
Context-Dependent Role of Glucocorticoid Receptor Alpha and Beta in Breast Cancer Cell Behaviour
 , , ,
, , ,
Abstract
:1. Introduction

2. Materials and Methods
2.1. Samples and Validation Datasets
2.2. Cell Cultures, Treatments, and Transfections
2.3. Western Blot
2.4. Immunohistochemistry
2.5. Immunocytochemistry
2.6. Cell Viability, Proliferation, Live–Dead Cell Ratio, and Cell Migration
2.7. Statistical Methods
3. Results
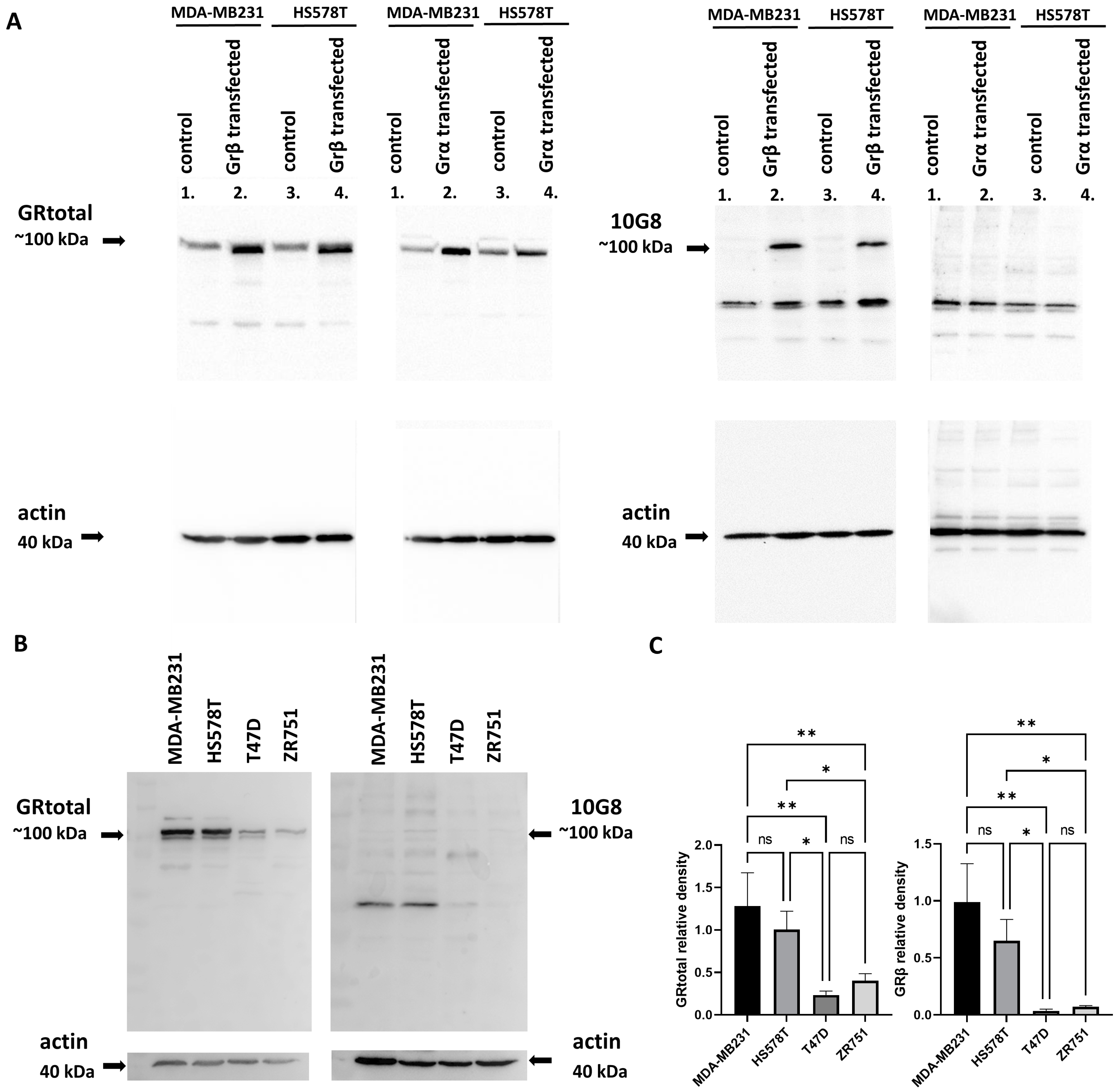
3.1. Characterization of Glucocorticoid Receptor Expression in Normal and Cancerous Breast Tissue and Breast Cancer Cell Lines
3.2. Different Glucocorticoid Receptor Isoforms in Breast Cancer
3.3. The Opposite Effect of Glucocorticoid Receptor Expression in Breast Cancer Cell Viability, Proliferation, Cell Death, and Migration Depending on Hormone Receptor Status
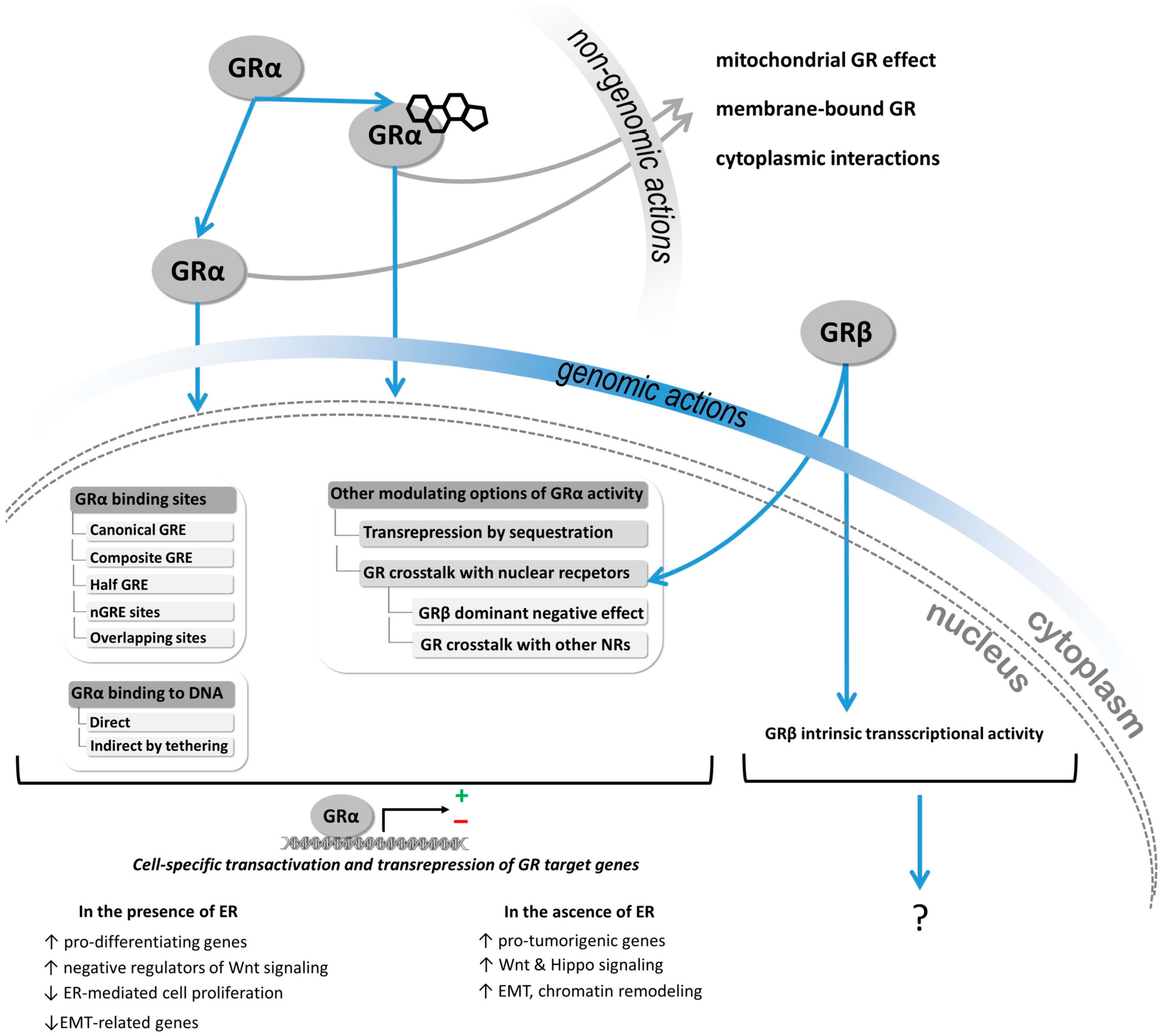
4. Discussion
4.1. Challenging Detection of GR Expression in Breast Cancer
4.2. GR Expression in Breast Cancer in the Context of ER
4.3. The Role of GRβ Isoform in GR Action in Breast Cancer
4.4. The Effect of Ligand Availability on GR Action on Breast Cancer Cell Behavior
4.5. GR Activity Signature in Breast Cancer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Brown, J.; Morikawa, A.; Method, M. Breast Cancer-Specific Mortality in Early Breast Cancer as Defined by High-Risk Clinical and Pathologic Characteristics. PLoS ONE 2022, 17, e0264637. [Google Scholar] [CrossRef]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO–ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Vanzulli, S.I.; Lanari, C. Hormone Receptors in Breast Cancer: More than Estrogen Receptors. Medicina 2019, 79, 540–545. [Google Scholar] [PubMed]
- Noureddine, L.M.; Trédan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4446. [Google Scholar] [CrossRef] [PubMed]
- Obradović, M.M.S.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Münst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids Promote Breast Cancer Metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef]
- Zhidkova, E.M.; Lylova, E.S.; Savinkova, A.V.; Mertsalov, S.A.; Kirsanov, K.I.; Belitsky, G.A.; Yakubovskaya, M.G.; Lesovaya, E.A. A Brief Overview of the Paradoxical Role of Glucocorticoids in Breast Cancer. Breast Cancer 2020, 14, 1178223420974667. [Google Scholar] [CrossRef]
- Mayayo-Peralta, I.; Zwart, W.; Prekovic, S. Duality of Glucocorticoid Action in Cancer: Tumor-Suppressor or Oncogene? Endocr. Relat. Cancer 2021, 28, R157–R171. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive Overview of the Structure and Regulation of the Glucocorticoid Receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Chrousos, G.P.; Kino, T. Intracellular Glucocorticoid Signaling: A Formerly Simple System Turns Stochastic. Sci. STKE 2005, 2005, pe48. [Google Scholar] [CrossRef]
