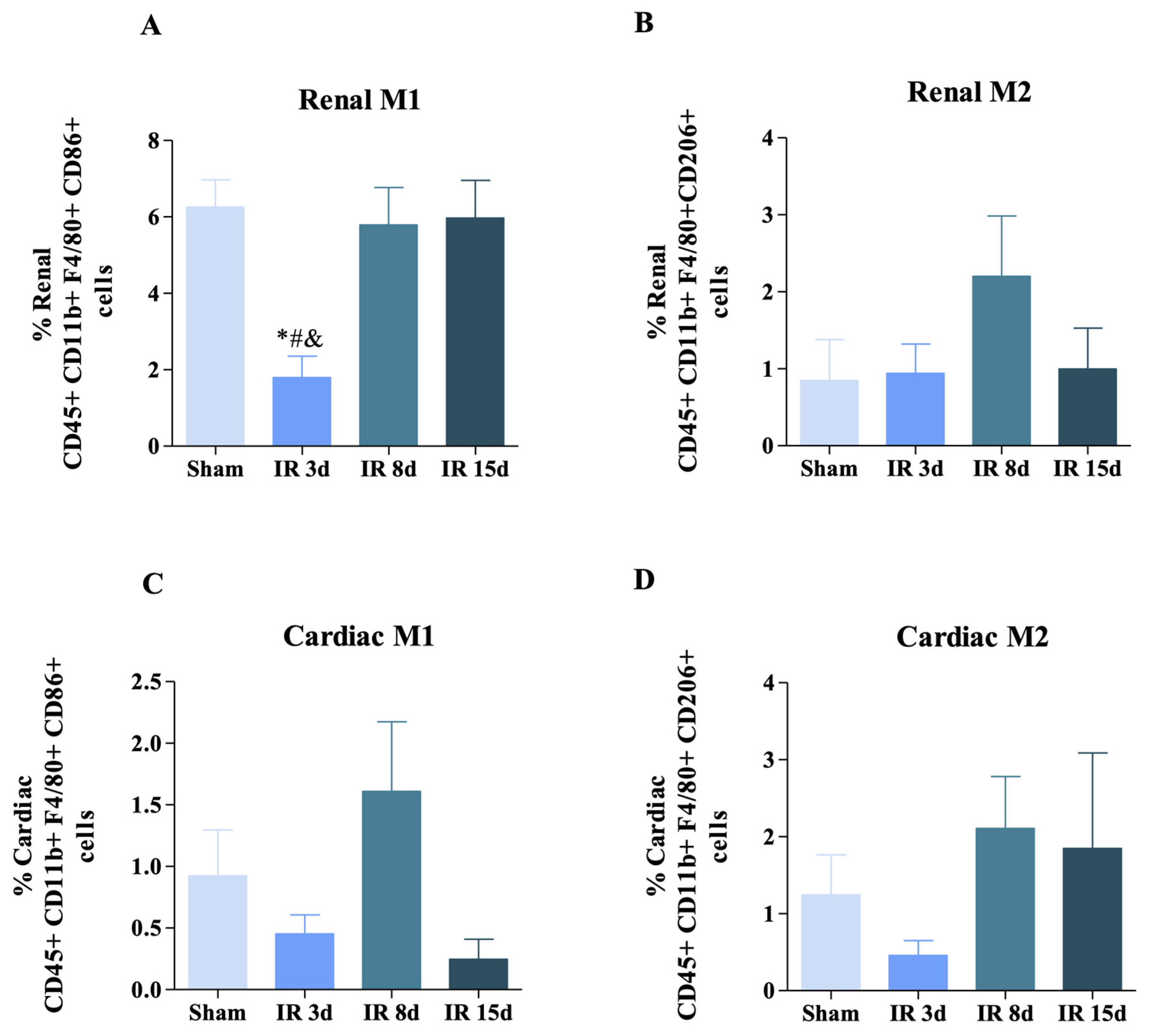
3.2. Renal Ischemic Injury Induces Macrophage Populations in the Renal and Cardiac Tissue
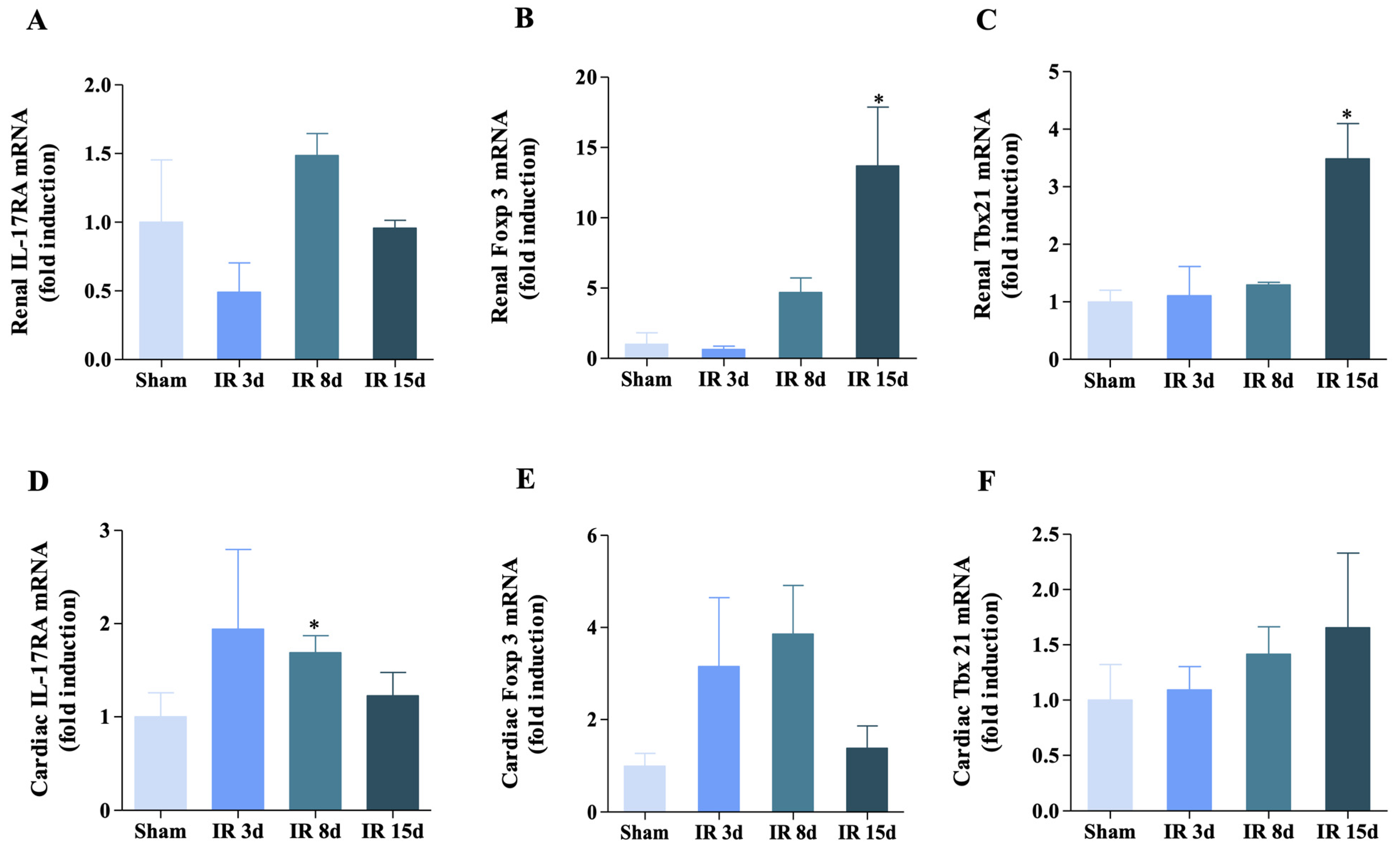
3.3. Gene Expression of IL-17RA, Foxp3, and Tbx21 Was Modified for Renal I/R in the Renal and Cardiac Tissue
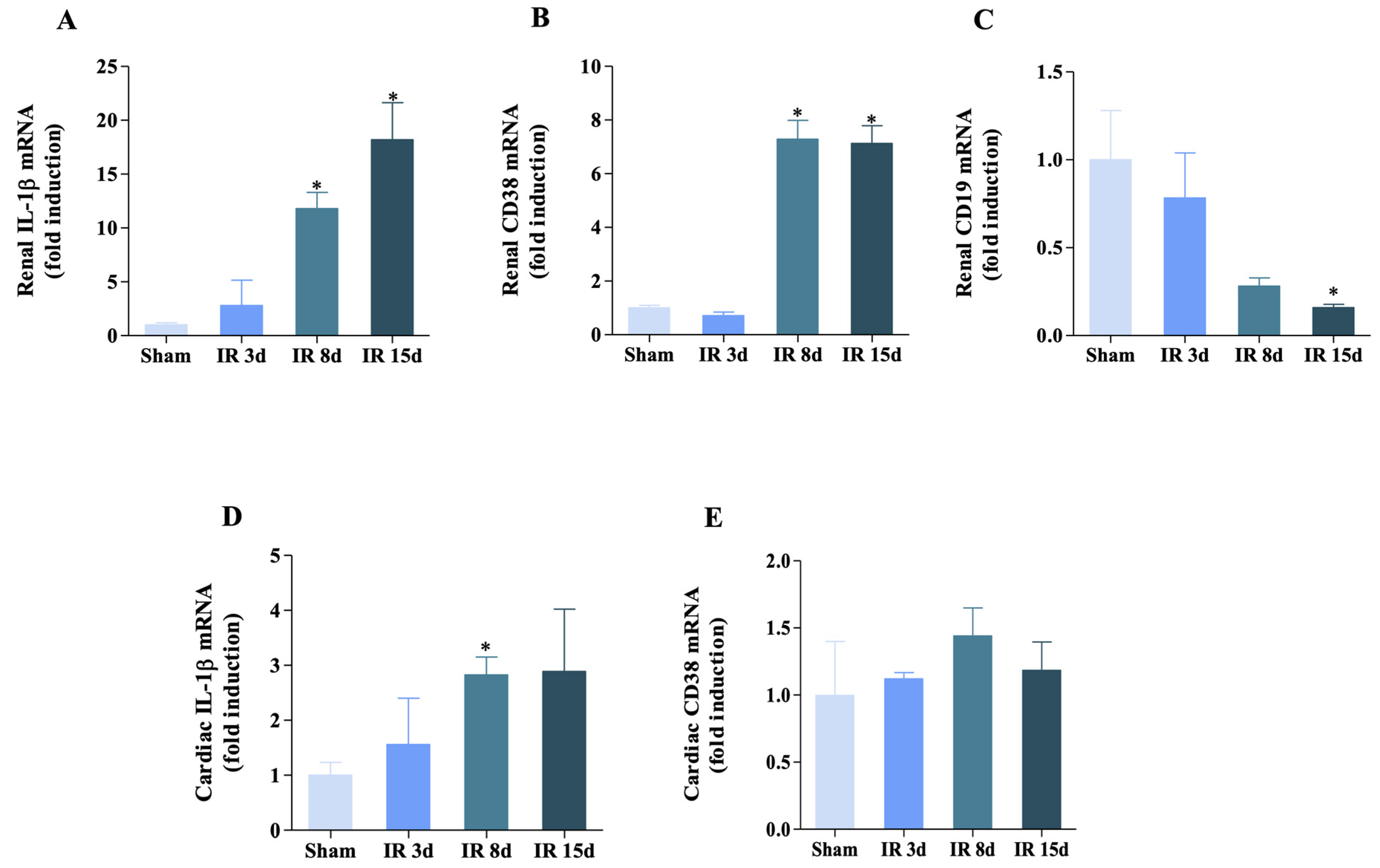
3.4. Inflammatory Status Is Modulated in Both Heart and Kidney
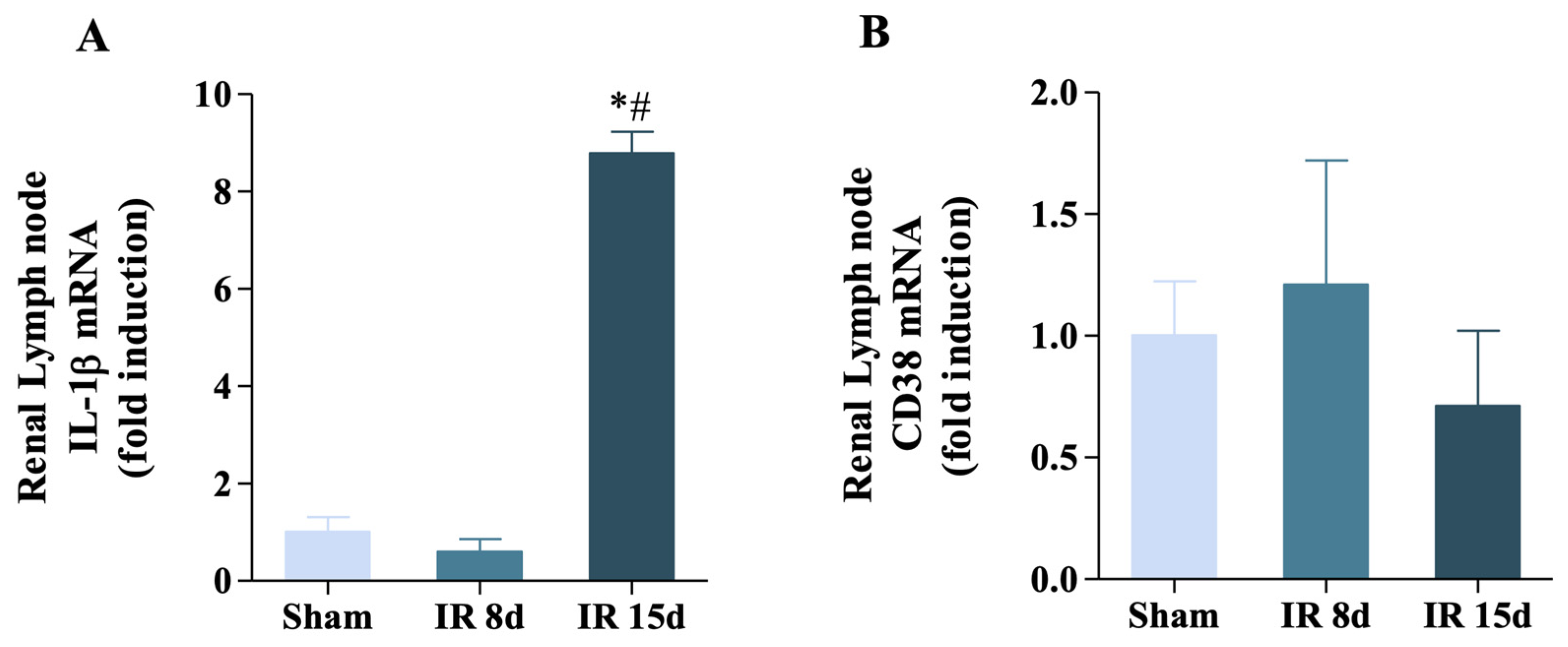
3.5. Renal Ischemia/Reperfusion Was Able to Increase IL-1β Expression but Not CD38 in the Renal Lymph Node
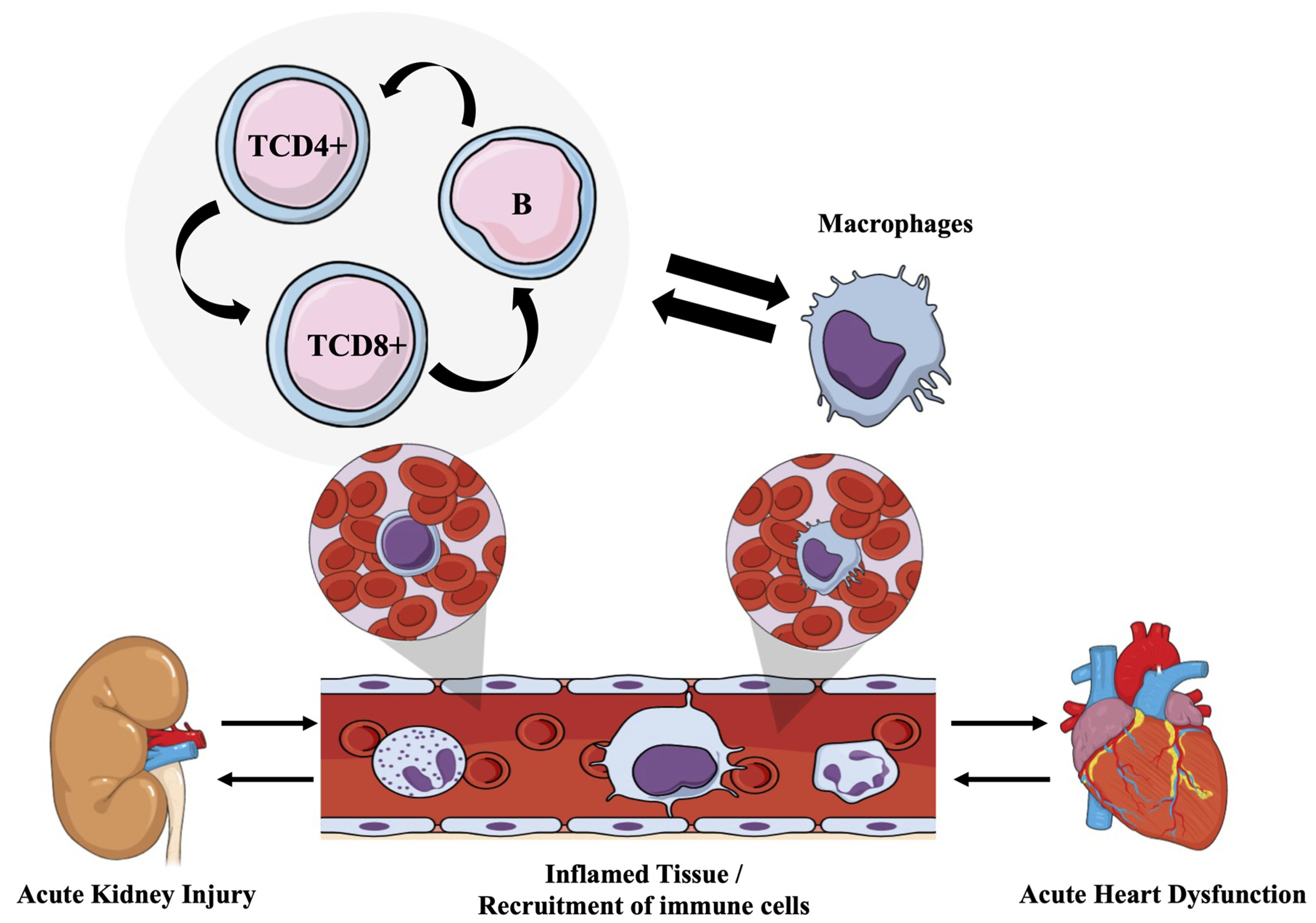
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santos-Zas, I.; Lemarie, J.; Tedgui, A.; Ait-Oufella, H. Adaptive Immune Responses Contribute to Post-ischemic Cardiac Remodeling. Front. Cardiovasc. Med. 2018, 5, 198. [Google Scholar] [CrossRef]
- Alarcon, M.M.L.; Trentin-Sonoda, M.; Panico, K.; Schleier, Y.; Duque, T.; Moreno-Loaiza, O.; de Yurre, A.R.; Ferreira, F.; Caio-Silva, W.; Coury, P.R.; et al. Cardiac arrhythmias after renal I/R depend on IL-1beta. J. Mol. Cell. Cardiol. 2019, 131, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Neres-Santos, R.S.; Junho, C.V.C.; Panico, K.; Caio-Silva, W.; Pieretti, J.C.; Tamashiro, J.A.; Seabra, A.B.; Ribeiro, C.A.J.; Carneiro-Ramos, M.S. Mitochondrial Dysfunction in Cardiorenal Syndrome 3: Renocardiac Effect of Vitamin C. Cells 2021, 10, 3029. [Google Scholar] [CrossRef]
- Fu, Y.; Xiang, Y.; Li, H.; Chen, A.; Dong, Z. Inflammation in kidney repair: Mechanism and therapeutic potential. Pharmacol. Ther. 2022, 237, 108240. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015, 11, 88–101. [Google Scholar] [CrossRef]
