
The Crosstalk between Microbiome and Mitochondrial Homeostasis in Neurodegeneration

Abstract
:1. Introduction
2. Animal Models Employed to Investigate the Communication between Host-Microbes, Mitochondria and Neuronal Function
3. Bacterial Metabolites Affecting Mitochondrial Function and Dynamics
4. Mitochondria in the Gut–Brain Axis
5. Neurodegenerative Disorders: Mitochondria and Microbiome

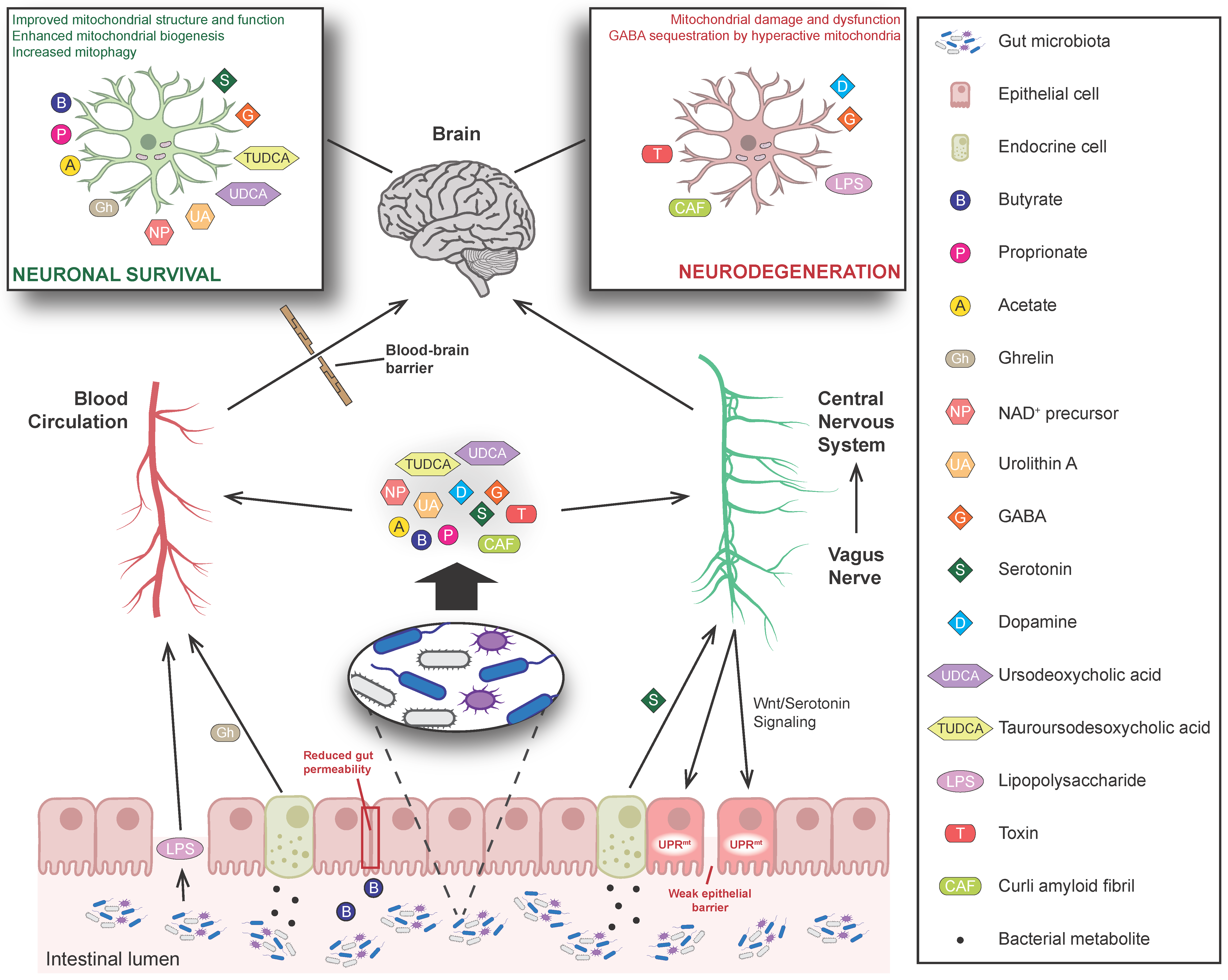{kind=link}
| Microbiome Metabolite | Mitochondrial Effect | Neuronal Effect | Human Disorder | Reference | |
|---|---|---|---|---|---|
| Lipopolysaccharide (LPS) | Enhanced mitochondrial fission and fragmentation Increased ROS production Downregulated mitophagy | Microglia inflammation | PD, AD | [55,56,57,58] | |
| Short Chain Fatty Acids (SCFAs) | Butyrate | Modified mitochondrial activity Increased ATP production Enhanced mitochondrial biogenesis | Increased neuronal function and proliferation Decreased brain inflammation Restricted action of ceramides | PD | [135,151,152,153,154,155,214] |
| Upregulated mtDNA copy number Increased mitochondrial biogenesis Increased oxidative stress | Increased neural stem cell self-renewal, differentiation and viability | ALS | [70,188,189,190,191] | ||
| Propionate | Altered mitochondrial morphology Restored mitochondrial respiration | Increased Treg cell suppressive capacity | MS | [148,149] | |
| Acetate | Altered mitochondrial mass Rectified function of electron transport complex II | Reduced microglial phagocytosis of Aβ Increased Aβ deposition Microglia maturation | AD | [150] | |
| Urolithin A (UA) | Restricted mitochondria ER-interactions Reduced Ca2+ influx from the ER Reduced mtROS accumulation | Suppressed Tau phosphorylation Decreased APP and BACE-1 expression Reduced Aβ production and cognitive impairment | AD | [165,166] | |
| Induced mitophagy | Enhanced microglial Aβ plaque clearance Reduced neuroinflammation Abolished Tau hyperphosphorylation | AD | [97] | ||
| Induced mitophagy Enhanced mitochondrial biogenesis | Reduced loss of dopaminergic neurons Ameliorated behavioral deficits and neuroinflammation | AD | [206,207] | ||
| NAD+ precursors | Induced mitophagy | Enhanced microglial Aβ plaque clearance Suppressed neuroinflammation Abolished Tau hyperphosphorylation | AD | [97,167,168,169] | |
| Protected mitochondrial function | Reduced neurodegeneration Improved motor function | PD | [204,205] | ||
| Altered expression patterns of mitochondrial genes Increased mitochondrial integrity and function | Enhanced motor neuron survival Decreased glial activation | ALS | [176,180,181,182,183,184,185] | ||
| Hormones | Ghrelin | Inhibited mitochondrial depolarization and ROS generation | Neuroprotection Improved cognitive function | AD | [170,171,172] |
| Restored mitochondrial function Reduced apoptosis | Reduced dopamine depletion and dopaminergic neuronal loss | PD | [210,211] | ||
| Neurotransmitters | Dopamine | Decreased mitochondrial respiration Induced mitochondrial depolarization | Dysfunction of the striatum | Schizophrenia | [156,157] |
| Serotonin | Increased mitochondrial biogenesis and function (respiration and ATP production) | Reduced neurotoxic effect of oxidative stress | PD | [158] | |
| GABA | Increased mitochondrial activity | Reduced GABAergic signaling Defective social behavior | Autism, Schizophrenia | [159] | |
| Secondary bile acids | Ursodeoxycholic acid (UDCA) | Upregulated mitophagy Reduced apoptosis | Rescued dopaminergic neurons Improved motor function | PD | [208,209] |
| Tauroursodesoxy cholic acid (TUDCA) | |||||
| Antibiotics | Rapamycin | Mitochondrial impairment | Augmented motor neuron degeneration Induced apoptosis | ALS | [177] |
| Minocycline | Inhibition of cytochrome c release Reduction of Ca2+ overload Changes in transmembrane potential | Neuroprotection Reduced neuroinflammation | ALS, PD | [178,179,215,216] | |
6. Concluding Remarks and Potential Therapeutic Interventions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wiley, N.C.; Dinan, T.G.; Ross, R.P.; Stanton, C.; Clarke, G.; Cryan, J.F. The microbiota-gut-brain axis as a key regulator of neural function and the stress response: Implications for human and animal health. J. Anim. Sci. 2017, 95, 3225–3246. [Google Scholar]
- Rangaraju, V.; Lewis, T.L., Jr.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Drerup, C.M. Axonal Transport and Mitochondrial Function in Neurons. Front. Cell. Neurosci. 2019, 13, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunissen, F.; West, P.K.; Brennan, S.; Petrović, B.; Hooshmand, K.; Akkari, P.A.; Keon, M.; Guennewig, B. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl. Neurodegener. 2021, 10, 46. [Google Scholar] [CrossRef]
- Rani, L.; Mondal, A.C. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: Pathogenic and therapeutic implications. Mitochondrion 2020, 50, 25–34. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in health, aging and diseases: The epigenetic perspective. Biogerontology 2015, 16, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, A.P.; Bidkhori, G.; Shoaie, S.; Clarke, E.; Morrin, H.; Hye, A.; Williams, G.; Ballard, C.; Francis, P.; Aarsland, D. Postmortem Cortical Transcriptomics of Lewy Body Dementia Reveal Mitochondrial Dysfunction and Lack of Neuroinflammation. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2020, 28, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; Liévens, J.C. Mitochondrial quality control in amyotrophic lateral sclerosis: Towards a common pathway? Neural Regen. Res. 2017, 12, 1052–1061. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [Green Version]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Neish, A.S. Redox signaling mediated by the gut microbiota. Free. Radic. Res. 2013, 47, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef] [Green Version]
