
The Role and Mechanism of Transglutaminase 2 in Regulating Hippocampal Neurogenesis after Traumatic Brain Injury
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Blade Penetrating Stab Wound to the Hippocampus
2.3. Tamoxifen Induction and BrdU Labeling
2.4. Cell Cultures and Collection of Conditioned Medium
2.5. Immunostaining
2.6. Quantitative RT-PCR
2.7. Western Blot
2.8. Lentivirus Production and In Vivo Grafting
2.9. Proliferation, Differentiation, Self-Renewal, and Migration Analyses of Cultured NSPCs
2.10. RNA Sequencing
2.11. Statistical Analysis
3. Results
3.1. Tgm2 Is Upregulated in NSPCs following TBI
3.2. Deletion of TGM2 in NSPCs Inhibits Hippocampal Neurogenesis
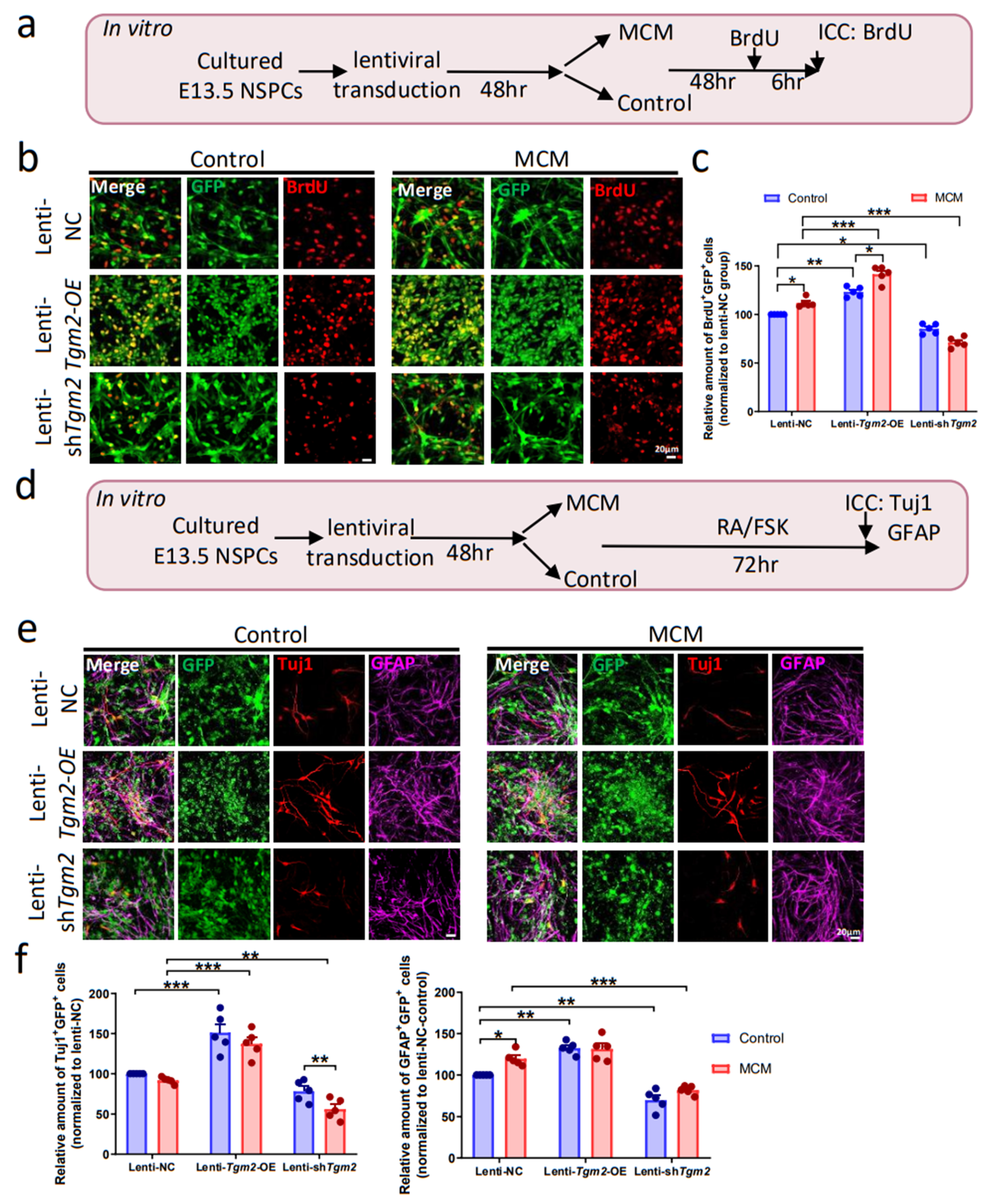
3.3. Overexpression of TGM2 in NSPCs Promotes Neurogenesis In Vitro
3.4. Overexpression of TGM2 Promotes NSPCs Migration In Vitro
3.5. Overexpression of TGM2 Enhance Adult Neurogenesis after TBI
3.6. TGM2 Regulates the Expression of Genes Associated with Proliferation and Differentiation of NSPCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Masel, B.E.; DeWitt, D.S. Traumatic brain injury: A disease process, not an event. J. Neurotrauma. 2010, 27, 1529–1540. [Google Scholar] [CrossRef]
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef]
- Dixon, K.J. Pathophysiology of Traumatic Brain Injury. Phys. Med. Rehabil. Clin. N. Am. 2017, 28, 215–225. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Weston, N.M.; Sun, D. The Potential of Stem Cells in Treatment of Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2018, 18, 1. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transpl. 2017, 26, 1118–1130. [Google Scholar] [CrossRef]
- Faiz, M.; Sachewsky, N.; Gascon, S.; Bang, K.W.; Morshead, C.M.; Nagy, A. Adult Neural Stem Cells from the Subventricular Zone Give Rise to Reactive Astrocytes in the Cortex after Stroke. Cell Stem Cell 2015, 17, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Agoston, D.V. Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J. Neurotrauma. 2010, 27, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Yu, A.; Wang, X.; Gao, X.; Chen, J. Post-Injury Treatment of 7,8-Dihydroxyflavone Promotes Neurogenesis in the Hippocampus of the Adult Mouse. J. Neurotrauma. 2016, 33, 2055–2064. [Google Scholar] [CrossRef]
- Sun, D.; Bullock, M.R.; Altememi, N.; Zhou, Z.; Hagood, S.; Rolfe, A.; McGinn, M.J.; Hamm, R.; Colello, R.J. The effect of epidermal growth factor in the injured brain after trauma in rats. J. Neurotrauma. 2010, 27, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Redell, J.B.; Maynard, M.E.; Underwood, E.L.; Vita, S.M.; Dash, P.K.; Kobori, N. Traumatic brain injury and hippocampal neurogenesis: Functional implications. Exp. Neurol. 2020, 331, 113372. [Google Scholar] [CrossRef] [PubMed]
