
Transcriptional Regulation during Aberrant Activation of NF-κB Signalling in Cancer

Abstract
:1. Introduction
2. NF-κB: One of the Key Factors Linking Inflammation in Cancer
3. Factors Contributing to the Hyperactivation of NF-κB Pathway in Cancers
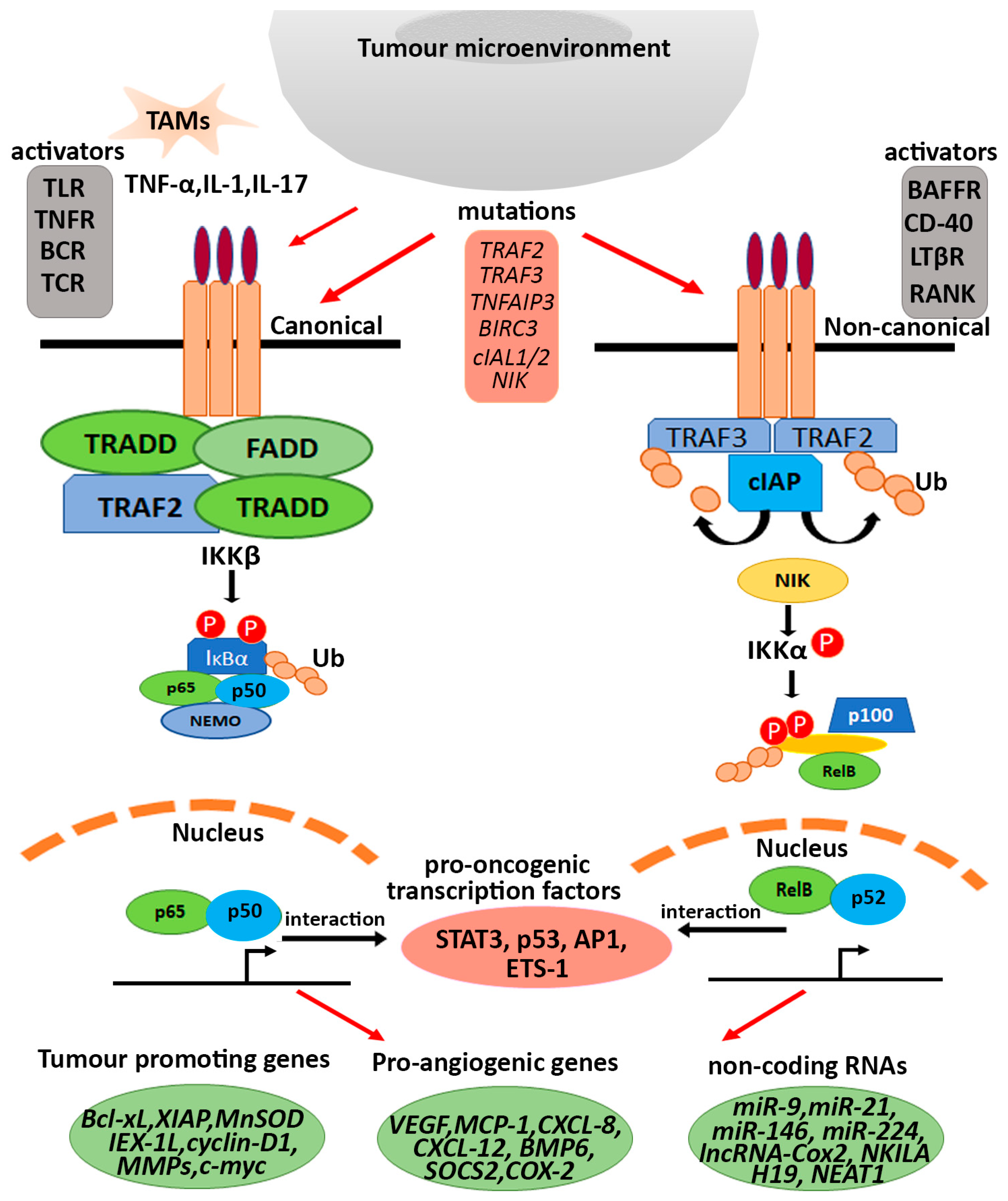
3.1. Cancer Associated Immune Signalling Molecules
3.2. Intrinsic Mutations and Post Translational Modifications (PTMs)

{kind=link}
{kind=link}
| Factors/Regulators of NF-κB Pathway | Type of Mutation/Chromosomal Translocation | Cancer Type | Response | References |
|---|---|---|---|---|
| TRAF3 | Bi-allelic deletion at 14q32 | MM | Increased p52/p100 ratios | [91,177] |
| TRAF2 | Bi-allelic deletion at 9q34 | MM | Increased p52/p100 ratios | [91,177] |
| CYLD | Bi-allelic deletion at 16q12 | MM | Increased p52/p100 ratios | [91,177] |
| cIAP1/cIAP2 | Bi-allelic deletion at 11q22 | MM | Increased p52/p100 ratios | [91,177] |
| NIK | t(17;22) translocation, IgH translocation or amplification | MM | Overexpression of NIK | [93] |
| LTBR | amplification of the entire 12p chromosome arm | MM | Activatory | [91] |
| NF-κB2 | t(10;14)(q24;q32) t(10q24) | MALT Lymphoma, DLBCL | Activatory, Enhanced expression of NF-κB2 gene and protein | [15,91,153] |
| AP12-MALT1 | t(11;18)(q21;q21) | B-cell Lymphoma, MALT lymphoma | Activatory | [154,155] |
| c-Rel | t(2p 13-15) | DLBCL, B-cell lymphoma, Follicular large cell lymphoma | Enhanced amplification of c-Rel gene | [120,121,149,159,161] |
| RelA | (11q13) site with t(11;14)(q13;q32) | NHL, Diffuse large cell lymphoma, Squamous carcinoma of head and neck, Breast cancer | Activatory | [159,162] |
| Bcl3 | t(14;19)(q32;q13.1) | B-cell leukaemia | Activatory | [163] |
3.3. Epigenetic Modification in the Component(s) of NF-κB Pathway
4. Double Edged Role of NF-κB from Immunosurveillance to Pro-Tumorigenic Functions
5. Aberrant NF-κB Activation Driven Expression of Tumour Promoting Genes
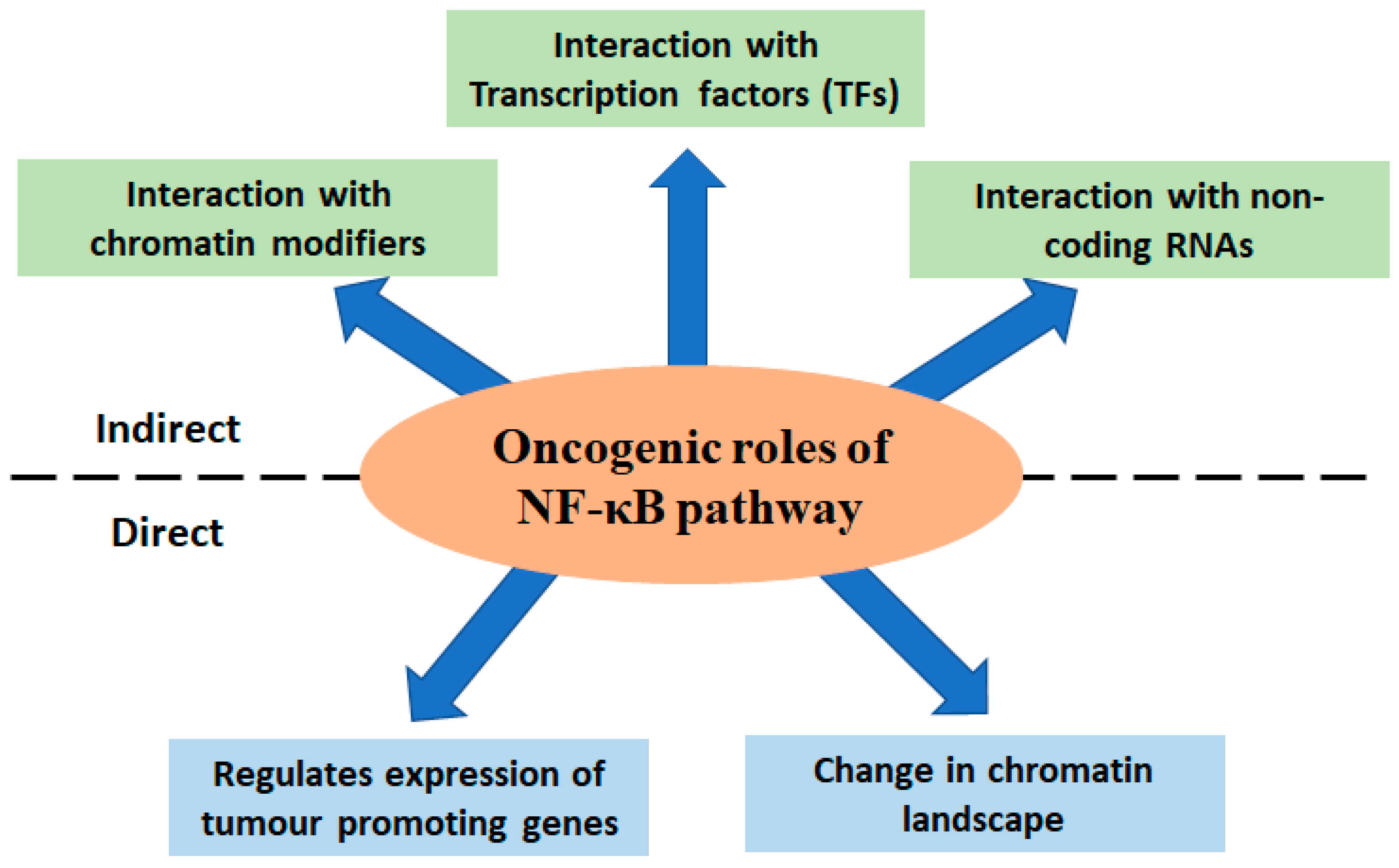
6. Different Modes of Deregulated NF-κB Signalling in Cancer
6.1. Interaction with Transcription Factors
6.2. Effect of Pro Tumorigenic Non-Coding RNAs
7. Role of NF-κB Signalling in Shaping the Cancer Cell Chromatin Landscape
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; Ghosh, S. Signal transduction through NF-κB. Immunol. Today 1998, 19, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-κB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational biogenesis of NF-κB p50 by the 26S proteasome. Cell 1998, 92, 819–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caamano, J.; Hunter, C.A. NF-κB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sif, S.; Gilmore, T.D. NF-kappa B p100 is one of the high-molecular-weight proteins complexed with the v-Rel oncoprotein in transformed chicken spleen cells. J. Virol. 1993, 67, 7612–7617. [Google Scholar] [CrossRef] [PubMed]
- Ryseck, R.-P.; Bull, P.; Takamiya, M.; Bours, V.; Siebenlist, U.; Dobrzanski, P.; Bravo, R. RelB, a new Rel family transcription activator that can interact with p50-NF-kappa B. Mol. Cell. Biol. 1992, 12, 674–684. [Google Scholar] [PubMed] [Green Version]
- Chen, I.; Wilhelmsen, K.; Temin, H.M. Structure and expression of c-rel, the cellular homolog to the oncogene of reticuloendotheliosis virus strain T. J. Virol. 1983, 45, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmsen, K.C.; Eggleton, K.; Temin, H.M. Nucleic acid sequences of the oncogene v-rel in reticuloendotheliosis virus strain T and its cellular homolog, the proto-oncogene c-rel. J. Virol. 1984, 52, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Gifford, A.M.; Riviere, L.R.; Tempst, P.; Nolan, G.P.; Baltimore, D. Cloning of the p50 DNA binding subunit of NF-κB: Homology to rel and dorsal. Cell 1990, 62, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Duyne, G.V.; Ghosh, S.; Sigler, P.B. Structure of NF-κB p50 homodimer bound to a κB site. Nature 1995, 373, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Villalobos, J.; Burd, P.R.; Kelly, K.; Siebenlist, U. Cloning of a mitogen-inducible gene encoding a κB DNA-binding protein with homology to the rel oncogene and to cell-cycle motifs. Nature 1990, 348, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Hatada, E.N.; Hohmann, H.-P.; Haiker, M.; Bartsch, C.; Röthlisberger, U.; Lahm, H.-W.; Schlaeger, E.J.; Van Loon, A.; Scheidereit, C. Cloning of the DNA-binding subunit of human nuclear factor kappa B: The level of its mRNA is strongly regulated by phorbol ester or tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1991, 88, 966–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, A.; Chang, C.-C.; Lombardi, L.; Salina, M.; Corradini, P.; Maiolo, A.T.; Chaganti, R.; Dalla-Favera, R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell 1991, 67, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.M.; Liptay, S.; Betts, J.C.; Nabel, G.J. Structural and functional analysis of NF-kappa B. Determinants of DNA binding specificity and protein interaction. J. Biol. Chem. 1994, 269, 32162–32167. [Google Scholar] [CrossRef]
- MERCURIO, F.; DIDONATO, J.; ROSETTE, C.; Karin, M. Molecular cloning and characterization of a novel rel/NF-χB family member displaying structural and functional homology to NF-χB p50/p105. DNA Cell Biol. 1992, 11, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Burd, P.; Brown, K.; Villalobos, J.; Park, S.; Ryseck, R.P.; Bravo, R.; Kelly, K.; Siebenlist, U. A novel mitogen-inducible gene product related to p50/p105-NF-kappa B participates in transactivation through a kappa B site. Mol. Cell. Biol. 1992, 12, 685–695. [Google Scholar]
- Ruben, S.M.; Dillon, P.J.; Schreck, R.; Henkel, T.; Chen, C.-H.; Maher, M.; Baeuerle, P.A.; Rosen, C.A. Isolation of a rel-related Human cDNAThat Potentially Encodes the 65-kD Subunit of NF-κB. Science 1991, 251, 1490–1493. [Google Scholar] [CrossRef]
- Ryseck, R.-P.; Novotny, J.; Bravo, R. Characterization of elements determining the dimerization properties of RelB and p50. Mol. Cell. Biol. 1995, 15, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Bours, V.; Azarenko, V.; Dejardin, E.; Siebenlist, U. Human RelB (I-Rel) functions as a kappa B site-dependent transactivating member of the family of Rel-related proteins. Oncogene 1994, 9, 1699–1702. [Google Scholar]