- Lucattini, R.; Likic, V.A.; Lithgow, T. Bacterial proteins predisposed for targeting to mitochondria. Mol. Biol. Evol. 2004, 21, 652–658. [Google Scholar] [CrossRef]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Chen, B.; Zhao, Z.; He, N.; Zeng, Z.; Wu, B.; Fukushima, Y.; Dai, M.; Huang, Q.; Xu, D.; et al. Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res. Cardiol. 2013, 108, 342. [Google Scholar] [CrossRef]
- Bauerly, K.; Harris, C.; Chowanadisai, W.; Graham, J.; Havel, P.J.; Tchaparian, E.; Satre, M.; Karliner, J.S.; Rucker, R.B. Altering pyrroloquinoline quinone nutritional status modulates mitochondrial, lipid, and energy metabolism in rats. PLoS ONE 2011, 6, e21779. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.B.; Chowanadisai, W.; Mishchuk, D.O.; Satre, M.A.; Slupsky, C.M.; Rucker, R.B. Dietary pyrroloquinoline quinone (PQQ) alters indicators of inflammation and mitochondrial-related metabolism in human subjects. J. Nutr. Biochem. 2013, 24, 2076–2084. [Google Scholar] [CrossRef] [Green Version]
- Gruber, J.; Kennedy, B.K. Microbiome and Longevity: Gut Microbes Send Signals to Host Mitochondria. Cell 2017, 169, 1168–1169. [Google Scholar] [CrossRef] [Green Version]
- Ballard, J.W.O.; Towarnicki, S.G. Mitochondria, the gut microbiome and ROS. Cell. Signal. 2020, 75, 109737. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Sivaramakrishnan, P.; Lin, C.J.; Neve, I.A.A.; He, J.; Tay, L.W.R.; Sowa, J.N.; Sizovs, A.; Du, G.; Wang, J.; et al. Microbial Genetic Composition Tunes Host Longevity. Cell 2017, 169, 1249–1262.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, P.; Darra, A.; Agrawal, A. Microbe-mitochondrion crosstalk and health: An emerging paradigm. Mitochondrion 2018, 39, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Saint-Georges-Chaumet, Y.; Edeas, M. Microbiota-mitochondria inter-talk: Consequence for microbiota-host interaction. Pathog. Dis. 2016, 74, ftv096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, H.; Rao, X.; Yu, Y.; Li, W.; Zheng, P.; Zhao, L.; Zhou, C.; Pu, J.; Yang, D.; et al. Comprehensive analysis of the lysine acetylome and succinylome in the hippocampus of gut microbiota-dysbiosis mice. J. Adv. Res. 2021, 30, 27–38. [Google Scholar] [CrossRef]
- Gnainsky, Y.; Zfanya, N.; Elgart, M.; Omri, E.; Brandis, A.; Mehlman, T.; Itkin, M.; Malitsky, S.; Adamski, J.; Soen, Y. Systemic Regulation of Host Energy and Oogenesis by Microbiome-Derived Mitochondrial Coenzymes. Cell Rep. 2021, 34, 108583. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Nagpal, J.; Cryan, J.F. Microbiota-brain interactions: Moving toward mechanisms in model organisms. Neuron 2021, 109, 3930–3953. [Google Scholar] [CrossRef]
- Cox, L.M.; Maghzi, A.H.; Liu, S.; Tankou, S.K.; Dhang, F.H.; Willocq, V.; Song, A.; Wasén, C.; Tauhid, S.; Chu, R.; et al. Gut Microbiome in Progressive Multiple Sclerosis. Ann. Neurol. 2021, 89, 1195–1211. [Google Scholar] [CrossRef]
- Zhang, F.; Berg, M.; Dierking, K.; Félix, M.A.; Shapira, M.; Samuel, B.S.; Schulenburg, H. Caenorhabditis elegans as a Model for Microbiome Research. Front. Microbiol. 2017, 8, 485. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef]
- Sokolowski, M.B. Drosophila: Genetics meets behaviour. Nat. Rev. Genet. 2001, 2, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Kitani-Morii, F.; Friedland, R.P.; Yoshida, H.; Mizuno, T. Drosophila as a Model for Microbiota Studies of Neurodegeneration. J. Alzheimer’s Dis. JAD 2021, 84, 479–490. [Google Scholar] [CrossRef]
- Clark, R.I.; Walker, D.W. Role of gut microbiota in aging-related health decline: Insights from invertebrate models. Cell. Mol. Life Sci. CMLS 2018, 75, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Wang, L.; Jiang, B. The Role of Gut Microbiota in Aging and Aging Related Neurodegenerative Disorders: Insights from Drosophila Model. Life 2021, 11, 855. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.A.; Lang, J.M.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host-microbe model system. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef] [PubMed]
- Jalali, D.; Guevarra, J.A.; Martinez, L.; Hung, L.; Vonhoff, F.J. Nutraceutical and Probiotic Approaches to Examine Molecular Interactions of the Amyloid Precursor Protein APP in Drosophila Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7022. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Prakash, S. A novel synbiotic delays Alzheimer’s disease onset via combinatorial gut-brain-axis signaling in Drosophila melanogaster. PLoS ONE 2019, 14, e0214985. [Google Scholar] [CrossRef] [Green Version]
- Westfall, S.; Lomis, N.; Prakash, S. Longevity extension in Drosophila through gut-brain communication. Sci. Rep. 2018, 8, 8362. [Google Scholar] [CrossRef]