- Villasana, L.E.; Westbrook, G.L.; Schnell, E. Neurologic impairment following closed head injury predicts post-traumatic neurogenesis. Exp. Neurol. 2014, 261, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Berninger, B.; Gotz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Luciano, D.; Jo, R.; Abdi, K.; Paez-Gonzalez, P.; Sheng, H.; Warner, D.S.; Liu, C.; Eroglu, C.; Kuo, C.T. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 2013, 497, 369–373. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [PubMed]
- Beninati, S.; Piacentini, M.; Bergamini, C.M. Transglutaminase 2, a double face enzyme. Amino Acids 2017, 49, 415–423. [Google Scholar] [CrossRef]
- Ientile, R.; Curro, M.; Caccamo, D. Transglutaminase 2 and neuroinflammation. Amino Acids 2015, 47, 19–26. [Google Scholar] [CrossRef]
- Min, B.; Chung, K.C. New insight into transglutaminase 2 and link to neurodegenerative diseases. BMB Rep. 2018, 51, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Bian, L.; Lv, D.; Bi, S.; Dai, Y.; Yang, K.; Lu, H.; Zhou, H.; Que, Y.; Wang, D.; et al. Enhanced neural differentiation of neural stem cells by sustained release of Shh from TG2 gene-modified EMSC co-culture in vitro. Amino Acids 2021, 53, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Que, Y.; Lv, D.; Bi, S.; Xu, Z.; Wang, D.; Zhang, Z. Overexpression of TG2 enhances the differentiation of ectomesenchymal stem cells into neuronlike cells and promotes functional recovery in adult rats following spinal cord injury. Mol. Med. Rep. 2019, 20, 2763–2773. [Google Scholar] [CrossRef] [PubMed]
- Tolentino, P.J.; DeFord, S.M.; Notterpek, L.; Glenn, C.C.; Pike, B.R.; Wang, K.K.; Hayes, R.L. Up-regulation of tissue-type transglutaminase after traumatic brain injury. J. Neurochem. 2002, 80, 579–588. [Google Scholar] [CrossRef]
- Kjell, J.; Fischer-Sternjak, J.; Thompson, A.J.; Friess, C.; Sticco, M.J.; Salinas, F.; Cox, J.; Martinelli, D.C.; Ninkovic, J.; Franze, K.; et al. Defining the Adult Neural Stem Cell Niche Proteome Identifies Key Regulators of Adult Neurogenesis. Cell Stem Cell 2020, 26, 277–293.e278. [Google Scholar] [CrossRef]
- Tucholski, J.; Johnson, G.V. Tissue transglutaminase directly regulates adenylyl cyclase resulting in enhanced cAMP-response element-binding protein (CREB) activation. J. Biol. Chem. 2003, 278, 26838–26843. [Google Scholar] [CrossRef]
- Bush, T.G.; Puvanachandra, N.; Horner, C.H.; Polito, A.; Ostenfeld, T.; Svendsen, C.N.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 1999, 23, 297–308. [Google Scholar] [CrossRef]