- Inoue, J.-I.; Kerr, L.D.; Kakizuka, A.; Verma, I.M. IκBγ, a 70 kd protein identical to the C-terminal half of p110 NF-κB: A new member of the IκB family. Cell 1992, 68, 1109–1120. [Google Scholar] [CrossRef]
- Gerondakis, S.; Morrice, N.; Richardson, I.; Wettenhall, R.; Fecondo, J.; Grumont, R. The activity of a 70 kilodalton I kappa B molecule identical to the carboxyl terminus of the p105 NF-kappa B precursor is modulated by protein kinase A. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1993, 4, 617–627. [Google Scholar]
- Grumont, R.J.; Gerondakis, S. Alternative splicing of RNA transcripts encoded by the murine p105 NF-kappa B gene generates I kappa B gamma isoforms with different inhibitory activities. Proc. Natl. Acad. Sci. USA 1994, 91, 4367–4371. [Google Scholar] [CrossRef] [Green Version]
- Scheidereit, C. IκB kinase complexes: Gateways to NF-κB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkB kinase complex (IKK) contains two kinase subunits, IKKa and IKKb, necessary for IkB phosphorylation and NF-kB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. The Rel/NF-κB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [CrossRef] [Green Version]
- Shindo, M.; Nakano, H.; Sakon, S.; Yagita, H. Assignment of (I kappa B) kinase (beta)(IKBKB) to human chromosome band 8p12--> p11 by in situ hybridization. Cytogenet. Genome Res. 1998, 82, 32. [Google Scholar] [CrossRef]
- Ling, L.; Cao, Z.; Goeddel, D.V. NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA 1998, 95, 3792–3797. [Google Scholar] [CrossRef] [Green Version]
- Zabel, U.; Henkel, T.; Silva, M.S.; Baeuerle, P.A. Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus. EMBO J. 1993, 12, 201–211. [Google Scholar] [CrossRef]
- Zabel, U.; Baeuerle, P.A. Purified human IκB can rapidly dissociate the complex of the NF-κB transcription factor with its cognate DNA. Cell 1990, 61, 255–265. [Google Scholar] [CrossRef]
- Arenzana-Seisdedos, F.; Turpin, P.; Rodriguez, M.; Thomas, D.; Hay, R.T.; Virelizier, J.-L.; Dargemont, C. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell Sci. 1997, 110, 369–378. [Google Scholar] [CrossRef]
- Zhang, Q.; Didonato, J.A.; Karin, M.; McKeithan, T.W. BCL3 encodes a nuclear protein which can alter the subcellular location of NF-kappa B proteins. Mol. Cell. Biol. 1994, 14, 3915–3926. [Google Scholar] [PubMed] [Green Version]
- Hatada, E.N.; Nieters, A.; Wulczyn, F.G.; Naumann, M.; Meyer, R.; Nucifora, G.; McKeithan, T.W.; Scheidereit, C. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc. Natl. Acad. Sci. USA 1992, 89, 2489–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmody, R.J.; Ruan, Q.; Palmer, S.; Hilliard, B.; Chen, Y.H. Negative regulation of toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science 2007, 317, 675–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, V.Y.-F.; Huang, W.; Asagiri, M.; Spann, N.; Hoffmann, A.; Glass, C.; Ghosh, G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2012, 2, 824–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Deng, L.; Wang, H.; Wang, V.Y.-F. Atypical IκB Bcl3 enhances the generation of the NF-κB p52 homodimer. Front. Cell Dev. Biol. 2022, 10, 930619. [Google Scholar] [CrossRef] [PubMed]
- Totzke, G.; Essmann, F.; Pohlmann, S.; Lindenblatt, C.; Jänicke, R.U.; Schulze-Osthoff, K. A novel member of the IκB family, human IκB-ζ, inhibits transactivation of p65 and its DNA binding. J. Biol. Chem. 2006, 281, 12645–12654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, S.; Ito, H.; Miyajima, A. IκBη, a nuclear IκB protein, positively regulates the NF-κB–mediated expression of proinflammatory cytokines. Proc. Natl. Acad. Sci. USA 2010, 107, 11924–11929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, E.; Schmitz, I.; Marissen, W.E.; Osborn, S.L.; Touma, M.; Sasada, T.; Reche, P.A.; Tibaldi, E.V.; Hussey, R.E.; Kruisbeek, A.M. Peptide-induced negative selection of thymocytes activates transcription of an NF-κB inhibitor. Mol. Cell 2002, 9, 637–648. [Google Scholar] [CrossRef]
- Schuster, M.; Glauben, R.; Plaza-Sirvent, C.; Schreiber, L.; Annemann, M.; Floess, S.; Kühl, A.A.; Clayton, L.K.; Sparwasser, T.; Schulze-Osthoff, K. IκBNS protein mediates regulatory T cell development via induction of the Foxp3 transcription factor. Immunity 2012, 37, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: Molecular basis and biological significance. Oncogene 2006, 25, 6731–6748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barré, B.; Coqueret, O.; Perkins, N.D. Regulation of activity and function of the p52 NF-κB subunit following DNA damage. Cell Cycle 2010, 9, 4795–4804. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Osborn, L.; Kunkel, S.; Nabel, G.J. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 1989, 86, 2336–2340. [Google Scholar] [CrossRef] [Green Version]
- Duh, E.J.; Maury, W.J.; Folks, T.M.; Fauci, A.S.; Rabson, A.B. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. USA 1989, 86, 5974–5978. [Google Scholar] [CrossRef] [Green Version]
- Lowenthal, J.W.; Ballard, D.W.; Böhnlein, E.; Greene, W.C. Tumor necrosis factor alpha induces proteins that bind specifically to kappa B-like enhancer elements and regulate interleukin 2 receptor alpha-chain gene expression in primary human T lymphocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 2331–2335. [Google Scholar] [CrossRef] [Green Version]
- Awane, M.; Andres, P.G.; Li, D.J.; Reinecker, H.-C. NF-κB-inducing kinase is a common mediator of IL-17-, TNF-α-, and IL-1β-induced chemokine promoter activation in intestinal epithelial cells. J. Immunol. 1999, 162, 5337–5344. [Google Scholar] [CrossRef]