- Batista, L.L.; Malta, S.M.; Guerra Silva, H.C.; Borges, L.D.F.; Rocha, L.O.; da Silva, J.R.; Rodrigues, T.S.; Venturini, G.; Padilha, K.; da Costa Pereira, A.; et al. Kefir metabolites in a fly model for Alzheimer’s disease. Sci. Rep. 2021, 11, 11262. [Google Scholar] [CrossRef]
- Ho, L.; Zhao, D.; Ono, K.; Ruan, K.; Mogno, I.; Tsuji, M.; Carry, E.; Brathwaite, J.; Sims, S.; Frolinger, T.; et al. Heterogeneity in gut microbiota drive polyphenol metabolism that influences α-synuclein misfolding and toxicity. J. Nutr. Biochem. 2019, 64, 170–181. [Google Scholar] [CrossRef]
- Tan, F.H.P.; Liu, G.; Lau, S.A.; Jaafar, M.H.; Park, Y.H.; Azzam, G.; Li, Y.; Liong, M.T. Lactobacillus probiotics improved the gut microbiota profile of a Drosophila melanogaster Alzheimer’s disease model and alleviated neurodegeneration in the eye. Benef. Microbes 2020, 11, 79–89. [Google Scholar] [CrossRef]
- Carregosa, D.; Mota, S.; Ferreira, S.; Alves-Dias, B.; Loncarevic-Vasiljkovic, N.; Crespo, C.L.; Menezes, R.; Teodoro, R.; Santos, C.N.D. Overview of Beneficial Effects of (Poly)phenol Metabolites in the Context of Neurodegenerative Diseases on Model Organisms. Nutrients 2021, 13, 2940. [Google Scholar] [CrossRef]
- Walsh, D.; McCarthy, J.; O’Driscoll, C.; Melgar, S. Pattern recognition receptors--molecular orchestrators of inflammation in inflammatory bowel disease. Cytokine Growth Factor Rev. 2013, 24, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chang, M. Roles of PRR-Mediated Signaling Pathways in the Regulation of Oxidative Stress and Inflammatory Diseases. Int. J. Mol. Sci. 2021, 22, 7688. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emre, Y.; Nübel, T. Uncoupling protein UCP2: When mitochondrial activity meets immunity. FEBS Lett. 2010, 584, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Cui, L.; Gao, J.; Zhu, M.; Zhang, Y.; Zhang, H.L. Gut Microbial Metabolites in Parkinson’s Disease: Implications of Mitochondrial Dysfunction in the Pathogenesis and Treatment. Mol. Neurobiol. 2021, 58, 3745–3758. [Google Scholar] [CrossRef]
- Harland, M.; Torres, S.; Liu, J.; Wang, X. Neuronal Mitochondria Modulation of LPS-Induced Neuroinflammation. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 1756–1765. [Google Scholar] [CrossRef]
- Park, J.; Min, J.S.; Kim, B.; Chae, U.B.; Yun, J.W.; Choi, M.S.; Kong, I.K.; Chang, K.T.; Lee, D.S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, X.; Chen, H.; Feng, J.; Yan, M.; Yu, Y. Dexmedetomidine alleviates LPS-induced apoptosis and inflammation in macrophages by eliminating damaged mitochondria via PINK1 mediated mitophagy. Int. Immunopharmacol. 2019, 73, 471–481. [Google Scholar] [CrossRef]
- Lebreton, A.; Stavru, F.; Cossart, P. Organelle targeting during bacterial infection: Insights from Listeria. Trends Cell Biol. 2015, 25, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Rudel, T.; Kepp, O.; Kozjak-Pavlovic, V. Interactions between bacterial pathogens and mitochondrial cell death pathways. Nat. Rev. Microbiol. 2010, 8, 693–705. [Google Scholar] [CrossRef]
- Liu, H.; Bao, W.; Lin, M.; Niu, H.; Rikihisa, Y. Ehrlichia type IV secretion effector ECH0825 is translocated to mitochondria and curbs ROS and apoptosis by upregulating host MnSOD. Cell. Microbiol. 2012, 14, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Holscher, H.D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumeng, L.; Davis, E.J. The oxidation of acetate by liver mitochondria. FEBS Lett. 1973, 29, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Kiviluoma, K.T.; Peuhkurinen, K.J.; Hassinen, I.E. Adenine nucleotide transport and adenosine production in isolated rat heart mitochondria during acetate metabolism. Biochim. Et Biophys. Acta 1989, 974, 274–281. [Google Scholar] [CrossRef]
- Jiang, L.; Gulanski, B.I.; De Feyter, H.M.; Weinzimer, S.A.; Pittman, B.; Guidone, E.; Koretski, J.; Harman, S.; Petrakis, I.L.; Krystal, J.H.; et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Investig. 2013, 123, 1605–1614. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Nepelska, M.; de Wouters, T.; Jacouton, E.; Béguet-Crespel, F.; Lapaque, N.; Doré, J.; Arulampalam, V.; Blottière, H.M. Commensal gut bacteria modulate phosphorylation-dependent PPARγ transcriptional activity in human intestinal epithelial cells. Sci. Rep. 2017, 7, 43199. [Google Scholar] [CrossRef] [Green Version]
- Lamichane, S.; Dahal Lamichane, B.; Kwon, S.M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. Int. J. Mol. Sci. 2018, 19, 949. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Singhal, R.; Shah, Y.M. Oxygen battle in the gut: Hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J. Biol. Chem. 2020, 295, 10493–10505. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Plotegher, N.; Bubacco, L.; Greggio, E.; Civiero, L. Ceramides in Parkinson’s Disease: From Recent Evidence to New Hypotheses. Front. Neurosci. 2019, 13, 330. [Google Scholar] [CrossRef]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef]