- He, B.D.; Liu, C.M.; Teng, Z.Q. A Mouse Model of Neurodegeneration Induced by Blade Penetrating Stab Wound to the Hippocampus. Biology 2022, 11, 1365. [Google Scholar] [CrossRef]
- Liu, P.P.; Tang, G.B.; Xu, Y.J.; Zeng, Y.Q.; Zhang, S.F.; Du, H.Z.; Teng, Z.Q.; Liu, C.M. MiR-203 Interplays with Polycomb Repressive Complexes to Regulate the Proliferation of Neural Stem/Progenitor Cells. Stem Cell Rep. 2017, 9, 190–202. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Deng, Y.S.; Dai, S.K.; Mi, T.W.; Li, R.Y.; Liu, P.P.; Liu, C.; He, B.D.; He, X.C.; Du, H.Z.; et al. Loss of microglial EED impairs synapse density, learning, and memory. Mol. Psychiatry 2022, 27, 2999–3009. [Google Scholar] [CrossRef]
- Liu, C.; Teng, Z.Q.; Santistevan, N.J.; Szulwach, K.E.; Guo, W.; Jin, P.; Zhao, X. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell 2010, 6, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Xu, Y.J.; Dai, S.K.; Du, H.Z.; Wang, Y.Y.; Li, X.G.; Teng, Z.Q.; Liu, C.M. Polycomb Protein EED Regulates Neuronal Differentiation through Targeting SOX11 in Hippocampal Dentate Gyrus. Stem Cell Rep. 2019, 13, 115–131. [Google Scholar] [CrossRef]
- Liu, C.; Dai, S.K.; Sun, Z.; Wang, Z.; Liu, P.P.; Du, H.Z.; Yu, S.; Liu, C.M.; Teng, Z.Q. GA-binding protein GABPbeta1 is required for the proliferation of neural stem/progenitor cells. Stem Cell Res. 2019, 39, 101501. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dai, S.K.; Shi, R.X.; He, X.C.; Wang, Y.Y.; He, B.D.; Sun, X.W.; Du, H.Z.; Liu, C.M.; Teng, Z.Q. Transcriptional profiling of microglia in the injured brain reveals distinct molecular features underlying neurodegeneration. Glia 2021, 69, 1292–1306. [Google Scholar] [CrossRef] [PubMed]
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539. [Google Scholar] [CrossRef]
- Walther, D.J.; Stahlberg, S.; Vowinckel, J. Novel roles for biogenic monoamines: From monoamines in transglutaminase-mediated post-translational protein modification to monoaminylation deregulation diseases. FEBS J. 2011, 278, 4740–4755. [Google Scholar] [CrossRef]
- Hummerich, R.; Thumfart, J.O.; Findeisen, P.; Bartsch, D.; Schloss, P. Transglutaminase-mediated transamidation of serotonin, dopamine and noradrenaline to fibronectin: Evidence for a general mechanism of monoaminylation. FEBS Lett. 2012, 586, 3421–3428. [Google Scholar] [CrossRef]
- Wilson, N.M.; Titus, D.J.; Oliva, A.A., Jr.; Furones, C.; Atkins, C.M. Traumatic Brain Injury Upregulates Phosphodiesterase Expression in the Hippocampus. Front. Syst. Neurosci. 2016, 10, 5. [Google Scholar] [CrossRef]