- Lu, N.Z.; Cidlowski, J.A. Translational Regulatory Mechanisms Generate N-Terminal Glucocorticoid Receptor Isoforms with Unique Transcriptional Target Genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Voss, T.C.; Sung, M.-H.; Baek, S.; John, S.; Hawkins, M.; Grøntved, L.; Schiltz, R.L.; Hager, G.L. Reprogramming the Chromatin Landscape: Interplay of the Estrogen and Glucocorticoid Receptors at the Genomic Level. Cancer Res. 2013, 73, 5130–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abduljabbar, R.; Negm, O.H.; Lai, C.-F.; Jerjees, D.A.; Al-Kaabi, M.; Hamed, M.R.; Tighe, P.J.; Buluwela, L.; Mukherjee, A.; Green, A.R.; et al. Clinical and Biological Significance of Glucocorticoid Receptor (GR) Expression in Breast Cancer. Breast Cancer Res. Treat. 2015, 150, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the Glucocorticoid Receptor Is Associated with Poor Prognosis in Estrogen Receptor-Negative Breast Cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Lan, X.; Wu, D.; Sunkel, B.; Ye, Z.; Huang, J.; Liu, Z.; Clinton, S.K.; Jin, V.X.; Wang, Q. Ligand-Dependent Genomic Function of Glucocorticoid Receptor in Triple-Negative Breast Cancer. Nat. Commun. 2015, 6, 8323. [Google Scholar] [CrossRef] [Green Version]
- West, D.C.; Kocherginsky, M.; Tonsing-Carter, E.Y.; Dolcen, D.N.; Hosfield, D.J.; Lastra, R.R.; Sinnwell, J.P.; Thompson, K.J.; Bowie, K.R.; Harkless, R.V.; et al. Discovery of a Glucocorticoid Receptor (GR) Activity Signature Using Selective GR Antagonism in ER-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 3433–3446. [Google Scholar] [CrossRef] [Green Version]
- Ritter, H.D.; Mueller, C.R. Expression Microarray Identifies the Unliganded Glucocorticoid Receptor as a Regulator of Gene Expression in Mammary Epithelial Cells. BMC Cancer 2014, 14, 275. [Google Scholar] [CrossRef] [Green Version]
- Cairat, M.; Al Rahmoun, M.; Gunter, M.J.; Heudel, P.-E.; Severi, G.; Dossus, L.; Fournier, A. Use of Systemic Glucocorticoids and Risk of Breast Cancer in a Prospective Cohort of Postmenopausal Women. BMC Med. 2021, 19, 186. [Google Scholar] [CrossRef]
- Shi, W.; Wang, D.; Yuan, X.; Liu, Y.; Guo, X.; Li, J.; Song, J. Glucocorticoid Receptor–IRS-1 Axis Controls EMT and the Metastasis of Breast Cancers. J. Mol. Cell Biol. 2019, 11, 1042–1055. [Google Scholar] [CrossRef] [Green Version]
- Groeneweg, F.L.; van Royen, M.E.; Fenz, S.; Keizer, V.I.P.; Geverts, B.; Prins, J.; de Kloet, E.R.; Houtsmuller, A.B.; Schmidt, T.S.; Schaaf, M.J.M. Quantitation of Glucocorticoid Receptor DNA-Binding Dynamics by Single-Molecule Microscopy and FRAP. PLoS ONE 2014, 9, e90532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Ramírez, P.; Tliba, O. Glucocorticoid Receptor β (GRβ): Beyond Its Dominant-Negative Function. Int. J. Mol. Sci. 2021, 22, 3649. [Google Scholar] [CrossRef] [PubMed]
- Butz, H.; Patócs, A. Mechanisms behind Context-Dependent Role of Glucocorticoids in Breast Cancer Progression. Cancer Metastasis Rev. 2022, 41, 803–832. [Google Scholar] [CrossRef] [PubMed]
- Duma, D.; Jewell, C.M.; Cidlowski, J.A. Multiple Glucocorticoid Receptor Isoforms and Mechanisms of Post-Translational Modification. J. Steroid Biochem. Mol. Biol. 2006, 102, 11–21. [Google Scholar] [CrossRef]