- Elewaut, D.; DiDonato, J.A.; Kim, J.M.; Truong, F.; Eckmann, L.; Kagnoff, M.F. NF-κB is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J. Immunol. 1999, 163, 1457–1466. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. Biol. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Coope, H.; Atkinson, P.; Huhse, B.; Belich, M.; Janzen, J.; Holman, M.; Klaus, G.; Johnston, L.; Ley, S. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.-W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Yan, M.; Seshasayee, D.; Wang, H.; Lee, W.; French, D.M.; Grewal, I.S.; Cochran, A.G.; Gordon, N.C.; Yin, J. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity 2002, 17, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.-C.J.S. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.-C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Xiao, G.; Fong, A.; Sun, S.-C. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Cvijic, M.E.; Fong, A.; Harhaj, E.W.; Uhlik, M.T.; Waterfield, M.; Sun, S.C. Retroviral oncoprotein Tax induces processing of NF-κB2/p100 in T cells: Evidence for the involvement of IKKα. EMBO J. 2001, 20, 6805–6815. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, G.; Wang, V.Y.-F. Origin of the functional distinctiveness of NF-κB/p52. Front. Cell Dev. Biol. 2021, 9, 3338. [Google Scholar] [CrossRef]
- Miyamoto, S.; Schmitt, M.J.; Verma, I.M. Qualitative changes in the subunit composition of kappa B-binding complexes during murine B-cell differentiation. Proc. Natl. Acad. Sci. USA 1994, 91, 5056–5060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lernbecher, T.; Müller, U.; Wirth, T. Distinct NF-κB/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature 1993, 365, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.W.; Rey, F.A.; Sodeoka, M.; Verdine, G.L.; Harrison, S.C. Structure of the NF-κB p50 homodimer bound to DNA. Nature 1995, 373, 311–317. [Google Scholar] [CrossRef]
- Cramer, P.; Larson, C.J.; Verdine, G.L.; Müller, C.W. Structure of the human NF-κB p52 homodimer-DNA complex at 2.1 Å resolution. EMBO J. 1997, 16, 7078–7090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-Q.; Ghosh, S.; Ghosh, G. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Biol. 1998, 5, 67–73. [Google Scholar] [CrossRef]
- Huang, D.-B.; Vu, D.; Ghosh, G. NF-κB RelB forms an intertwined homodimer. Structure 2005, 13, 1365–1373. [Google Scholar] [CrossRef] [Green Version]
- Driessler, F.; Venstrom, K.; Sabat, R.; Asadullah, K.; Schottelius, A.J.C. Molecular mechanisms of interleukin-10-mediated inhibition of NF-κ B activity: A role for p50. Clin. Exp. Immunol. 2004, 135, 64–73. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Oakley, F.; Lin, F.; Packham, G.; Mann, D.A.; Mann, J. The NF-κB p50: p50: HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010, 53, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.K.; Dunn, L.L.; Rice, N.R. The PEST-like sequence of I kappa B alpha is responsible for inhibition of DNA binding but not for cytoplasmic retention of c-Rel or RelA homodimers. Mol. Cell. Biol. 1995, 15, 872–882. [Google Scholar] [CrossRef] [Green Version]
- Latimer, M.; Ernst, M.K.; Dunn, L.L.; Drutskaya, M.; Rice, N.R. The N-terminal domain of IκBα masks the nuclear localization signal (s) of p50 and c-Rel homodimers. Mol. Cell. Biol. 1998, 18, 2640–2649. [Google Scholar] [CrossRef] [Green Version]
- Basak, S.; Shih, V.F.-S.; Hoffmann, A. Generation and activation of multiple dimeric transcription factors within the NF-κB signaling system. Mol. Cell. Biol. 2008, 28, 3139–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.E.; Huang, D.-B.; Chen, Y.-Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Zhang, M.; Harhaj, E.W.; Sun, S.-C. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004, 279, 26243–26250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, S.J.; Albrecht, J.-C.; Giehler, F.; Kieser, A.; Sticht, H.; Biesinger, B. Noncanonical NF-κB activation by the oncoprotein Tio occurs through a nonconserved TRAF3-binding motif. Sci. Signal. 2013, 6, ra27. [Google Scholar] [CrossRef]
- He, J.Q.; Zarnegar, B.; Oganesyan, G.; Saha, S.K.; Yamazaki, S.; Doyle, S.E.; Dempsey, P.W.; Cheng, G. Rescue of TRAF3-null mice by p100 NF-κB deficiency. J. Exp. Med. 2006, 203, 2413–2418. [Google Scholar] [CrossRef] [Green Version]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.-c.; Mak, T.W. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.-L.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.D.; Hostager, B.S.; Bishop, G.A. Differential signaling and tumor necrosis factor receptor–associated factor (Traf) degradation mediated by Cd40 and the Epstein-Barr virus oncoprotein latent membrane protein 1 (Lmp1). J. Exp. Med. 2001, 193, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.-H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Zhang, M.; Sun, S.-C. β-TrCP binding and processing of NF-κB2/p100 involve its phosphorylation at serines 866 and 870. Cell. Signal. 2006, 18, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, J.C.; Nabel, G.J. Differential regulation of NF-kappaB2 (p100) processing and control by amino-terminal sequences. Mol. Cell. Biol. 1996, 16, 6363–6371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Lewis, C.E. NF-κB as a central regulator of macrophage function in tumors. J. Leukoc. Biol. 2010, 88, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejardin, E. The alternative NF-κB pathway from biochemistry to biology: Pitfalls and promises for future drug development. J Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef]
- Espinosa, L.; Bigas, A.; Mulero, M.C. Alternative nuclear functions for NF-κB family members. Am. J. Cancer Res. 2011, 1, 446. [Google Scholar] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer Res. 2013, 12, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yılmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative dissection and modeling of the NF-κB p100-p105 module reveals interdependent precursor proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Calado, D.P.; Wang, Z.; Fröhler, S.; Köchert, K.; Qian, Y.; Koralov, S.B.; Schmidt-Supprian, M.; Sasaki, Y.; Unitt, C. An oncogenic role for alternative NF-κB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep. 2015, 11, 715–726. [Google Scholar] [CrossRef] [Green Version]
- SHELDON, W.H.; JAMES, D.F. Cirrhosis following infectious hepatitis: A report of five cases, in two of which there was superimposed primary liver cell carcinoma. Arch. Intern. Med. 1948, 81, 666–689. [Google Scholar] [CrossRef]