- Bigler, E.D.; Blatter, D.D.; Anderson, C.V.; Johnson, S.C.; Gale, S.D.; Hopkins, R.O.; Burnett, B. Hippocampal volume in normal aging and traumatic brain injury. AJNR Am. J. Neuroradiol. 1997, 18, 11–23. [Google Scholar]
- Tomaiuolo, F.; Carlesimo, G.A.; Di Paola, M.; Petrides, M.; Fera, F.; Bonanni, R.; Formisano, R.; Pasqualetti, P.; Caltagirone, C. Gross morphology and morphometric sequelae in the hippocampus, fornix, and corpus callosum of patients with severe non-missile traumatic brain injury without macroscopically detectable lesions: A T1 weighted MRI study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1314–1322. [Google Scholar] [CrossRef]
- Rola, R.; Mizumatsu, S.; Otsuka, S.; Morhardt, D.R.; Noble-Haeusslein, L.J.; Fishman, K.; Potts, M.B.; Fike, J.R. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp. Neurol. 2006, 202, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, S.; Teramoto, T.; Whalen, M.J.; Irizarry, M.C.; Takagi, Y.; Qiu, J.; Harada, J.; Waeber, C.; Breakefield, X.O.; Moskowitz, M.A. FGF-2 regulates neurogenesis and degeneration in the dentate gyrus after traumatic brain injury in mice. J. Clin. Investig. 2003, 112, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Jhaveri, D.J.; Bartlett, P.F. The therapeutic potential of endogenous hippocampal stem cells for the treatment of neurological disorders. Front. Cell. Neurosci. 2013, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Morganti-Kossman, M.C.; Lenzlinger, P.M.; Hans, V.; Stahel, P.; Csuka, E.; Ammann, E.; Stocker, R.; Trentz, O.; Kossmann, T. Production of cytokines following brain injury: Beneficial and deleterious for the damaged tissue. Mol. Psychiatry 1997, 2, 133–136. [Google Scholar] [CrossRef]
- Sung, P.S.; Lin, P.Y.; Liu, C.H.; Su, H.C.; Tsai, K.J. Neuroinflammation and Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 701. [Google Scholar] [CrossRef]
- Whitney, N.P.; Eidem, T.M.; Peng, H.; Huang, Y.; Zheng, J.C. Inflammation mediates varying effects in neurogenesis: Relevance to the pathogenesis of brain injury and neurodegenerative disorders. J. Neurochem. 2009, 108, 1343–1359. [Google Scholar] [CrossRef]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Sato, K. Effects of Microglia on Neurogenesis. Glia 2015, 63, 1394–1405. [Google Scholar] [CrossRef]
- Lin, C.Y.; Tsai, P.H.; Kandaswami, C.C.; Chang, G.D.; Cheng, C.H.; Huang, C.J.; Lee, P.P.; Hwang, J.J.; Lee, M.T. Role of tissue transglutaminase 2 in the acquisition of a mesenchymal-like phenotype in highly invasive A431 tumor cells. Mol. Cancer 2011, 10, 87. [Google Scholar] [CrossRef]