- Elkashif, A.; Bingham, V.; Haddock, P.; Humphries, M.P.; McQuaid, S.; Mullan, P.B.; McCarthy, H.O.; Buckley, N.E. Glucocorticoid Receptor Expression Predicts Good Outcome in Response to Taxane-Free, Anthracycline-Based Therapy in Triple Negative Breast Cancer. J. Oncol. 2020, 2020, 3712825. [Google Scholar] [CrossRef]
- West, D.C.; Pan, D.; Tonsing-Carter, E.Y.; Hernandez, K.M.; Pierce, C.F.; Styke, S.C.; Bowie, K.R.; Garcia, T.I.; Kocherginsky, M.; Conzen, S.D. GR and ER Coactivation Alters the Expression of Differentiation Genes and Associates with Improved ER+ Breast Cancer Outcome. Mol. Cancer Res. 2016, 14, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Xin, Z.; Cai, Y.; Dang, L.T.; Burke, H.M.S.; Revote, J.; Charitakis, N.; Bienroth, D.; Nim, H.T.; Li, Y.-F.; Ramialison, M. MonaGO: A Novel Gene Ontology Enrichment Analysis Visualisation System. BMC Bioinform. 2022, 23, 69. [Google Scholar] [CrossRef]
- Nagy, Z.; Marta, A.; Butz, H.; Liko, I.; Racz, K.; Patocs, A. Modulation of the Circadian Clock by Glucocorticoid Receptor Isoforms in the H295R Cell Line. Steroids 2016, 116, 20–27. [Google Scholar] [CrossRef]
- Nagy, Z.; Acs, B.; Butz, H.; Feldman, K.; Marta, A.; Szabo, P.M.; Baghy, K.; Pazmany, T.; Racz, K.; Liko, I.; et al. Overexpression of GRß in Colonic Mucosal Cell Line Partly Reflects Altered Gene Expression in Colonic Mucosa of Patients with Inflammatory Bowel Disease. J. Steroid Biochem. Mol. Biol. 2016, 155, 76–84. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Smith, M.S.; Fitzsimmons, M.D. Detecting Steroidal Effects on Immediate Early Gene Expression in the Hypothalamus. Neuroprotocols 1992, 1, 52–66. [Google Scholar] [CrossRef]
- Krokker, L.; Szabó, B.; Németh, K.; Tóháti, R.; Sarkadi, B.; Mészáros, K.; Patócs, A.; Butz, H. Three Dimensional Cell Culturing for Modeling Adrenal and Pituitary Tumors. Pathol. Oncol. Res. 2021, 27, 640676. [Google Scholar] [CrossRef] [PubMed]
- Szabó, B.; Németh, K.; Mészáros, K.; Krokker, L.; Likó, I.; Saskői, É.; Németh, K.; Szabó, P.T.; Szücs, N.; Czirják, S.; et al. Aspirin Mediates Its Antitumoral Effect Through Inhibiting PTTG1 in Pituitary Adenoma. J. Clin. Endocrinol. Metab. 2022, 107, 3066–3079. [Google Scholar] [CrossRef] [PubMed]
- Braunschweiger, P.G.; Schiffer, L.M. Antiproliferative Effects of Corticosteroids in C3H/HeJ Mammary Tumors and Implications for Sequential Combination Chemotherapy. Cancer Res. 1981, 41, 3324–3330. [Google Scholar] [PubMed]
- Kornegoor, R.; Verschuur-Maes, A.H.J.; Buerger, H.; Hogenes, M.C.H.; de Bruin, P.C.; Oudejans, J.J.; van der Groep, P.; Hinrichs, B.; van Diest, P.J. Molecular Subtyping of Male Breast Cancer by Immunohistochemistry. Mod. Pathol. 2012, 25, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galliher-Beckley, A.J.; Williams, J.G.; Cidlowski, J.A. Ligand-Independent Phosphorylation of the Glucocorticoid Receptor Integrates Cellular Stress Pathways with Nuclear Receptor Signaling. Mol. Cell. Biol. 2011, 31, 4663–4675. [Google Scholar] [CrossRef] [Green Version]