- Di Lullo, L.; Bellasi, A.; Barbera, V.; Russo, D.; Russo, L.; Di Iorio, B.; Cozzolino, M.; Ronco, C. Pathophysiology of the cardio-renal syndromes types 1–5: An uptodate. Indian Heart J. 2017, 69, 255–265. [Google Scholar] [CrossRef]
- Meng, X.; Jin, J.; Lan, H.Y. Driving role of macrophages in transition from acute kidney injury to chronic kidney disease. Chin. Med. J. 2022, 135, 757–766. [Google Scholar] [CrossRef]
- Baek, J.H. The Impact of Versatile Macrophage Functions on Acute Kidney Injury and Its Outcomes. Front. Physiol. 2019, 10, 1016. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jang, H.R. Role of T cells in ischemic acute kidney injury and repair. Korean J. Intern. Med. 2022, 37, 534–550. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Li, L.; Okusa, M.D. Inflammation in acute kidney injury. Nephron Exp. Nephrol. 2008, 109, e102–e107. [Google Scholar] [CrossRef]
- Breton, S.; Battistone, M.A. Unexpected Participation of Intercalated Cells in Renal Inflammation and Acute Kidney Injury. Nephron 2022, 146, 268–273. [Google Scholar] [CrossRef]
- Ascon, D.B.; Lopez-Briones, S.; Liu, M.; Ascon, M.; Savransky, V.; Colvin, R.B.; Soloski, M.J.; Rabb, H. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J. Immunol. 2006, 177, 3380–3387. [Google Scholar] [CrossRef]
- Hochegger, K.; Schatz, T.; Eller, P.; Tagwerker, A.; Heininger, D.; Mayer, G.; Rosenkranz, A.R. Role of α/β and γ/δ T cells in renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2007, 293, F741–F747. [Google Scholar] [CrossRef]
- Liu, M.; Chien, C.C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774. [Google Scholar] [CrossRef]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef]