- Walshe, J.; Wolff, H. Primary Carcinoma of the Liver following Viral Hepatitis. Report of Two Cases. Lancet 1952, 2, 1007–1010. [Google Scholar] [CrossRef]
- Steiner, P.; Davies, J. Cirrhosis and primary liver carcinoma in Uganda Africans. Br. J. Cancer 1957, 11, 523. [Google Scholar] [CrossRef] [Green Version]
- Higginson, J. The geographical pathology of primary liver cancer. Cancer Res. 1963, 23, 1624–1633. [Google Scholar]
- Pisani, P.; Parkin, D.M.; Muñoz, N.; Ferlay, J.J.C.E. Cancer and infection: Estimates of the attributable fraction in 1990. Cancer Epidemiol. Prev. Biomark. 1997, 6, 387–400. [Google Scholar]
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2001, 249, 61–74. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Gerondakis, S. The c-Rel transcription factor in development and disease. Genes Cancer Cell 2011, 2, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, T.D. The Rel/NF-κB/IκB signal transduction pathway and cancer. In Signal Transduction Cancer; Spinger: Berlin/Heidelberg, Germany, 2004; pp. 241–265. [Google Scholar] [CrossRef]
- Shattuck-Brandt, R.L.; Richmond, A. Enhanced degradation of I-κBα contributes to endogenous activation of NF-κB in Hs294T melanoma cells. Cancer Res. 1997, 57, 3032–3039. [Google Scholar] [PubMed]
- Charalambous, M.; Lightfoot, T.; Speirs, V.; Horgan, K.; Gooderham, N. Expression of COX-2, NF-κB-p65, NF-κB-p50 and IKKα in malignant and adjacent normal human colorectal tissue. Br. J. Cancer 2009, 101, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.S. Selective activation of NF-κB subunits in human breast cancer: Potential roles for NF-κB2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poma, P.; Labbozzetta, M.; D’Alessandro, N.; Notarbartolo, M. NF-κB is a potential molecular drug target in triple-negative breast cancers. Omics: A J. Integr. Biol. 2017, 21, 225–231. [Google Scholar] [CrossRef]
- Devanaboyina, M.; Kaur, J.; Whiteley, E.; Lin, L.; Einloth, K.; Morand, S.; Stanbery, L.; Hamouda, D.; Nemunaitis, J. NF-κB Signaling in Tumor Pathways Focusing on Breast and Ovarian Cancer. Oncol. Rev. 2022, 16, 10568. [Google Scholar] [CrossRef]
- Saitoh, Y.; Bruyn, V.J.M.; Uota, S.; Hasegawa, A.; Yamamoto, N.; Imoto, I.; Inazawa, J.; Yamaoka, S. Overexpression of NF-κB inducing kinase underlies constitutive NF-κB activation in lung cancer cells. Lung Cancer 2010, 70, 263–270. [Google Scholar] [CrossRef]
- Yang, J.; Splittgerber, R.; Yull, F.E.; Kantrow, S.; Ayers, G.D.; Karin, M.; Richmond, A. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J. Clin. Investig. 2010, 120, 2563–2574. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor κB activity is required for survival of activated B cell–like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Eluard, B.; Nuan-Aliman, S.; Faumont, N.; Collares, D.; Bordereaux, D.; Montagne, A.; Martins, I.; Cagnard, N.; Caly, M.; Taoui, O. The alternative RelB NF-κB subunit is a novel critical player in diffuse large B-cell lymphoma. Blood J. Am. Soc. Hematol. 2022, 139, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Morisaki, T.; Sasaki, N.; Nakano, K.; Mibu, R.; Tanaka, M.; Katano, M. Increased nuclear factor-kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res. 2004, 24, 675–682. [Google Scholar] [PubMed]
- Li, W.; Tan, D.; Zenali, M.J.; Brown, R.E. Constitutive activation of nuclear factor-kappa B (NF-kB) signaling pathway in fibrolamellar hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2010, 3, 238. [Google Scholar]
- Lee, D.W.; Ramakrishnan, D.; Valenta, J.; Parney, I.F.; Bayless, K.J.; Sitcheran, R. The NF-κB RelB protein is an oncogenic driver of mesenchymal glioma. PLoS ONE 2013, 8, e57489. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Wang, K.; Huang, R.; Liu, Y.; Zhang, Y.; Hu, H. RELB: A novel prognostic marker for glioblastoma as identified by population-based analysis. Oncol. Lett. 2019, 18, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, F.; Monjanel, H.; Fabre, C.; Billot, K.; Sapharikas, E.; Chereau, F.; Bordereaux, D.; Molina, T.J.; Avet-Loiseau, H.; Baud, V. Frequent engagement of RelB activation is critical for cell survival in multiple myeloma. PLoS ONE 2013, 8, e59127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Bergsagel, P.L.; Kuehl, W.M. Classical and/or alternative NF-κB pathway activation in multiple myeloma. Blood J. Am. Soc. Hematol. 2010, 115, 3541–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joos, S.; Otaño-Joos, M.I.; Ziegler, S.; Bruderlein, S.; Du Manoir, S.; Bentz, M.; Moller, P.; Lichter, P. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 1996, 87, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Barth, T.F.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Möller, P. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2003, 101, 3681–3686. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Cherry, E.; Lee, D.; Jung, J.; Sitcheran, R. AI-06NON-CANONICAL NF-kB SIGNALING DRIVES THE AGGRESSIVE INVASIVENESS OF GLIOBLASTOMA. Neuro-Oncol. 2014, 16, v2. [Google Scholar] [CrossRef] [Green Version]
- Bours, V.; Dejardin, E.; Goujon-Letawe, F.; Merville, M.-P.; Castronovo, V. The NF-κB transcription factor and cancer: High expression of NF-κB-and IκB-related proteins in tumor cell lines. Biochem. Pharmacol. 1994, 47, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Franzoso, G.; Azarenko, V.; Park, S.; Kanno, T.; Brown, K.; Siebenlist, U. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell 1993, 72, 729–739. [Google Scholar] [CrossRef]
- Krappmann, D.; Emmerich, F.; Kordes, U.; Scharschmidt, E.; Dörken, B.; Scheidereit, C. Molecular mechanisms of constitutive NF-κB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene 1999, 18, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, S.A.; Schattner, E.J.; Cesarman, E. Inhibition of NF-κB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood J. Am. Soc. Hematol. 2000, 96, 2537–2542. [Google Scholar]