- Li, M.; Song, D.; Chen, X.; Wang, X.; Xu, L.; Yang, M.; Yang, J.; Kalvakolanu, D.V.; Wei, X.; Liu, X.; et al. RSL3 triggers glioma stem cell differentiation via the Tgm2/AKT/ID1 signaling axis. Biochim. Biophys. Acta. Mol. Basis Dis. 2022, 1868, 166529. [Google Scholar] [CrossRef]
- Shimada, J.; Suzuki, Y.; Kim, S.J.; Wang, P.C.; Matsumura, M.; Kojima, S. Transactivation via RAR/RXR-Sp1 interaction: Characterization of binding between Sp1 and GC box motif. Mol. Endocrinol. 2001, 15, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Tatsukawa, H.; Fukaya, Y.; Frampton, G.; Martinez-Fuentes, A.; Suzuki, K.; Kuo, T.F.; Nagatsuma, K.; Shimokado, K.; Okuno, M.; Wu, J.; et al. Role of transglutaminase 2 in liver injury via cross-linking and silencing of transcription factor Sp1. Gastroenterology 2009, 136, 1783–1795.e1710. [Google Scholar] [CrossRef] [PubMed]
- Tatsukawa, H.; Kojima, S. Recent advances in understanding the roles of transglutaminase 2 in alcoholic steatohepatitis. Cell Biol. Int. 2010, 34, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.D.; Wang, H.W.; Cai, X.J. Transcription factor Sp1 ameliorates sepsis-induced myocardial injury via ZFAS1/Notch signaling in H9C2 cells. Cytokine 2021, 140, 155426. [Google Scholar] [CrossRef] [PubMed]
- Tatsukawa, H.; Hitomi, K. Role of Transglutaminase 2 in Cell Death, Survival, and Fibrosis. Cells 2021, 10, 1842. [Google Scholar] [CrossRef] [PubMed]
- Sarang, Z.; Molnar, P.; Nemeth, T.; Gomba, S.; Kardon, T.; Melino, G.; Cotecchia, S.; Fesus, L.; Szondy, Z. Tissue transglutaminase (TG2) acting as G protein protects hepatocytes against Fas-mediated cell death in mice. Hepatology 2005, 42, 578–587. [Google Scholar] [CrossRef]
- Filiano, A.J.; Bailey, C.D.; Tucholski, J.; Gundemir, S.; Johnson, G.V. Transglutaminase 2 protects against ischemic insult, interacts with HIF1beta, and attenuates HIF1 signaling. FASEB J. 2008, 22, 2662–2675. [Google Scholar] [CrossRef]
- Gundemir, S.; Johnson, G.V. Intracellular localization and conformational state of transglutaminase 2: Implications for cell death. PLoS ONE 2009, 4, e6123. [Google Scholar] [CrossRef]
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Physiol. 2012, 227, 1130–1137. [Google Scholar] [CrossRef]
- Melino, G.; Annicchiarico-Petruzzelli, M.; Piredda, L.; Candi, E.; Gentile, V.; Davies, P.J.; Piacentini, M. Tissue transglutaminase and apoptosis: Sense and antisense transfection studies with human neuroblastoma cells. Mol. Cell. Biol. 1994, 14, 6584–6596. [Google Scholar] [CrossRef]