- Jang, H.R.; Gandolfo, M.T.; Ko, G.J.; Satpute, S.R.; Racusen, L.; Rabb, H. B cells limit repair after ischemic acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 654–665. [Google Scholar] [CrossRef]
- Linfert, D.; Chowdhry, T.; Rabb, H. Lymphocytes and ischemia-reperfusion injury. Transplant. Rev. 2009, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Burne-Taney, M.J.; Yokota, N.; Rabb, H. Persistent renal and extrarenal immune changes after severe ischemic injury. Kidney Int. 2005, 67, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xia, N.; Cheng, X. Regulatory T Cells in Chronic Heart Failure. Front. Immunol. 2021, 12, 732794. [Google Scholar] [CrossRef]
- Rurik, J.G.; Aghajanian, H.; Epstein, J.A. Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ. Res. 2021, 128, 1766–1779. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rosenzweig, R.; Asalla, S.; Nehra, S.; Prabhu, S.D.; Bansal, S.S. TNFR1 Contributes to Activation-Induced Cell Death of Pathological CD4(+) T Lymphocytes During Ischemic Heart Failure. JACC Basic Transl. Sci. 2022, 7, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Reyes, A.M.; Youker, K.A.; Trevino, A.R.; Celis, R.; Hamilton, D.J.; Flores-Arredondo, J.H.; Orrego, C.M.; Bhimaraj, A.; Estep, J.D.; Torre-Amione, G. Full Expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis. J. Am. Heart Assoc. 2016, 5, e002484. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Lin, Q.Y.; Wang, L.; Xie, X.; Zhang, Y.L.; Li, H.H. Rituximab prevents and reverses cardiac remodeling by depressing B cell function in mice. Biomed. Pharmacother. 2019, 114, 108804. [Google Scholar] [CrossRef] [PubMed]