- Gudz, T.I.; Tserng, K.Y.; Hoppel, C.L. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 1997, 272, 24154–24158. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.; Andriamihaja, M.; Lan, A.; Khodorova, N.; Audebert, M.; Blouin, J.M.; Grauso, M.; Lancha, L.; Benetti, P.H.; Benamouzig, R.; et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: The adaptive response. Free. Radic. Biol. Med. 2016, 93, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottawea, W.; Chiang, C.K.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, S.; Singh, S.; Taniere, P.; Langman, M.J.; Eggo, M.C. Sulfide-detoxifying enzymes in the human colon are decreased in cancer and upregulated in differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G288–G296. [Google Scholar] [CrossRef] [Green Version]
- Magnúsdóttir, S.; Ravcheev, D.; de Crécy-Lagard, V.; Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 2015, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Jackson, T.M.; Rawling, J.M.; Roebuck, B.D.; Kirkland, J.B. Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP-ribose) levels but do not affect diethylnitrosamine-induced altered hepatic foci in Fischer-344 rats. J. Nutr. 1995, 125, 1455–1461. [Google Scholar]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [Green Version]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 2019, 24, 405–418.e7. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef]
- Chellappa, K.; McReynolds, M.R.; Lu, W.; Zeng, X.; Makarov, M.; Hayat, F.; Mukherjee, S.; Bhat, Y.R.; Lingala, S.R.; Shima, R.T.; et al. NAD precursors cycle between host tissues and the gut microbiome. Cell Metab. 2022, 34, 1947–1959.e5. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Beloborodova, N.; Bairamov, I.; Olenin, A.; Shubina, V.; Teplova, V.; Fedotcheva, N. Effect of phenolic acids of microbial origin on production of reactive oxygen species in mitochondria and neutrophils. J. Biomed. Sci. 2012, 19, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesper, M.; Nonnast, A.B.H.; Kumowski, N.; Stoehr, R.; Schuett, K.; Marx, N.; Kappel, B.A. Gut-Derived Metabolite Indole-3-Propionic Acid Modulates Mitochondrial Function in Cardiomyocytes and Alters Cardiac Function. Front. Med. 2021, 8, 648259. [Google Scholar] [CrossRef] [PubMed]
- Behera, J.; Ison, J.; Voor, M.J.; Tyagi, N. Probiotics Stimulate Bone Formation in Obese Mice via Histone Methylations. Theranostics 2021, 11, 8605–8623. [Google Scholar] [CrossRef] [PubMed]
- Janssens, Y.; Wynendaele, E.; Verbeke, F.; Debunne, N.; Gevaert, B.; Audenaert, K.; Van DeWiele, C.; De Spiegeleer, B. Screening of quorum sensing peptides for biological effects in neuronal cells. Peptides 2018, 101, 150–156. [Google Scholar] [CrossRef] [PubMed]
- De Spiegeleer, A.; Elewaut, D.; Van Den Noortgate, N.; Janssens, Y.; Debunne, N.; Van Langenhove, S.; Govindarajan, S.; De Spiegeleer, B.; Wynendaele, E. Quorum sensing molecules as a novel microbial factor impacting muscle cells. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165646. [Google Scholar] [CrossRef]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Hartsough, L.A.; Park, M.; Kotlajich, M.V.; Lazar, J.T.; Han, B.; Lin, C.J.; Musteata, E.; Gambill, L.; Wang, M.C.; Tabor, J.J. Optogenetic control of gut bacterial metabolism to promote longevity. eLife 2020, 9, e56849. [Google Scholar] [CrossRef]
- Lin, C.J.; Wang, M.C. Microbial metabolites regulate host lipid metabolism through NR5A-Hedgehog signalling. Nat. Cell Biol. 2017, 19, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Josephson, H.; Ntzouni, M.; Skoglund, C.; Linder, S.; Turkina, M.V.; Vikström, E. Pseudomonas aeruginosa N-3-Oxo-Dodecanoyl-Homoserine Lactone Impacts Mitochondrial Networks Morphology, Energetics, and Proteome in Host Cells. Front. Microbiol. 2020, 11, 1069. [Google Scholar] [CrossRef]
- Schwarzer, C.; Fu, Z.; Shuai, S.; Babbar, S.; Zhao, G.; Li, C.; Machen, T.E. Pseudomonas aeruginosa homoserine lactone triggers apoptosis and Bak/Bax-independent release of mitochondrial cytochrome C in fibroblasts. Cell. Microbiol. 2014, 16, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Yoshida, K.; Ikegame, M.; Okamura, H. Quorum sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone: An all-rounder in mammalian cell modification. J. Oral Biosci. 2020, 62, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, P.A.; Tamaki, F.K.; Cardoso, A.R.; Marana, S.R.; Kowaltowski, A.J. H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol. 2015, 4, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.; Zieba, J.K.; Foegeding, N.J.; Torres, T.P.; Shelton, C.D.; Shealy, N.G.; Byndloss, A.J.; Cevallos, S.A.; Gertz, E.; Tiffany, C.R.; et al. High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science 2021, 373, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.R.; Kakimoto, P.A.; Kowaltowski, A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: Role of very long chain acyl-CoA dehydrogenases. PLoS ONE 2013, 8, e77088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, M.; Künstner, A.; Schilf, P.; Sünderhauf, A.; Rupp, J.; Jöhren, O.; Schwaninger, M.; Sina, C.; Baines, J.F.; Ibrahim, S.M. Mitochondrial gene polymorphism is associated with gut microbial communities in mice. Sci. Rep. 2017, 7, 15293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Coarfa, C.; Qin, X.; Bonnen, P.E.; Milosavljevic, A.; Versalovic, J.; Aagaard, K. mtDNA haplogroup and single nucleotide polymorphisms structure human microbiome communities. BMC Genom. 2014, 15, 257. [Google Scholar] [CrossRef] [Green Version]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Kiely, B.; Cryan, J.F.; Dinan, T.G. Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience 2010, 170, 1179–1188. [Google Scholar] [CrossRef]