- Tucholski, J.; Roth, K.A.; Johnson, G.V. Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J. Neurochem. 2006, 97, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.E. Mechanisms of traumatic brain injury: Biomechanical, structural and cellular considerations. Crit. Care Nurs. Q. 2000, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.T.; Liu, C.M.; Teng, Z.Q. Mouse model of voluntary movement deficits induced by needlestick injuries to the primary motor cortex. J. Neurosci. Methods 2022, 365, 109380. [Google Scholar] [CrossRef] [PubMed]
- Blaya, M.O.; Wasserman, J.M.; Pieper, A.A.; Sick, T.J.; Bramlett, H.M.; Dietrich, W.D. Neurotherapeutic capacity of P7C3 agents for the treatment of Traumatic Brain Injury. Neuropharmacology 2019, 145, 268–282. [Google Scholar] [CrossRef]
- Zhang, Z.; Ishrat, S.; O’Bryan, M.; Klein, B.; Saraswati, M.; Robertson, C.; Kannan, S. Pediatric Traumatic Brain Injury Causes Long-Term Deficits in Adult Hippocampal Neurogenesis and Cognition. J. Neurotrauma. 2020, 37, 1656–1667. [Google Scholar] [CrossRef]
- Ibrahim, S.; Hu, W.; Wang, X.; Gao, X.; He, C.; Chen, J. Traumatic Brain Injury Causes Aberrant Migration of Adult-Born Neurons in the Hippocampus. Sci. Rep. 2016, 6, 21793. [Google Scholar] [CrossRef]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef]
- Keillor, J.W.; Apperley, K.Y.; Akbar, A. Inhibitors of tissue transglutaminase. Trends Pharmacol. Sci. 2015, 36, 32–40. [Google Scholar] [CrossRef]
- Penumatsa, K.C.; Toksoz, D.; Warburton, R.R.; Kharnaf, M.; Preston, I.R.; Kapur, N.K.; Khosla, C.; Hill, N.S.; Fanburg, B.L. Transglutaminase 2 in pulmonary and cardiac tissue remodeling in experimental pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L752–L762. [Google Scholar] [CrossRef]
- Dafik, L.; Albertelli, M.; Stamnaes, J.; Sollid, L.M.; Khosla, C. Activation and inhibition of transglutaminase 2 in mice. PLoS ONE 2012, 7, e30642. [Google Scholar] [CrossRef]
- Shinde, A.V.; Su, Y.; Palanski, B.A.; Fujikura, K.; Garcia, M.J.; Frangogiannis, N.G. Pharmacologic inhibition of the enzymatic effects of tissue transglutaminase reduces cardiac fibrosis and attenuates cardiomyocyte hypertrophy following pressure overload. J. Mol. Cell. Cardiol. 2018, 117, 36–48. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′–3′) | |
|---|---|---|
| Angpt1 | forward | CACATAGGGTGCAGCAACCA |
| reverse | CGTCGTGTTCTGGAAGAATGA | |
| Apoe | forward | CTGACAGGATGCCTAGCCG |
| reverse | CGCAGGTAATCCCAGAAGC | |
| Aqp4 | forward | CTTTCTGGAAGGCAGTCTCAG |
| reverse | CCACACCGAGCAAAACAAAGAT | |
| Cnp | forward | ACGAGTGCAAGACGCTATTCA |
| reverse | GGTGCCGTCGTGGTACTTC | |
| Dcx | forward | CATTTTGACGAACGAGACAAAGC |
| reverse | TGGAAGTCCATTCATCCGTGA | |