- Ballard, D.W.; Bohnlein, E.; Lowenthal, J.W.; Wano, Y.; Franza, B.R.; Greene, W.C. HTLV-I tax induces cellular proteins that activate the κB element in the IL-2 receptor α gene. Science 1988, 241, 1652–1655. [Google Scholar] [CrossRef]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Förster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J. A role for NF-κB in organ specific cancer and cancer stem cells. Cancers 2019, 11, 655. [Google Scholar] [CrossRef] [Green Version]
- Watnick, R.S. The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C (high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Chefetz, I.; Holmberg, J.C.; Alvero, A.B.; Visintin, I.; Mor, G. Inhibition of Aurora-A kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting NFĸB pathway. Cell Cycle 2011, 10, 2206–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.-C.; Harhaj, E.W. Receptors and adaptors for NF-kB signaling. In NF-Kb/Rel Transcription Factor Fam; Springer: Berlin/Heidelberg, Germany, 2006; pp. 24–26. [Google Scholar]
- Novack, D.V.; Yin, L.; Hagen-Stapleton, A.; Schreiber, R.D.; Goeddel, D.V.; Ross, F.P.; Teitelbaum, S.L. The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 2003, 198, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Munroe, M.E.; Bishop, G.A. Role of tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) in distinct and overlapping CD40 and TNF receptor 2/CD120b-mediated B lymphocyte activation. J. Biol. Chem. 2004, 279, 53222–53231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauert, H.; Wicovsky, A.; Müller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Nakayama, M.; Nakano, H.; Yagita, H.; Yamamoto, N.; Yamaoka, S. TWEAK induces NF-κB2 p100 processing and long lasting NF-κB activation. J. Biol. Chem. 2003, 278, 36005–36012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbmann, C.; Leeb, S.; Vogl, D.; Maendel, M.; Herfarth, H.; Schölmerich, J.; Falk, W.; Rogler, G. Inducible CD40 expression mediates NFκB activation and cytokine secretion in human colonic fibroblasts. Gut 2003, 52, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Colpaert, S.; D’Haens, G.R.; Kasran, A.; de Boer, M.; Rutgeerts, P.; Geboes, K.; Ceuppens, J.L. Hyperexpression of CD40 ligand (CD154) in inflammatory bowel disease and its contribution to pathogenic cytokine production. J. Immunol. 1999, 163, 4049–4057. [Google Scholar] [CrossRef]
- Danese, S.; Scaldaferri, F.; Vetrano, S.; Stefanelli, T.; Graziani, C.; Repici, A.; Ricci, R.; Straface, G.; Sgambato, A.; Malesci, A. Critical role of the CD40–CD40-ligand pathway in regulating mucosal inflammation-driven angiogenesis in inflammatory bowel disease. Gut 2007, 56, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Blake, M.L.; Tometsko, M.; Miller, R.; Jones, J.C.; Dougall, W.C. RANK expression on breast cancer cells promotes skeletal metastasis. Clin. Exp. Metastasis 2014, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.P.; Miller, R.E.; Jones, J.C.; Zhang, J.; Keller, E.T.; Dougall, W.C. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008, 68, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Deaglio, S.; Dominguez-Sola, D.; Rasi, S.; Vaisitti, T.; Agostinelli, C.; Spina, V.; Bruscaggin, A.; Monti, S.; Cerri, M. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood J. Am. Soc. Hematol. 2011, 118, 4930–4934. [Google Scholar]
- Hyeon, J.; Lee, B.; Shin, S.-H.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Park, W.-Y.; Ko, Y.-H. Targeted deep sequencing of gastric marginal zone lymphoma identified alterations of TRAF3 and TNFAIP3 that were mutually exclusive for MALT1 rearrangement. Mod. Pathol. 2018, 31, 1418–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtois, G.; Gilmore, T. Mutations in the NF-κ B signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [Green Version]
- Fracchiolla, N.; Lombardi, L.; Salina, M.; Migliazza, A.; Baldini, L.; Berti, E.; Cro, L.; Polli, E.; Maiolo, A.; Neri, A. Structural alterations of the NF-kappa B transcription factor lyt-10 in lymphoid malignancies. Oncogene 1993, 8, 2839–2845. [Google Scholar]
- Barth, T.F.; Döhner, H.; Werner, C.A.; Stilgenbauer, S.; Schlotter, M.; Pawlita, M.; Lichter, P.; Möller, P.; Bentz, M. Characteristic pattern of chromosomal gains and losses in primary large B-cell lymphomas of the gastrointestinal tract. Blood J. Am. Soc. Hematol. 1998, 91, 4321–4330. [Google Scholar]
- Lu, D.; Thompson, J.; Gorski, G.; Rice, N.; Mayer, M.; Yunis, J. Alterations at the rel locus in human lymphoma. Oncogene 1991, 6, 1235–1241. [Google Scholar]
- Kalaitzidis, D.; Gilmore, T.D. Genomic organization and expression of the rearranged REL proto-oncogene in the human B-cell lymphoma cell line RC-K8. Genes Chromosom. Cancer 2002, 34, 129–135. [Google Scholar] [CrossRef]
- Zhang, M.; Xu-Monette, Z.Y.; Li, L.; Manyam, G.C.; Visco, C.; Tzankov, A.; Wang, J.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A. RelA NF-κB subunit activation as a therapeutic target in diffuse large B-cell lymphoma. Aging 2016, 8, 3321. [Google Scholar] [CrossRef] [Green Version]
- Willis, T.G.; Jadayel, D.M.; Du, M.-Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.J.C. Bcl10 is involved in t (1; 14)(p22; q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Akagi, T.; Motegi, M.; Tamura, A.; Suzuki, R.; Hosokawa, Y.; Suzuki, H.; Ota, H.; Nakamura, S.; Morishima, Y.; Taniwaki, M. A novel gene, MALT1 at 18q21, is involved in t (11; 18)(q21; q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene 1999, 18, 5785–5794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, P.C.; Yonezumi, M.; Inohara, N.; McAllister-Lucas, L.M.; Abazeed, M.E.; Chen, F.F.; Yamaoka, S.; Seto, M.; Núñez, G. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-κB signaling pathway. J. Biol. Chem. 2001, 276, 19012–19019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uren, A.G.; O’Rourke, K.; Aravind, L.A.; Pisabarro, M.T.; Seshagiri, S.; Koonin, E.V.; Dixit, V.M. Identification of paracaspases and metacaspases: Two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol. Cell 2000, 6, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Zhang, J.; Lombardi, L.; Neri, A.; Dalla-Favera, R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol. Cell. Biol. 1995, 15, 5180–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Carrasco, D.; Claudio, E.; Ryseck, R.