- Michael, Y.L.; Carlson, N.E.; Chlebowski, R.T.; Aickin, M.; Weihs, K.L.; Ockene, J.K.; Bowen, D.J.; Ritenbaugh, C. Influence of Stressors on Breast Cancer Incidence in the Women’s Health Initiative. Health Psychol. 2009, 28, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Nordeen, S.K. Overlapping but Distinct Profiles of Gene Expression Elicited by Glucocorticoids and Progestins. Recent Prog. Horm. Res. 2003, 58, 199–226. [Google Scholar] [CrossRef]
- Leo, J.C.L.; Guo, C.; Woon, C.T.; Aw, S.E.; Lin, V.C.L. Glucocorticoid and Mineralocorticoid Crosstalk with Progesterone Receptor to Induce Focal Adhesion and Growth Inhibition in Breast Cancer Cells. Endocrinology 2004, 145, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.M.; Harris, J.M.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L.; et al. Androgen Receptor Inhibits Estrogen Receptor-Alpha Activity and Is Prognostic in Breast Cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.-B.; Tang, C.-H.; Huang, Y.; Fang, H.; Yu, Z.-Q.; Wu, L.-M.; Liu, R.-Y. Alternative Splicing in Exon 9 of Glucocorticoid Receptor Pre-MRNA Is Regulated by SRp40. Mol. Biol. Rep. 2010, 37, 1427–1433. [Google Scholar] [CrossRef]
- Proverbs-Singh, T.; Feldman, J.L.; Morris, M.J.; Autio, K.A.; Traina, T.A. Targeting the Androgen Receptor in Prostate and Breast Cancer: Several New Agents in Development. Endocr. Relat. Cancer 2015, 22, R87–R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for Estrogen Receptor Expression in Human Cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An Expanded Reference Database of Human and Mouse Transcriptional Regulatory Interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Gong, H.; Jarzynka, M.J.; Cole, T.J.; Lee, J.H.; Wada, T.; Zhang, B.; Gao, J.; Song, W.-C.; DeFranco, D.B.; Cheng, S.-Y.; et al. Glucocorticoids Antagonize Estrogens by Glucocorticoid Receptor-Mediated Activation of Estrogen Sulfotransferase. Cancer Res. 2008, 68, 7386–7393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, R.J. Interactions between Estrogen and Growth Factor Receptors in Human Breast Cancers and the Tumor-Associated Vasculature. Breast J. 2003, 9, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Growth Factor Receptor Crosstalk with Estrogen Receptor as a Mechanism for Tamoxifen Resistance in Breast Cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef]
- Moutsatsou, P.; Papavassiliou, A.G. The Glucocorticoid Receptor Signalling in Breast Cancer. J. Cell. Mol. Med. 2008, 12, 145–163. [Google Scholar] [CrossRef] [Green Version]
- Enuka, Y.; Feldman, M.E.; Chowdhury, A.; Srivastava, S.; Lindzen, M.; Sas-Chen, A.; Massart, R.; Cheishvili, D.; Suderman, M.J.; Zaltsman, Y.; et al. Epigenetic Mechanisms Underlie the Crosstalk between Growth Factors and a Steroid Hormone. Nucleic Acids Res. 2017, 45, 12681–12699. [Google Scholar] [CrossRef] [Green Version]