- Feitoza, C.Q.; Goncalves, G.M.; Semedo, P.; Cenedeze, M.A.; Pinheiro, H.S.; Beraldo, F.C.; dos Santos, O.F.; Teixeira Vde, P.; dos Reis, M.A.; Mazzali, M.; et al. Inhibition of COX 1 and 2 prior to renal ischemia/reperfusion injury decreases the development of fibrosis. Mol. Med. 2008, 14, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef]
- Dinarello, C.A. The role of the interleukin-1-receptor antagonist in blocking inflammation mediated by interleukin-1. N. Engl. J. Med. 2000, 343, 732–734. [Google Scholar] [CrossRef]
- Sandoval-Montes, C.; Santos-Argumedo, L. CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J. Leukoc. Biol. 2005, 77, 513–521. [Google Scholar] [CrossRef]
- Choe, C.U.; Lardong, K.; Gelderblom, M.; Ludewig, P.; Leypoldt, F.; Koch-Nolte, F.; Gerloff, C.; Magnus, T. CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLoS ONE 2011, 6, e19046. [Google Scholar] [CrossRef]
- Miyazawa, S.; Watanabe, H.; Miyaji, C.; Hotta, O.; Abo, T. Leukocyte accumulation and changes in extra-renal organs during renal ischemia reperfusion in mice. J. Lab. Clin. Med. 2002, 139, 269–278. [Google Scholar] [CrossRef]
- Chen, J.; Hartono, J.R.; John, R.; Bennett, M.; Zhou, X.J.; Wang, Y.; Wu, Q.; Winterberg, P.D.; Nagami, G.T.; Lu, C.Y. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 2011, 80, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Noel, S.; Kurzhagen, J.T.; Sadasivam, M.; Pierorazio, P.M.; Arend, L.J.; Hamad, A.R.; Rabb, H. CD4(+) T Cell-Derived NGAL Modifies the Outcome of Ischemic Acute Kidney Injury. J. Immunol. 2020, 204, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Ito, M.; Komai, K.; Iizuka-Koga, M.; Matsuo, K.; Nakayama, T.; Yoshie, O.; Amano, K.; Nishimasu, H.; Nureki, O.; et al. Kidney GATA3(+) regulatory T cells play roles in the convalescence stage after antibody-mediated renal injury. Cell. Mol. Immunol. 2021, 18, 1249–1261. [Google Scholar] [CrossRef]