- Lyte, M. Microbial endocrinology: Host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes 2014, 5, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, E.; Ross, R.P.; O’Toole, P.W.; Fitzgerald, G.F.; Stanton, C. γ-Aminobutyric acid production by culturable bacteria from the human intestine. J. Appl. Microbiol. 2012, 113, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [Green Version]
- Leeuwendaal, N.K.; Cryan, J.F.; Schellekens, H. Gut peptides and the microbiome: Focus on ghrelin. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 243–252. [Google Scholar] [CrossRef]
- Del Rio, D.; Zimetti, F.; Caffarra, P.; Tassotti, M.; Bernini, F.; Brighenti, F.; Zini, A.; Zanotti, I. The Gut Microbial Metabolite Trimethylamine-N-Oxide Is Present in Human Cerebrospinal Fluid. Nutrients 2017, 9, 1053. [Google Scholar] [CrossRef] [Green Version]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Konjar, Š.; Pavšič, M.; Veldhoen, M. Regulation of Oxygen Homeostasis at the Intestinal Epithelial Barrier Site. Int. J. Mol. Sci. 2021, 22, 9170. [Google Scholar] [CrossRef]
- MacFabe, D.F.; Cain, D.P.; Rodriguez-Capote, K.; Franklin, A.E.; Hoffman, J.E.; Boon, F.; Taylor, A.R.; Kavaliers, M.; Ossenkopp, K.P. Neurobiological effects of intraventricular propionic acid in rats: Possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav. Brain Res. 2007, 176, 149–169. [Google Scholar] [CrossRef]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23, 19260. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, J.; Du, J.; Wang, F.; Fang, R.; Yu, C.; Xiong, J.; Chen, W.; Lu, Z.; Liu, J. Clostridium butyricum exerts a neuroprotective effect in a mouse model of traumatic brain injury via the gut-brain axis. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2018, 30, e13260. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.; Wang, F.; Ding, G.; Chen, W.; Fang, R.; Yao, Y.; Pang, M.; Lu, Z.Q.; Liu, J. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 2016, 1642, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Val-Laillet, D.; Guérin, S.; Coquery, N.; Nogret, I.; Formal, M.; Romé, V.; Le Normand, L.; Meurice, P.; Randuineau, G.; Guilloteau, P.; et al. Oral sodium butyrate impacts brain metabolism and hippocampal neurogenesis, with limited effects on gut anatomy and function in pigs. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 2160–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyles, L.; Snelling, T.; Umlai, U.K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matt, S.M.; Allen, J.M.; Lawson, M.A.; Mailing, L.J.; Woods, J.A.; Johnson, R.W. Butyrate and Dietary Soluble Fiber Improve Neuroinflammation Associated With Aging in Mice. Front. Immunol. 2018, 9, 1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.S.; Chen, P.S.; Chuang, D.M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Kaszaki, J.; Erces, D.; Varga, G.; Szabó, A.; Vécsei, L.; Boros, M. Kynurenines and intestinal neurotransmission: The role of N-methyl-D-aspartate receptors. J. Neural Transm. 2012, 119, 211–223. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Markey, S.P.; Heyes, M.P. Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse. Neuroscience 1992, 51, 25–39. [Google Scholar] [CrossRef]
- Benakis, C.; Brea, D.; Caballero, S.; Faraco, G.; Moore, J.; Murphy, M.; Sita, G.; Racchumi, G.; Ling, L.; Pamer, E.G.; et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 2016, 22, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.K.; Janowitz, C.; Metea, C.; Asquith, M.; Karstens, L.; Rosenbaum, J.T.; Lin, P. Short chain fatty acids ameliorate immune-mediated uveitis partially by altering migration of lymphocytes from the intestine. Sci. Rep. 2017, 7, 11745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.C.; Cao, Z.S.; Chang, K.M.; Juang, J.L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef]
- Raval, U.; Harary, J.M.; Zeng, E.; Pasinetti, G.M. The dichotomous role of the gut microbiome in exacerbating and ameliorating neurodegenerative disorders. Expert Rev. Neurother. 2020, 20, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinto, M.F.; Empadinhas, N.; Cardoso, S.M. The neuromicrobiology of Parkinson’s disease: A unifying theory. Ageing Res. Rev. 2021, 70, 101396. [Google Scholar] [CrossRef] [PubMed]
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16. [Google Scholar] [CrossRef]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Erny, D.; Dokalis, N.; Mezö, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276.e7. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Mitochondria could be a potential key mediator linking the intestinal microbiota to depression. J. Cell. Biochem. 2020, 121, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, M.W.; Alim, I.; Bultman, S.J.; Ratan, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Valvassori, S.S.; Varela, R.B.; Ferreira, C.L.; Rochi, N.; Benedet, J.; Scaini, G.; Kapczinski, F.; Streck, E.L.; Zugno, A.I.; et al. Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav. Pharmacol. 2011, 22, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.; Ojha, U.; Bhurtel, S.; Bing, G.; Choi, D.Y. Lipopolysaccharide-induced functional and structural injury of the mitochondria in the nigrostriatal pathway. Neurosci. Res. 2017, 114, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [Green Version]