- Perez Kerkvliet, C.; Dwyer, A.R.; Diep, C.H.; Oakley, R.H.; Liddle, C.; Cidlowski, J.A.; Lange, C.A. Glucocorticoid Receptors Are Required Effectors of TGFβ1-Induced P38 MAPK Signaling to Advanced Cancer Phenotypes in Triple-Negative Breast Cancer. Breast Cancer Res. 2020, 22, 39. [Google Scholar] [CrossRef]
- Strehl, C.; Buttgereit, F. Unraveling the Functions of the Membrane-Bound Glucocorticoid Receptors: First Clues on Origin and Functional Activity: Unraveling the Secret of the MGRs. Ann. N.Y. Acad. Sci. 2014, 1318, 1–6. [Google Scholar] [CrossRef]
- Deng, Q.; Riquelme, D.; Trinh, L.; Low, M.J.; Tomić, M.; Stojilkovic, S.; Aguilera, G. Rapid Glucocorticoid Feedback Inhibition of ACTH Secretion Involves Ligand-Dependent Membrane Association of Glucocorticoid Receptors. Endocrinology 2015, 156, 3215–3227. [Google Scholar] [CrossRef] [Green Version]
- Schiller, B.J.; Chodankar, R.; Watson, L.C.; Stallcup, M.R.; Yamamoto, K.R. Glucocorticoid Receptor Binds Half Sites as a Monomer and Regulates Specific Target Genes. Genome Biol. 2014, 15, 418. [Google Scholar] [CrossRef]
- Fürst, R.; Schroeder, T.; Eilken, H.M.; Bubik, M.F.; Kiemer, A.K.; Zahler, S.; Vollmar, A.M. MAPK Phosphatase-1 Represents a Novel Anti-inflammatory Target of Glucocorticoids in the Human Endothelium. FASEB J. 2007, 21, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernocchi, S.; Battello, N.; Schmitz, S.; Revets, D.; Billing, A.M.; Turner, J.D.; Muller, C.P. Membrane Glucocorticoid Receptor Activation Induces Proteomic Changes Aligning with Classical Glucocorticoid Effects. Mol. Cell. Proteom. 2013, 12, 1764–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, M.; Oasa, S.; Yamamoto, J.; Mikuni, S.; Kinjo, M. A Quantitative Study of Internal and External Interactions of Homodimeric Glucocorticoid Receptor Using Fluorescence Cross-Correlation Spectroscopy in a Live Cell. Sci. Rep. 2017, 7, 4336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leehy, K.A.; Regan Anderson, T.M.; Daniel, A.R.; Lange, C.A.; Ostrander, J.H. Modifications to Glucocorticoid and Progesterone Receptors Alter Cell Fate in Breast Cancer. J. Mol. Endocrinol. 2016, 56, R99–R114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dattilo, V.; Amato, R.; Perrotti, N.; Gennarelli, M. The Emerging Role of SGK1 (Serum- and Glucocorticoid-Regulated Kinase 1) in Major Depressive Disorder: Hypothesis and Mechanisms. Front. Genet. 2020, 11, 826. [Google Scholar] [CrossRef]
- Hermes, G.L.; Delgado, B.; Tretiakova, M.; Cavigelli, S.A.; Krausz, T.; Conzen, S.D.; McClintock, M.K. Social Isolation Dysregulates Endocrine and Behavioral Stress While Increasing Malignant Burden of Spontaneous Mammary Tumors. Proc. Natl. Acad. Sci. USA 2009, 106, 22393–22398. [Google Scholar] [CrossRef] [Green Version]