- Gandolfo, M.T.; Jang, H.R.; Bagnasco, S.M.; Ko, G.J.; Agreda, P.; Satpute, S.R.; Crow, M.T.; King, L.S.; Rabb, H. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009, 76, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, Y.; Feng, T.; Li, H.; Xiong, Y. Gal-9/Tim-3 signaling pathway activation suppresses the generation of Th17 cells and promotes the induction of Foxp3+regulatory T cells in renal ischemia-reperfusion injury. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Madjid, M.; Awan, I.; Willerson, J.T.; Casscells, S.W. Leukocyte count and coronary heart disease: Implications for risk assessment. J. Am. Coll. Cardiol. 2004, 44, 1945–1956. [Google Scholar] [CrossRef]
- Rieckmann, M.; Delgobo, M.; Gaal, C.; Buchner, L.; Steinau, P.; Reshef, D.; Gil-Cruz, C.; Horst, E.N.T.; Kircher, M.; Reiter, T.; et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J. Clin. Investig. 2019, 129, 4922–4936. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.S.; Ruhrberg, C.; Cantley, L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Harris, D.C.; Wang, Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology 2015, 30, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Luebbe, J.; Kilian, C.; Riedel, J.H.; Hiekmann, S.; Asada, N.; Ginsberg, P.; Robben, L.; Song, N.; Kaffke, A.; et al. IL-17 Receptor C Signaling Controls CD4(+) TH17 Immune Responses and Tissue Injury in Immune-Mediated Kidney Diseases. J. Am. Soc. Nephrol. 2021, 32, 3081–3098. [Google Scholar] [CrossRef]