- Czerniczyniec, A.; Bustamante, J.; Lores-Arnaiz, S. Dopamine modifies oxygen consumption and mitochondrial membrane potential in striatal mitochondria. Mol. Cell. Biochem. 2010, 341, 251–257. [Google Scholar] [CrossRef]
- Brenner-Lavie, H.; Klein, E.; Zuk, R.; Gazawi, H.; Ljubuncic, P.; Ben-Shachar, D. Dopamine modulates mitochondrial function in viable SH-SY5Y cells possibly via its interaction with complex I: Relevance to dopamine pathology in schizophrenia. Biochim. Biophys. Acta 2008, 1777, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Tiwari, P.; Ghai, U.; Gupta, S.; Figueiredo, D.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; et al. Serotonin regulates mitochondrial biogenesis and function in rodent cortical neurons via the 5-HT(2A) receptor and SIRT1-PGC-1α axis. Proc. Natl. Acad. Sci. USA 2019, 116, 11028–11037. [Google Scholar] [CrossRef] [Green Version]
- Kanellopoulos, A.K.; Mariano, V.; Spinazzi, M.; Woo, Y.J.; McLean, C.; Pech, U.; Li, K.W.; Armstrong, J.D.; Giangrande, A.; Callaerts, P.; et al. Aralar Sequesters GABA into Hyperactive Mitochondria, Causing Social Behavior Deficits. Cell 2020, 180, 1178–1197.e20. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, X.; Chen, P.; Liu, L.; Xin, N.; Tian, Y.; Dillin, A. The Mitochondrial Unfolded Protein Response Is Mediated Cell-Non-autonomously by Retromer-Dependent Wnt Signaling. Cell 2018, 174, 870–883.e17. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Keita, Å.V.; Phan, V.; McKay, C.M.; Schoultz, I.; Lee, J.; Murphy, M.P.; Fernando, M.; Ronaghan, N.; Balce, D.; et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am. J. Pathol. 2014, 184, 2516–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.Q.; Haikal, C.; Li, W.; Li, J.Y. Gut Inflammation in Association With Pathogenesis of Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.H.; Song, X.X.; Liu, X.L.; Chen, S.D.; Tang, H.D. Inflammatory pathways in Alzheimer’s disease mediated by gut microbiota. Ageing Res. Rev. 2021, 68, 101317. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered Gut Microbiota Related to Inflammatory Responses in Patients With Huntington’s Disease. Front. Immunol. 2020, 11, 603594. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Bacskai, B.J.; Calvo-Rodriguez, M. Mitochondria-ER contacts and glucose: The powerhouse of Alzheimer’s disease? Cell Calcium 2021, 97, 102434. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202. [Google Scholar] [CrossRef]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; He, H.J.; Xiong, X.; Zhou, S.; Wang, W.W.; Feng, L.; Han, R.; Xie, C.L. NAD+ in Alzheimer’s Disease: Molecular Mechanisms and Systematic Therapeutic Evidence Obtained in vivo. Front. Cell Dev. Biol. 2021, 9, 668491. [Google Scholar] [CrossRef]
- Martins, I.; Gomes, S.; Costa, R.O.; Otvos, L.; Oliveira, C.R.; Resende, R.; Pereira, C.M. Leptin and ghrelin prevent hippocampal dysfunction induced by Aβ oligomers. Neuroscience 2013, 241, 41–51. [Google Scholar] [CrossRef]
- Moon, M.; Choi, J.G.; Nam, D.W.; Hong, H.S.; Choi, Y.J.; Oh, M.S.; Mook-Jung, I. Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-β1-42 oligomer-injected mice. J. Alzheimer’s Dis. JAD 2011, 23, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, M.; Cha, M.Y.; Mook-Jung, I. Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimer’s Dis. JAD 2014, 41, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Forloni, G. Doxycycline for Alzheimer’s Disease: Fighting β-Amyloid Oligomers and Neuroinflammation. Front. Pharmacol. 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yulug, B.; Hanoglu, L.; Ozansoy, M.; Isık, D.; Kilic, U.; Kilic, E.; Schabitz, W.R. Therapeutic role of rifampicin in Alzheimer’s disease. Psychiatry Clin. Neurosci. 2018, 72, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santa-Cecília, F.V.; Leite, C.A.; Del-Bel, E.; Raisman-Vozari, R. The Neuroprotective Effect of Doxycycline on Neurodegenerative Diseases. Neurotox. Res. 2019, 35, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Stavrovskaya, I.G.; Drozda, M.; Kim, B.Y.; Ona, V.; Li, M.; Sarang, S.; Liu, A.S.; Hartley, D.M.; Wu, D.C.; et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002, 417, 74–78. [Google Scholar] [CrossRef]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golko-Perez, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. A Novel Iron Chelator-Radical Scavenger Ameliorates Motor Dysfunction and Improves Life Span and Mitochondrial Biogenesis in SOD1(G93A) ALS Mice. Neurotox. Res. 2017, 31, 230–244. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. TEM 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, R.; Ren, J.M.; Cadman, K.S.; Moore, I.K.; Perret, P.; Pypaert, M.; Young, L.H.; Semenkovich, C.F.; Shulman, G.I. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1340–E1346. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Scarpulla, R.C. NRF-1: A trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990, 4, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virbasius, C.A.; Virbasius, J.V.; Scarpulla, R.C. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 1993, 7, 2431–2445. [Google Scholar] [CrossRef] [PubMed]