-P.; Bravo, R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J. Exp. Med. 1997, 186, 999–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houldsworth, J.; Mathew, S.; Rao, P.H.; Dyomina, K.; Louie, D.C.; Parsa, N.; Offit, K.; Chaganti, R. REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood 1996, 87, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kralova, J.; Schatzle, J.; Bargmann, W.; Bose, H. Transformation of avian fibroblasts overexpressing the c-rel proto-oncogene and a variant of c-rel lacking 40 C-terminal amino acids. J. Virol. 1994, 68, 2073–2083. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.H.; Houldsworth, J.; Dyomina, K.; Parsa, N.Z.; Cigudosa, J.C.; Louie, D.C.; Popplewell, L.; Offit, K.; Jhanwar, S.C.; Chaganti, R. Chromosomal and gene amplification in diffuse large B-cell lymphoma. J. Am. Soc. Hematol. 1998, 92, 234–240. [Google Scholar] [CrossRef]
- Mathew, S.; Murty, V.; Dalla-Favera, R.; Chaganti, R. Chromosomal localization of genes encoding the transcription factors, c-rel, NF-kBp50, NF-cBp65, and lyt-10 by fluorescence in situ. Oncogene 1993, 8, 191–193. [Google Scholar] [PubMed]
- Ohno, H.; Takimoto, G.; McKeithan, T.W. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 1990, 60, 991–997. [Google Scholar] [CrossRef]
- Rylatt, D.B.; Aitken, A.; Bilham, T.; Condon, G.D.; Embi, N.; Cohen, P. Glycogen synthase from rabbit skeletal muscle: Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur. J. Biochem. 1980, 107, 529–537. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional insights from cell biology and animal models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.-S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3β participates in nuclear factor κB–mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ougolkov, A.V.; Bone, N.D.; Fernandez-Zapico, M.E.; Kay, N.E.; Billadeau, D.D. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor κB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood J. Am. Soc. Hematol. 2007, 110, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-κB, and glucose regulation. Cancer Res. 2008, 68, 6643–6651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, M.; Kawano, Y.; Zhu, H.; Waxman, J.; Kypta, R.M. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene 2004, 23, 7882–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakoori, A.; Ougolkov, A.; Yu, Z.W.; Zhang, B.; Modarressi, M.H.; Billadeau, D.D.; Mai, M.; Takahashi, Y.; Minamoto, T. Deregulated GSK3β activity in colorectal cancer: Its association with tumor cell survival and proliferation. Biochem. Biophys. Res. Commun. 2005, 334, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Osthoff, K.; Ferrari, D.; Riehemann, K.; Wesselborg, S. Regulation of NF-κB activation by MAP kinase cascades. Immunobiology 1997, 198, 35–49. [Google Scholar] [CrossRef]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of gadd45 β by NF-κ B downregulates pro-apoptotic JNK signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T. Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Wilde, G.; Van Damme, P.; Vanden Berghe, W.; Haegeman, G. Transcriptional activation of the NF-κB p65 subunit by mitogen-and stress-activated protein kinase-1 (MSK1). EMBO J. 2003, 22, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghe, W.V.; Plaisance, S.; Boone, E.; De Bosscher, K.; Schmitz, M.L.; Fiers, W.; Haegeman, G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 1998, 273, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsagel, P.L.; Carpten, J.; Chesi, M.; VanWier, S.; Keats, J.J.; Sebag, M.; Chng, W.-J.; Schop, R.; Baker, A.; Chng, T.-H. Promiscuous Mutations Frequently Activate the Non-Canonical NFkB Pathway in Multiple Myeloma (MM). Blood 2006, 108, 109. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, J.; Sun, T.; Li, N.; Zhang, L.; Ren, J.; Yuan, H.; Kan, S.; Pan, Q.; Li, X. Epithelial EZH2 serves as an epigenetic determinant in experimental colitis by inhibiting TNFα-mediated inflammation and apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3796–E3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Ito, T.; Shimizu, T.; Ishida, T.; Semba, K.; Watanabe, S.; Yamaguchi, N.; Inoue, J.i. Epigenetic alteration of the NF-κB-inducing kinase (NIK) gene is involved in enhanced NIK expression in basal-like breast cancer. Cancer Sci. 2010, 101, 2391–2397. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Goncalves, R.M.; Klafke, K.; de Souza, P.O.; Dillenburg, F.C.; Carro, L.; Gelain, D.P.; Moreira, J.C.F. Inflammatory landscape of human brain tumors reveals an NFκB dependent cytokine pathway associated with mesenchymal glioblastoma. Cancer Lett. 2017, 390, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Hierarchies of NF-κB target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P.; Döppler, H.; Ferran, C.; Grey, S.T.; Toker, A. Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochem. J. 2005, 387, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Storz, P.; Döppler, H.; Toker, A. Protein kinase Cδ selectively regulates protein kinase D-dependent activation of NF-κB in oxidative stress signaling. Mol. Cell. Biol. 2004, 24, 2614–2626. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Matsumura, I.; Ezoe, S.; Satoh, Y.; Sakamaki, T.; Albanese, C.; Machii, T.; Pestell, R.G.; Kanakura, Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-κB activity that facilitates MnSOD-mediated ROS elimination. Mol. Cell 2002, 9, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Lorbeer, F.K.; Hockemeyer, D. TERT promoter mutations and telomeres during tumorigenesis. Curr. Opin. Genet. Dev. Biol. 2020, 60, 56–62. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, Q.-L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.-Y. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef]