- Lee, J.W.; Bae, E.; Kwon, S.H.; Yu, M.Y.; Cha, R.H.; Lee, H.; Kim, D.K.; Lee, J.P.; Ye, S.K.; Yoo, J.Y.; et al. Transcriptional modulation of the T helper 17/interleukin 17 axis ameliorates renal ischemia-reperfusion injury. Nephrol. Dial. Transplant. 2019, 34, 1481–1498. [Google Scholar] [CrossRef]
- Sampani, E.; Vagiotas, L.; Daikidou, D.V.; Nikolaidou, V.; Xochelli, A.; Kasimatis, E.; Lioulios, G.; Dimitriadis, C.; Fylaktou, A.; Papagianni, A.; et al. End stage renal disease has an early and continuous detrimental effect on regulatory T cells. Nephrology 2022, 27, 281–287. [Google Scholar] [CrossRef]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef]
- Liao, Y.H.; Xia, N.; Zhou, S.F.; Tang, T.T.; Yan, X.X.; Lv, B.J.; Nie, S.F.; Wang, J.; Iwakura, Y.; Xiao, H.; et al. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J. Am. Coll. Cardiol. 2012, 59, 420–429. [Google Scholar] [CrossRef]
- Yan, X.; Shichita, T.; Katsumata, Y.; Matsuhashi, T.; Ito, H.; Ito, K.; Anzai, A.; Endo, J.; Tamura, Y.; Kimura, K.; et al. Deleterious effect of the IL-23/IL-17A axis and γδT cells on left ventricular remodeling after myocardial infarction. J. Am. Heart Assoc. 2012, 1, e004408. [Google Scholar] [CrossRef]
- Li, H.; Chen, C.; Wang, D.W. Inflammatory Cytokines, Immune Cells, and Organ Interactions in Heart Failure. Front. Physiol. 2021, 12, 695047. [Google Scholar] [CrossRef]