- Harlan, B.A.; Killoy, K.M.; Pehar, M.; Liu, L.; Auwerx, J.; Vargas, M.R. Evaluation of the NAD+ biosynthetic pathway in ALS patients and effect of modulating NAD+ levels in hSOD1-linked ALS mouse models. Exp. Neurol. 2020, 327, 113219. [Google Scholar] [CrossRef] [PubMed]
- Labarre, A.; Guitard, E.; Tossing, G.; Forest, A.; Bareke, E.; Labrecque, M.; Tétreault, M.; Ruiz, M.; Alex Parker, J. Fatty acids derived from the probiotic Lacticaseibacillus rhamnosus HA-114 suppress age-dependent neurodegeneration. Commun. Biol. 2022, 5, 1340. [Google Scholar] [CrossRef]
- Niccolai, E.; Di Pilato, V.; Nannini, G.; Baldi, S.; Russo, E.; Zucchi, E.; Martinelli, I.; Menicatti, M.; Bartolucci, G.; Mandrioli, J.; et al. The Gut Microbiota-Immunity Axis in ALS: A Role in Deciphering Disease Heterogeneity? Biomedicines 2021, 9, 753. [Google Scholar] [CrossRef]
- Nicholson, K.; Bjornevik, K.; Abu-Ali, G.; Chan, J.; Cortese, M.; Dedi, B.; Jeon, M.; Xavier, R.; Huttenhower, C.; Ascherio, A.; et al. The human gut microbiota in people with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 186–194. [Google Scholar] [CrossRef]
- Ribeiro, M.F.; Santos, A.A.; Afonso, M.B.; Rodrigues, P.M.; Sá Santos, S.; Castro, R.E.; Rodrigues, C.M.P.; Solá, S. Diet-dependent gut microbiota impacts on adult neurogenesis through mitochondrial stress modulation. Brain Commun. 2020, 2, fcaa165. [Google Scholar]
- Tang, Y.; Chen, Y.; Jiang, H.; Nie, D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ. 2011, 18, 602–618. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Brantner, C.A.; Chiaramello, A. Epigenetic modifiers promote mitochondrial biogenesis and oxidative metabolism leading to enhanced differentiation of neuroprogenitor cells. Cell Death Dis. 2018, 9, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Zhu, W.; Hosoda, W.; Sen, J.M.; Mattson, M.P. Chronic Mild Gut Inflammation Accelerates Brain Neuropathology and Motor Dysfunction in α-Synuclein Mutant Mice. NeuroMolecular Med. 2019, 21, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarrese, P.; Falzano, L.; Fabbri, A.; Gambardella, L.; Frank, C.; Geny, B.; Popoff, M.R.; Malorni, W.; Fiorentini, C. Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of ATP-sensitive mitochondrial potassium channels. J. Biol. Chem. 2007, 282, 9029–9041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes-Costa, D.; Magalhães, J.D.; G-Fernandes, M.; Cardoso, S.M.; Empadinhas, N. Microbial BMAA and the Pathway for Parkinson’s Disease Neurodegeneration. Front. Aging Neurosci. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, A.R.; Munoz-Pinto, M.F.; Nunes-Costa, D.; Candeias, E.; Silva, D.F.; Magalhães, J.D.; Pereira-Santos, A.R.; Ferreira, I.L.; Alarico, S.; Tiago, I.; et al. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut 2021, 72, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Danilchanka, O.; Mekalanos, J.J. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 2014, 16, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, S.M.; Empadinhas, N. The Microbiome-Mitochondria Dance in Prodromal Parkinson’s Disease. Front. Physiol. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.; Forsyth, C.B.; Shaikh, M.; Voigt, R.M.; Engen, P.A.; Ramirez, V.; Keshavarzian, A. Diet in Parkinson’s Disease: Critical Role for the Microbiome. Front. Neurol. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Yamane, S.; Inagaki, N. Regulation of glucagon-like peptide-1 sensitivity by gut microbiota dysbiosis. J. Diabetes Investig. 2018, 9, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lau, C.Y.; Ma, F.; Zheng, C. Genome-wide screen identifies curli amyloid fibril as a bacterial component promoting host neurodegeneration. Proc. Natl. Acad. Sci. USA 2021, 118, e2106504118. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Li, X.; Gao, H.; Feng, Z.; Zhao, L.; Jia, X.; Zhang, H.; Liu, J. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson’s disease model. Neuropharmacology 2022, 207, 108963. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, J.; Qiu, J.; Wang, L.; Zhuo, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A protects dopaminergic neurons in experimental models of Parkinson’s disease by promoting mitochondrial biogenesis through the SIRT1/PGC-1α signaling pathway. Food Funct. 2022, 13, 375–385. [Google Scholar] [CrossRef]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2016, 53, 810–817. [Google Scholar] [CrossRef]
- Fonseca, I.; Gordino, G.; Moreira, S.; Nunes, M.J.; Azevedo, C.; Gama, M.J.; Rodrigues, E.; Rodrigues, C.M.P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Protects Against Mitochondrial Dysfunction and Cell Death via Mitophagy in Human Neuroblastoma Cells. Mol. Neurobiol. 2017, 54, 6107–6119. [Google Scholar] [CrossRef]
- Jiang, H.; Li, L.J.; Wang, J.; Xie, J.X. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 2008, 212, 532–537. [Google Scholar] [CrossRef]