| Dll1 | forward | CAGGACCTTCTTTCGCGTATG |
| reverse | AAGGGGAATCGGATGGGGTT | |
| Dll3 | forward | CTGGTGTCTTCGAGCTACAAAT |
| reverse | TGCTCCGTATAGACCGGGAC | |
| Erbb4 | forward | TCCCCCAGGCTTTCAACATAC |
| reverse | GCTGTGTCCAATTTCACTCCTA | |
| Fgf2 | forward | GCGACCCACACGTCAAACTA |
| reverse | TCCCTTGATAGACACAACTCCTC | |
| Gfap | forward | CCCTGGCTCGTGTGGATTT |
| reverse | GACCGATACCACTCCTCTGTC | |
| Gpr17 | forward | CACCCTGTCAAGTCCCTCAAG |
| reverse | GTGGGCTGACTAGCAGTGG | |
| Hey1 | forward | GCGCGGACGAGAATGGAAA |
| reverse | TCAGGTGATCCACAGTCATCTG | |
| Hey2 | forward | AAGCGCCCTTGTGAGGAAAC |
| reverse | GGTAGTTGTCGGTGAATTGGAC | |
| Hes5 | forward | AGTCCCAAGGAGAAAAACCGA |
| reverse | GCTGTGTTTCAGGTAGCTGAC | |
| Itga2 | forward | TGTCTGGCGTATAATGTTGGC |
| reverse | CTTGTGGGTTCGTAAGCTGCT | |
| Mapt | forward | CGCTGGGCATGTGACTCAA |
| reverse | TTTCTTCTCGTCATTTCCTGTCC | |
| Neurod1 | forward | ATGACCAAATCATACAGCGAGAG |
| reverse | TCTGCCTCGTGTTCCTCGT | |
| Ntrk2 | forward | CTGGGGCTTATGCCTGCTG |
| reverse | AGGCTCAGTACACCAAATCCTA | |
| Omf | forward | CTTCCTGCCTGTTCATCCTTC |
| reverse | ATCCAGGGTTCTCAGATTGGT | |
| Plp1 | forward | CCAGAATGTATGGTGTTCTCCC |
| reverse | GGCCCATGAGTTTAAGGACG | |
| Ppp2R2B | forward | ACGGGAGAGTTACTAGCGAC |
| reverse | GTAAGCTGCGTTTTGTTGAGG | |
| Pdgfrb | forward | TTCCAGGAGTGATACCAGCTT |
| reverse | AGGGGGCGTGATGACTAGG | |
| Slcla2 | forward | ACAATATGCCCAAGCAGGTAGA |
| reverse | CTTTGGCTCATCGGAGCTGA | |
| Sox10 | forward | CGGACGATGACAAGTTCCCC |
| reverse | GTGAGGGTACTGGTCGGCT | |
| Tgm2 | forward | CGCAACAGGGCTTCATCTAC |
| reverse | CCCGACTACGGTTCTTCAGGA | |
| Tle1 | forward | CCAGTACCTCTCACGCCTCA |
| reverse | GCCCACTCAGAGCACTAGAC | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, R.-X.; Liu, C.; Xu, Y.-J.; Wang, Y.-Y.; He, B.-D.; He, X.-C.; Du, H.-Z.; Hu, B.; Jiao, J.; Liu, C.-M.; et al. The Role and Mechanism of Transglutaminase 2 in Regulating Hippocampal Neurogenesis after Traumatic Brain Injury. Cells 2023, 12, 558. https://doi.org/10.3390/cells12040558
Shi R-X, Liu C, Xu Y-J, Wang Y-Y, He B-D, He X-C, Du H-Z, Hu B, Jiao J, Liu C-M, et al. The Role and Mechanism of Transglutaminase 2 in Regulating Hippocampal Neurogenesis after Traumatic Brain Injury. Cells. 2023; 12(4):558. https://doi.org/10.3390/cells12040558
Chicago/Turabian StyleShi, Ruo-Xi, Cong Liu, Ya-Jie Xu, Ying-Ying Wang, Bao-Dong He, Xuan-Cheng He, Hong-Zhen Du, Baoyang Hu, Jianwei Jiao, Chang-Mei Liu, and et al. 2023. "The Role and Mechanism of Transglutaminase 2 in Regulating Hippocampal Neurogenesis after Traumatic Brain Injury" Cells 12, no. 4: 558. https://doi.org/10.3390/cells12040558