- Kim, J.H.; Ha, I.S.; Hwang, C.I.; Lee, Y.J.; Kim, J.; Yang, S.H.; Kim, Y.S.; Cao, Y.A.; Choi, S.; Park, W.Y. Gene expression profiling of anti-GBM glomerulonephritis model: The role of NF-kappaB in immune complex kidney disease. Kidney Int. 2004, 66, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-κB signaling suppression. Cell. Signal. 2018, 42, 249–258. [Google Scholar] [CrossRef]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 2020, 12, 11325–11336. [Google Scholar] [CrossRef] [PubMed]
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Zhang, Q.; Wei, Q.; Lin, J.; Jia, J.; Zhang, J.; Yan, T.; Lv, Y.; Jiang, X.; et al. CD38 Causes Autophagic Flux Inhibition and Cardiac Dysfunction Through a Transcriptional Inhibition Pathway Under Hypoxia/Ischemia Conditions. Front. Cell Dev. Biol. 2020, 8, 191. [Google Scholar] [CrossRef]
- Boslett, J.; Hemann, C.; Christofi, F.L.; Zweier, J.L. Characterization of CD38 in the major cell types of the heart: Endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am. J. Physiol. Cell Physiol. 2018, 314, C297–C309. [Google Scholar] [CrossRef] [PubMed]
- Francipane, M.G. Renal organogenesis in the lymph node microenvironment. In Regenerative Nephrology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 17–25. [Google Scholar] [CrossRef]
- Schineis, P.; Runge, P.; Halin, C. Cellular traffic through afferent lymphatic vessels. Vasc. Pharmacol. 2019, 112, 31–41. [Google Scholar] [CrossRef]
- Lukacs-Kornek, V.; Burgdorf, S.; Diehl, L.; Specht, S.; Kornek, M.; Kurts, C. The kidney-renal lymph node-system contributes to cross-tolerance against innocuous circulating antigen. J. Immunol. 2008, 180, 706–715. [Google Scholar] [CrossRef]
- Duncan, L.M.; Meegan, L.S.; Unanue, E.R. IL-1 gene expression in lymphoid tissues. J. Immunol. 1991, 146, 565–571. [Google Scholar] [CrossRef]