- Dong, J.; Song, N.; Xie, J.; Jiang, H. Ghrelin antagonized 1-methyl-4-phenylpyridinium (MPP+)-induced apoptosis in MES23.5 cells. J. Mol. Neurosci. MN 2009, 37, 182–189. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Kahouli, I.; Dia, S.Y.; Singh, S.P.; Prakash, S. Microbiome, probiotics and neurodegenerative diseases: Deciphering the gut brain axis. Cell. Mol. Life Sci. CMLS 2017, 74, 3769–3787. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Tang, N.Y.; Kao, S.T.; Hsieh, C.L. Ferulic Acid Administered at Various Time Points Protects against Cerebral Infarction by Activating p38 MAPK/p90RSK/CREB/Bcl-2 Anti-Apoptotic Signaling in the Subacute Phase of Cerebral Ischemia-Reperfusion Injury in Rats. PLoS ONE 2016, 11, e0155748. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Dadar, M.; Anderson, G.; Chirumbolo, S.; Maes, M. Preventive treatments to slow substantia nigra damage and Parkinson’s disease progression: A critical perspective review. Pharmacol. Res. 2020, 161, 105065. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, E.M.; Sanz-Blasco, S.; Karachitos, A.; Bandez, M.J.; Fernandez-Gomez, F.J.; Perez-Alvarez, S.; de Mera, R.M.; Jordan, M.J.; Aguirre, N.; Galindo, M.F.; et al. Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem. Pharmacol. 2010, 79, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Hurkacz, M.; Dobrek, L.; Wiela-Hojeńska, A. Antibiotics and the Nervous System-Which Face of Antibiotic Therapy Is Real, Dr. Jekyll (Neurotoxicity) or Mr. Hyde (Neuroprotection)? Molecules 2021, 26, 7456. [Google Scholar] [CrossRef]
- Romo-Araiza, A.; Ibarra, A. Prebiotics and probiotics as potential therapy for cognitive impairment. Med. Hypotheses 2020, 134, 109410. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. Mitochondria and Antibiotics: For Good or for Evil? Biomolecules 2021, 11, 1050. [Google Scholar] [CrossRef]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Vander Wende, H.; Simko, M.; Klum, S.; Barfield, S.; Choi, H.; Pineda, V.V.; Kaeberlein, M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.H.; Wang, C.H.; Kuo, Y.W.; Ho, Y.J.; Chen, H.L. Fructo-oligosaccharide systemically diminished D-galactose-induced oxidative molecule damages in BALB/cJ mice. Br. J. Nutr. 2012, 107, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Krishna, G.; Muralidhara. Inulin supplementation during gestation mitigates acrylamide-induced maternal and fetal brain oxidative dysfunctions and neurotoxicity in rats. Neurotoxicol. Teratol. 2015, 49, 49–58. [Google Scholar] [CrossRef]
- Krishna, G.; Muralidhara. Oral supplements of inulin during gestation offsets rotenone-induced oxidative impairments and neurotoxicity in maternal and prenatal rat brain. Biomed. Pharmacother. 2018, 104, 751–762. [Google Scholar] [CrossRef]
- Srikantha, P.; Mohajeri, M.H. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 2115. [Google Scholar] [CrossRef] [Green Version]
- Morelli, L.; Capurso, L. FAO/WHO guidelines on probiotics: 10 years later. J. Clin. Gastroenterol. 2012, 46, S1–S2. [Google Scholar] [CrossRef]
- Huo, Y.; Lu, X.; Wang, X.; Chen, L.; Guo, H.; Zhang, M.; Li, Y. Bifidobacterium animalis subsp. lactis A6 Alleviates Obesity Associated with Promoting Mitochondrial Biogenesis and Function of Adipose Tissue in Mice. Molecules 2020, 25, 1490. [Google Scholar] [CrossRef] [Green Version]
- Mularczyk, M.; Bourebaba, Y.; Kowalczuk, A.; Marycz, K.; Bourebaba, L. Probiotics-rich emulsion improves insulin signalling in Palmitate/Oleate-challenged human hepatocarcinoma cells through the modulation of Fetuin-A/TLR4-JNK-NF-κB pathway. Biomed. Pharmacother. 2021, 139, 111560. [Google Scholar] [CrossRef]
- Nurrahma, B.A.; Tsao, S.P.; Wu, C.H.; Yeh, T.H.; Hsieh, P.S.; Panunggal, B.; Huang, H.Y. Probiotic Supplementation Facilitates Recovery of 6-OHDA-Induced Motor Deficit via Improving Mitochondrial Function and Energy Metabolism. Front. Aging Neurosci. 2021, 13, 668775. [Google Scholar] [CrossRef] [PubMed]
- Beltagy, D.M.; Nawar, N.F.; Mohamed, T.M.; Tousson, E.; El-Keey, M.M. Beneficial consequences of probiotic on mitochondrial hippocampus in Alzheimer’s disease. J. Complement. Integr. Med. 2021, 18, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Chunchai, T.; Thunapong, W.; Yasom, S.; Wanchai, K.; Eaimworawuthikul, S.; Metzler, G.; Lungkaphin, A.; Pongchaidecha, A.; Sirilun, S.; Chaiyasut, C.; et al. Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J. Neuroinflammation 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target Intestinal Microbiota to Alleviate Disease Progression in Amyotrophic Lateral Sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Razazan, A.; Karunakar, P.; Mishra, S.P.; Sharma, S.; Miller, B.; Jain, S.; Yadav, H. Activation of Microbiota Sensing—Free Fatty Acid Receptor 2 Signaling Ameliorates Amyloid-β Induced Neurotoxicity by Modulating Proteolysis-Senescence Axis. Front. Aging Neurosci. 2021, 13, 735933. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borbolis, F.; Mytilinaiou, E.; Palikaras, K. The Crosstalk between Microbiome and Mitochondrial Homeostasis in Neurodegeneration. Cells 2023, 12, 429. https://doi.org/10.3390/cells12030429
Borbolis F, Mytilinaiou E, Palikaras K. The Crosstalk between Microbiome and Mitochondrial Homeostasis in Neurodegeneration. Cells. 2023; 12(3):429. https://doi.org/10.3390/cells12030429
Chicago/Turabian StyleBorbolis, Fivos, Eirini Mytilinaiou, and Konstantinos Palikaras. 2023. "The Crosstalk between Microbiome and Mitochondrial Homeostasis in Neurodegeneration" Cells 12, no. 3: 429. https://doi.org/10.3390/cells12030429