- Savory, J.G.A.; Hsu, B.; Laquian, I.R.; Giffin, W.; Reich, T.; Haché, R.J.G.; Lefebvre, Y.A. Discrimination between NL1- and NL2-Mediated Nuclear Localization of the Glucocorticoid Receptor. Mol. Cell. Biol. 1999, 19, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Haché, R.J.; Tse, R.; Reich, T.; Savory, J.G.; Lefebvre, Y.A. Nucleocytoplasmic Trafficking of Steroid-Free Glucocorticoid Receptor. J. Biol. Chem. 1999, 274, 1432–1439. [Google Scholar] [CrossRef] [Green Version]
- Matthews, L.; Johnson, J.; Berry, A.; Trebble, P.; Cookson, A.; Spiller, D.; Rivers, C.; Norman, M.; White, M.; Ray, D. Cell Cycle Phase Regulates Glucocorticoid Receptor Function. PLoS ONE 2011, 6, e22289. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Type | Sample Number | Method | Availability |
|---|---|---|---|
| normal tissues (54 different types *) | 184 | IHC | https://www.proteinatlas.org/ (accessed on 11 November 2022) |
| normal breast | 459 | RNAseq | https://www.proteinatlas.org/ (accessed on 11 November 2022) |
| different cancer types (17 different types **) | 7931 | RNAseq | https://www.proteinatlas.org/ (accessed on 11 November 2022) |
| breast cancer | 16 | IHC | https://www.proteinatlas.org/ (accessed on 11 November 2022) |
| breast cancer cell lines | 86 | RNAseq | https://depmap.org/portal/ (accessed on 11 November 2022) |
| breast cancer | 4421 | RNAseq | http://bcgenex.ico.unicancer.fr/ (accessed on 11 November 2022) |
| breast cancer | 11,359 | microarray | http://bcgenex.ico.unicancer.fr (accessed on 11 November 2022) |
| Common GO-BP Terms | Significant Terms | Microarray (n = 10,455) | RNAseq (n = 4421) | ||
|---|---|---|---|---|---|
| p-Value | Associated Genes | p-Value | Associated Genes | ||
| positive regulation of cell migration | GO:0030335 | 2.8 × 10−5 | CAV1, CXCL12, IGF1, PECAM1, S1PR1 | 5.63 × 10−6 | CAV1, CDH5, CXCL12, DAB2, F10, F2R, FAM107A, FER, HGF, IGF1, KDR, PDGFD, PDGFRA, PECAM1, PPM1F, PRKCA, S1PR1, SEMA3G, SEMA5A, SPRY2, VSIR |
| positive regulation of smooth muscle cell proliferation | GO:0048661 | 1.18 × 10−4 | IGF1, S1PR1, TGFBR2 | 2.02 × 10−3 | CALCRL, IGF1, IL6R, PDGFD, S1PR1, TGFBR2, TLR4 |
| intracellular steroid hormone receptor signaling pathway | GO:0030518 | 1.3 × 10−4 | NR3C1, PLPP1 | 3.67 × 10−3 | NR3C1, NR3C2, PLPP1 |
| positive regulation of peptidyl-tyrosine phosphorylation | GO:0050731 | 3.9 × 10−4 | ENPP2, IGF1, PECAM1 | 3.53 × 10−4 | ANGPT4, BMP6, ENPP2, FGF7, HGF, IGF1, IL6R, NRP1, PECAM1, RELN |
| maintenance of blood–brain barrier | GO:0035633 | 1.11 × 10−3 | JAM2, PECAM1GO:0010634 | 2.61 × 10−6 | CDH5, CLDN5, DMD, JAM2, JAM3, LAMA2, MBP, PECAM1 |
| cellular response to transforming growth factor beta stimulus | GO:0071560 | 3.47 × 10−3 | CAV1, NR3C1 | 3.34 × 10−5 | ACVRL1, CAV1, FYN, MEF2C, NR3C1, PDE2A, PDE3A, PDGFD, ZFP36L2 |
| vasculogenesis | GO:0001570 | 3.98 × 10−3 | CAV1, TGFBR2 | 8.86 × 10−6 | CAV1, ENG, HEG1, KDR, MYOCD, QKI, SOX17, TGFBR2, TIE1, TMEM100 |
| glomerular endothelium development | GO:0072011 | 4.79 × 10−3 | PECAM1 | 2.63 × 10−3 | CD34, PECAM1 |
| regulation of membrane repolarization during action potential | GO:0098903 | 4.79 × 10−3 | CAV1 | 2.63 × 10−3 | CACNA2D1, CAV1 |