- Doisne, J.M.; Soulard, V.; Becourt, C.; Amniai, L.; Henrot, P.; Havenar-Daughton, C.; Blanchet, C.; Zitvogel, L.; Ryffel, B.; Cavaillon, J.M.; et al. Cutting edge: Crucial role of IL-1 and IL-23 in the innate IL-17 response of peripheral lymph node NK1.1- invariant NKT cells to bacteria. J. Immunol. 2011, 186, 662–666. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′->3′) | Reverse (5′->3′) |
| Ppia (cyclophilin A) | AGCATACAGGTCCTGGCATC | AGCTGTCCACAGTCGGAAAT |
| IL-17ra | GTGGCGGTTTTCCTTCAGCCACTTTGTG | GATGCTGTGTGTCCAAGGTCTCCACAGT |
| Foxp3 | AATAGTTCCTTCCCAGAGTTC | GGGTGGCATAGGTGAAAG |
| Tbx21 | TGTGTTAATCTCTGACCTGAA | CACCTGAGTCTTCTCTGTT |
| Cd19 | ACTAGCCTGGACTTCGTTAG | GGTTCTAGGTCGTCAGACTTAT |
| Cd38 | CGAAGGAGCTTCCAGTAACG | GGTTCTAGGTCGTCAGACTTAT |
| IL-1β | AGTTGACGGACCCCAAAAGA | GCTCTTGTTGATGTGCTGCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernier, I.C.S.; Neres-Santos, R.S.; Andrade-Oliveira, V.; Carneiro-Ramos, M.S. Immune Cells Are Differentially Modulated in the Heart and the Kidney during the Development of Cardiorenal Syndrome 3. Cells 2023, 12, 605. https://doi.org/10.3390/cells12040605
Vernier ICS, Neres-Santos RS, Andrade-Oliveira V, Carneiro-Ramos MS. Immune Cells Are Differentially Modulated in the Heart and the Kidney during the Development of Cardiorenal Syndrome 3. Cells. 2023; 12(4):605. https://doi.org/10.3390/cells12040605
Chicago/Turabian StyleVernier, Imara Caridad Stable, Raquel Silva Neres-Santos, Vinicius Andrade-Oliveira, and Marcela Sorelli Carneiro-Ramos. 2023. "Immune Cells Are Differentially Modulated in the Heart and the Kidney during the Development of Cardiorenal Syndrome 3" Cells 12, no. 4: 605. https://doi.org/10.3390/cells12040605