- Xu, X.; Li, Y.; Bharath, S.R.; Ozturk, M.B.; Bowler, M.W.; Loo, B.Z.L.; Tergaonkar, V.; Song, H. Structural basis for reactivating the mutant TERT promoter by cooperative binding of p52 and ETS1. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S. NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Lee, J.F.; Wang, S.H.; Chan, U.P.; Ip, P.C.; Lau, W.Y. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and Nf-kB in human colon cancer. Life Sci. 2002, 70, 2631–2646. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.X.; Ao, Z.; Prasad, K.; Wu, R.; Schlossman, S.F. IEX-1L, an apoptosis inhibitor involved in NF-κB-mediated cell survival. Science 1998, 281, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, F.; Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Beug, H.; Wirth, T. Epithelial-mesenchymal transition: NF-κB takes center stage. Cell Cycle 2004, 3, 1477–1480. [Google Scholar] [CrossRef] [Green Version]
- Duran, C.; Lee, D.; Jung, J.; Ravi, S.; Pogue, C.; Toussaint, L.; Bayless, K.; Sitcheran, R. NIK regulates MT1-MMP activity and promotes glioma cell invasion independently of the canonical NF-κB pathway. Oncogenesis 2016, 5, e231. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.-X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-κB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar] [PubMed]
- Yoshida, A.; Yoshida, S.; Ishibashi, T.; Kuwano, M.; Inomata, H. Suppression of retinal neovascularization by the NF-kappaB inhibitor pyrrolidine dithiocarbamate in mice. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1624–1629. [Google Scholar]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Textor, B.; Pein, O.T.; Licht, A.H.; Andrecht, S.; Sator-Schmitt, M.; Fusenig, N.E.; Angel, P.; Schorpp-Kistner, M. Critical role for NF-κB-induced JunB in VEGF regulation and tumor angiogenesis. EMBO J. 2007, 26, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Duyao, M.P.; Buckler, A.J.; Sonenshein, G.E. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc. Natl. Acad. Sci. USA 1990, 87, 4727–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duyao, M.; Kessler, D.; Spicer, D.; Bartholomew, C.; Cleveland, J.; Siekevitz, M.; Sonenshein, G. Transactivation of the c-myc promoter by human T cell leukemia virus type 1 tax is mediated by NF kappa B. J. Biol. Chem. 1992, 267, 16288–16291. [Google Scholar] [CrossRef]
- Kisseleva, T.; Song, L.; Vorontchikhina, M.; Feirt, N.; Kitajewski, J.; Schindler, C. NF-κB regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Investig. 2006, 116, 2955–2963. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.A.; Kawahara, T.L.; Sutphin, P.D.; Chang, H.Y.; Chi, J.-T.; Giaccia, A.J. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 2009, 15, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Rojo, F.; González-Pérez, A.; Furriol, J.; Nicolau, M.J.; Ferrer, J.; Burgués, O.; Sabbaghi, M.; González-Navarrete, I.; Cristobal, I.; Serrano, L. Non-canonical NF-κ B pathway activation predicts outcome in borderline oestrogen receptor positive breast carcinoma. Br. J. Cancer 2016, 115, 322–331. [Google Scholar] [CrossRef]
- Espinoza-Sánchez, N.A.; Győrffy, B.; Fuentes-Pananá, E.M.; Götte, M. Differential impact of classical and non-canonical NF-κB pathway-related gene expression on the survival of breast cancer patients. J. Cancer 2019, 10, 5191. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhou, J.; Zhou, P.; Xu, J.; Tang, Z.; Ma, H.; Guo, F. Prognostic significance of RelB overexpression in non-small cell lung cancer patients. Thorac. Cancer 2016, 7, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.K.; Peng, C.C.; Low, S.; Vijay, V.; Budiman, A.; Phang, B.H.; Lim, J.Q.; Jeyasekharan, A.D.; Lim, S.T.; Ong, C.K. Sustained activation of non-canonical NF-κB signalling drives glycolytic reprogramming in doxorubicin-resistant DLBCL. Leukemia 2022, 37, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Böhlig, L.; Rother, K. One function—Multiple mechanisms: The manifold activities of p53 as a transcriptional repressor. J. Biomed. Biotechnol. 2011, 2011, 464916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, N.D. Achieving transcriptional specificity with NF-κB. Int. J. Biochem. Cell Biol. 1997, 29, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-κB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohuslav, J.; Chen, L.-f.; Kwon, H.; Mu, Y.; Greene, W.C. p53 induces NF-κB activation by an IκB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J. Biol. Chem. 2004, 279, 26115–26125. [Google Scholar] [CrossRef] [Green Version]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-κB and p53. Mol. Cell. Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, C.A.; Singh, S.; Windle, B.; Sankala, H.M.; Graves, P.R.; Yeudall, W.A.; Deb, S.P.; Deb, S. p53 mutants induce transcription of NF-κB2 in H1299 cells through CBP and STAT binding on the NF-κB2 promoter and gain of function activity. Arch. Biochem. Biophys. 2012, 518, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-F.; Greene, W.C. Regulation of distinct biological activities of the NF-κB transcription factor complex by acetylation. J. Mol. Med. 2003, 81, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Jang, S.-M.; Kim, C.-H.; An, J.-H.; Kang, E.-J.; Choi, K.-H. New molecular bridge between RelA/p65 and NF-κB target genes via histone acetyltransferase TIP60 cofactor. J. Biol. Chem. 2012, 287, 7780–7791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, A.A.; Baltimore, D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; Von Werder, A.; Schmid, R. Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, H.; Shimizu, K.; Hayashi, Y.; Ohki, M. An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t (16; 21) chromosomal translocation. Cancer Res. 1994, 54, 2865–2868. [Google Scholar]