| signal transduction | GO:0007165 | 6.15 × 10−3 | CXCL12, IGF1, NR3C1, PECAM1, PLPP1, SPARCL1 | 9.17 × 10−3 | AKAP13, DLC1, KANK2, STARD8 |
| myotube cell development | GO:0014904 | 6.38 × 10−3 | IGF1 | 5.15 × 10−3 | IGF1, NFATC2 |
| monocyte extravasation | GO:0035696 | 6.38 × 10−3 | PECAM1 | 5.15 × 10−3 | CCR2, PDGFD |
| diapedesis | GO:0050904 | 6.38 × 10−3 | PECAM1 | 5.15 × 10−3 | FER, PECAM1 |
| myoblast proliferation | GO:0051450 | 6.38 × 10−3 | IGF1 | 1.04 × 10−4 | ATOH8, HGF, IGF1 |
| caveola assembly | GO:0070836 | 7.97 × 10−3 | CAV1 | 8.41 × 10−3 | CAV1, CAV2 |
| hematopoietic stem cell migration to bone marrow | GO:0097241 | 7.97 × 10−3 | JAM2 | 8.41 × 10−3 | JAM2, JAM3 |
| positive regulation of epithelial–mesenchymal transition involved in endocardial cushion formation | GO:1905007 | 7.97 × 10−3 | TGFBR2 | 8.41 × 10−3 | ENG, TGFBR2 |
| receptor-mediated endocytosis of virus by host cell | GO:0019065 | 9.55 × 10−3 | CAV1 | 1.13 × 10−5 | CAV1, CAV2, EPS15, PIKFYVE |
| cellular response to hyperoxia | GO:0071455 | 9.55 × 10−3 | CAV1 | 4.98 × 10−4 | CAV1, FAS, FOXO1 |
| neutrophil extravasation | GO:0072672 | 9.55 × 10−3 | PECAM1 | 4.98 × 10−4 | JAML, PECAM1, PIK3CG |
| Description | Significant Terms | p-Value | Associated Genes | Studies Microarray (n = 10,455), RNAseq (n = 4421) |
|---|---|---|---|---|
| sister chromatid cohesion | GO:0007062 | 1.37 × 10−3 | STAG3L3 | microarray |
| protein K29-linked ubiquitination | GO:0035519 | 1.17 × 10−5 | UBE2S, UBE2T | RNAseq |
| protein K27-linked ubiquitination | GO:0044314 | 1.17 × 10−5 | UBE2S, UBE2T | RNAseq |
| protein K6-linked ubiquitination | GO:0085020 | 2.81 × 10−5 | UBE2S, UBE2T | RNAseq |
| cell division | GO:0051301 | 2.36 × 10−4 | CDCA3, CDT1, SAC3D1, UBE2S | RNAseq |
| protein K11-linked ubiquitination | GO:0070979 | 3.13 × 10−4 | UBE2S, UBE2T | RNAseq |
| protein K63-linked ubiquitination | GO:0070534 | 7.24 × 10−4 | UBE2S, UBE2T | RNAseq |
| FAD biosynthetic process | GO:0006747 | 9.13 × 10−4 | FLAD1 | RNAseq |
| Golgi to transport vesicle transport | GO:0055108 | 9.13 × 10−4 | ARF1 | RNAseq |
| synaptic vesicle budding | GO:0070142 | 9.13 × 10−4 | ARF1 | RNAseq |
| negative regulation of protein localization to kinetochore | GO:1905341 | 9.13 × 10−4 | CDT1 | RNAseq |
| mitotic cleavage furrow ingression | GO:1990386 | 9.13 × 10−4 | ARF1 | RNAseq |
| positive regulation of DNA-dependent DNA replication | GO:2000105 | 9.13 × 10−4 | CDT1 | RNAseq |
| RNA phosphodiester bond hydrolysis, endonucleolytic | GO:0090502 | 1.39 × 10−3 | POP7, RNASEH2A | RNAseq |
| meiotic cell cycle | GO:0051321 | 1.72 × 10−3 | H2AX, PKMYT1 | RNAseq |
| DNA replication preinitiation complex assembly | GO:0071163 | 1.83 × 10−3 | CDT1 | RNAseq |
| response to sorbitol | GO:0072708 | 1.83 × 10−3 | CDT1 | RNAseq |
| lysosomal membrane organization | GO:0097212 | 1.83 × 10−3 | ARF1 | RNAseq |
| positive regulation of sodium ion transmembrane transport | GO:1902307 | 1.83 × 10−3 | ARF1 | RNAseq |