- Prasad, D.; Ouchida, M.; Lee, L.; Rao, V.N.; Reddy, E. TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t (16; 21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene 1994, 9, 3717–3729. [Google Scholar]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t (21; 22), fuses the EWS gene to another ETS–family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Dryden, N.H.; Sperone, A.; Martin-Almedina, S.; Hannah, R.L.; Birdsey, G.M.; Khan, S.T.; Layhadi, J.A.; Mason, J.C.; Haskard, D.O.; Göttgens, B. The transcription factor Erg controls endothelial cell quiescence by repressing activity of nuclear factor (NF)-κB p65. J. Biol. Chem. 2012, 287, 12331–12342. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cai, Y.; Shao, L.-j.; Siddiqui, J.; Palanisamy, N.; Li, R.; Ren, C.; Ayala, G.; Ittmann, M. Activation of NF-κB by TMPRSS2/ERG fusion isoforms through Toll-like receptor-4. Cancer Res. 2011, 71, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Kandagatla, P.; Singareddy, R.; Kropinski, A.; Sheng, S.; Cher, M.L.; Chinni, S.R. Androgens induce functional CXCR4 through ERG factor expression in TMPRSS2-ERG fusion-positive prostate cancer cells. Transl. Oncol. 2010, 3, 195-IN191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.; Li, Y. NF-κB/p52 augments ETS1 binding genome-wide to promote glioma progression. bioRxiv 2023. bioRxiv 2023.2001. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesler, A.; Xu, X.; Zheng, X.; Li, X.; Jiang, J.; Russo, J.J.; Ju, J. Identification of miR-215 mediated targets/pathways via translational immunoprecipitation expression analysis (TrIP-chip). Oncotarget 2015, 6, 24463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, J.; Shi, Y.; Tan, G.; Yang, C.H.; Fan, M.; Pfeffer, L.M.; Wu, Z.-H. DNA Damage Induces NF-κB-Dependent microRNA-21 up-Regulation and Promotes Breast Cancer Cell Invasion. J. Biol. Chem. 2012, 287, 21783–21795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, S.; Hu, T.; Liu, S.; He, Y.; Sun, S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 2009, 50, 490–499. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Scisciani, C.; Vossio, S.; Guerrieri, F.; Schinzari, V.; De Iaco, R.; de Meo, P.D.O.; Cervello, M.; Montalto, G.; Pollicino, T.; Raimondo, G. Transcriptional regulation of miR-224 upregulated in human HCCs by NFκB inflammatory pathways. J. Hepatol. 2012, 56, 855–861. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shan, C.; Kong, G.; Du, Y.; Ye, L.; Zhang, X. MicroRNA-520e suppresses growth of hepatoma cells by targeting the NF-κB-inducing kinase (NIK). Oncogene 2012, 31, 3607–3620. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Sun, L.; Liu, Q.; Gong, C.; Yao, Y.; Lv, X.; Lin, L.; Yao, H.; Su, F.; Li, D. A cytoplasmic NF-κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell 2015, 27, 370–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.; Zhao, J.; Yang, Y. Downregulation of lncRNA H19 inhibits the migration and invasion of melanoma cells by inactivating the NF-κB and PI3K/Akt signaling pathways. Mol. Med. Rep. 2018, 17, 7313–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, J.; Li, C.; Wang, X.; Dong, Y.; Shen, X.; Zhang, X. LncRNA H19 induced by helicobacter pylori infection promotes gastric cancer cell growth via enhancing NF-κB-induced inflammation. J. Inflamm. 2019, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, W.; Zhang, Y.; Liang, B. Long non-coding RNA-NEAT1 promotes cell migration and invasion via regulating miR-124/NF-κB pathway in cervical cancer. OncoTargets Ther. 2020, 13, 3265. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Zhang, Y.; Liu, W.; Huang, Y.; Shen, X.; Jing, R.; Pu, J.; Wang, X.; Ju, S.; Cong, H. LncRNA H19 overexpression induces bortezomib resistance in multiple myeloma by targeting MCL-1 via miR-29b-3p. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, H. LncRNA NEAT1 promotes dexamethasone resistance in multiple myeloma by targeting miR-193a/MCL1 pathway. J. Biochem. Mol. Toxicol. 2018, 32, e22008. [Google Scholar] [CrossRef]
- Tando, T.; Ishizaka, A.; Watanabe, H.; Ito, T.; Iida, S.; Haraguchi, T.; Mizutani, T.; Izumi, T.; Isobe, T.; Akiyama, T. Requiem protein links RelB/p52 and the Brm-type SWI/SNF complex in a noncanonical NF-κB pathway. J. Biol. Chem. 2010, 285, 21951–21960. [Google Scholar] [CrossRef] [Green Version]
- De Donatis, G.; Pape, E.; Pierron, A.; Cheli, Y.; Hofman, V.; Hofman, P.; Allegra, M.; Zahaf, K.; Bahadoran, P.; Rocchi, S. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene 2016, 35, 2735–2745. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kang, M.-J.; Han, H.-Y.; Lee, M.-G.; Jeong, S.-I.; Ryu, B.-K.; Ha, T.-K.; Her, N.-G.; Han, J.; Park, S.J. Epigenetic alteration of PRKCDBP in colorectal cancers and its implication in tumor cell resistance to TNFα-induced apoptosis. Clin. Cancer Res. 2011, 17, 7551–7562. [Google Scholar] [CrossRef] [Green Version]
- van Essen, D.; Zhu, Y.; Saccani, S. A feed-forward circuit controlling inducible NF-κB target gene activation by promoter histone demethylation. Mol. Cell 2010, 39, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Natoli, G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. Biol. 2002, 16, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargreaves, D.C.; Horng, T.; Medzhitov, R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 2009, 138, 129–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Factors of NF-κB Pathway and Alterations | Cancer Type | References |
|---|---|---|
RelA (p65)
| HCC, FLHCC, Breast cancer cell lines | [105,114,115] |
RelB
| GBM, MM, DLBCL | [113,116,117,118,119] |
c-Rel
| Breast tumours, Hodgkin lymphoma, DLBCL, GC-DLBCL, PMBL | [120,121,122] |
NF-κB–inducing kinase (NIK)
| GBM, MM, Hodgkin lymphoma | [110,119,123] |
IκBα
| Ovarian Carcinoma cell line, HCC, Melanoma | [104,105,109,124,125] |
IKK
| Lung carcinoma, Melanoma, Hodgkin lymphoma, ABC-DLBCL, PEL, ALT, HCC | [105,110,111,112,126,127,128] |
NF-κB1
| Breast tumours, Melanoma cells, Lung cancer, HCC, ALT, ABC-DLBCL | [104,105,110] |
NF-κB2
| Colon carcinoma cell line, MM, Breast cancer, Lung cancer | [106,107,110,119,124] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deka, K.; Li, Y. Transcriptional Regulation during Aberrant Activation of NF-κB Signalling in Cancer. Cells 2023, 12, 788. https://doi.org/10.3390/cells12050788
Deka K, Li Y. Transcriptional Regulation during Aberrant Activation of NF-κB Signalling in Cancer. Cells. 2023; 12(5):788. https://doi.org/10.3390/cells12050788
Chicago/Turabian StyleDeka, Kamalakshi, and Yinghui Li. 2023. "Transcriptional Regulation during Aberrant Activation of NF-κB Signalling in Cancer" Cells 12, no. 5: 788. https://doi.org/10.3390/cells12050788