| regulation of DNA replication origin binding | GO:1902595 | 1.83 × 10−3 | CDT1 | RNAseq |
| positive regulation of late endosome to lysosome transport | GO:1902824 | 1.83 × 10−3 | ARF1 | RNAseq |
| regulation of phospholipid metabolic process | GO:1903725 | 1.83 × 10−3 | ARF1 | RNAseq |
| double-strand break repair via homologous recombination | GO:0000724 | 2.20 × 10−3 | H2AX, RECQL4 | RNAseq |
| regulation of chromosome organization | GO:0033044 | 2.74 × 10−3 | CDT1 | RNAseq |
| deactivation of mitotic spindle assembly checkpoint | GO:1902426 | 2.74 × 10−3 | CDT1 | RNAseq |
| DNA replication | GO:0006260 | 3.46 × 10−3 | RECQL4, RNASEH2A | RNAseq |
| DNA replication, removal of RNA primer | GO:0043137 | 3.65 × 10−3 | RNASEH2A | RNAseq |
| dendritic spine organization | GO:0097061 | 3.65 × 10−3 | ARF1 | RNAseq |
| positive regulation of protein localization to kinetochore | GO:1905342 | 3.65 × 10−3 | CDT1 | RNAseq |
| regulation of receptor internalization | GO:0002090 | 4.56 × 10−3 | ARF1 | RNAseq |
| riboflavin metabolic process | GO:0006771 | 4.56 × 10−3 | FLAD1 | RNAseq |
| regulation of nuclear cell cycle DNA replication | GO:0033262 | 4.56 × 10−3 | CDT1 | RNAseq |
| positive regulation of ER to Golgi vesicle-mediated transport | GO:1902953 | 4.56 × 10−3 | ARF1 | RNAseq |
| free ubiquitin chain polymerization | GO:0010994 | 5.47 × 10−3 | UBE2S | RNAseq |
| regulation of DNA-dependent DNA replication initiation | GO:0030174 | 5.47 × 10−3 | CDT1 | RNAseq |
| regulation of Arp2/3 complex-mediated actin nucleation | GO:0034315 | 5.47 × 10−3 | ARF1 | RNAseq |
| kinetochore organization | GO:0051383 | 5.47 × 10−3 | CDT1 | RNAseq |
| mitotic cell cycle | GO:0000278 | 6.22 × 10−3 | CDT1, PKMYT1 | RNAseq |
| telomeric D-loop disassembly | GO:0061820 | 7.29 × 10−3 | RECQL4 | RNAseq |
| protein polyubiquitination | GO:0000209 | 8.93 × 10−3 | UBE2S, UBE2T | RNAseq |
| DNA replication checkpoint signaling | GO:0000076 | 9.10 × 10−3 | CDT1 | RNAseq |
| positive regulation of ubiquitin protein ligase activity | GO:1904668 | 9.10 × 10−3 | UBE2S | RNAseq |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butz, H.; Saskői, É.; Krokker, L.; Vereczki, V.; Alpár, A.; Likó, I.; Tóth, E.; Szőcs, E.; Cserepes, M.; Nagy, K.; et al. Context-Dependent Role of Glucocorticoid Receptor Alpha and Beta in Breast Cancer Cell Behaviour. Cells 2023, 12, 784. https://doi.org/10.3390/cells12050784
Butz H, Saskői É, Krokker L, Vereczki V, Alpár A, Likó I, Tóth E, Szőcs E, Cserepes M, Nagy K, et al. Context-Dependent Role of Glucocorticoid Receptor Alpha and Beta in Breast Cancer Cell Behaviour. Cells. 2023; 12(5):784. https://doi.org/10.3390/cells12050784
Chicago/Turabian StyleButz, Henriett, Éva Saskői, Lilla Krokker, Viktória Vereczki, Alán Alpár, István Likó, Erika Tóth, Erika Szőcs, Mihály Cserepes, Katalin Nagy, and et al. 2023. "Context-Dependent Role of Glucocorticoid Receptor Alpha and Beta in Breast Cancer Cell Behaviour" Cells 12, no. 5: 784. https://doi.org/10.3390/cells12050784