
Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis

Abstract
:1. Introduction
2. Gut Microbiota and Metabolites
2.1. Gut Microbiota and Short-Chain Fatty Acids
2.2. Gut Microbiota and Tryptophan Metabolites
2.3. Gut Microbiota and Bile Acid Metabolites
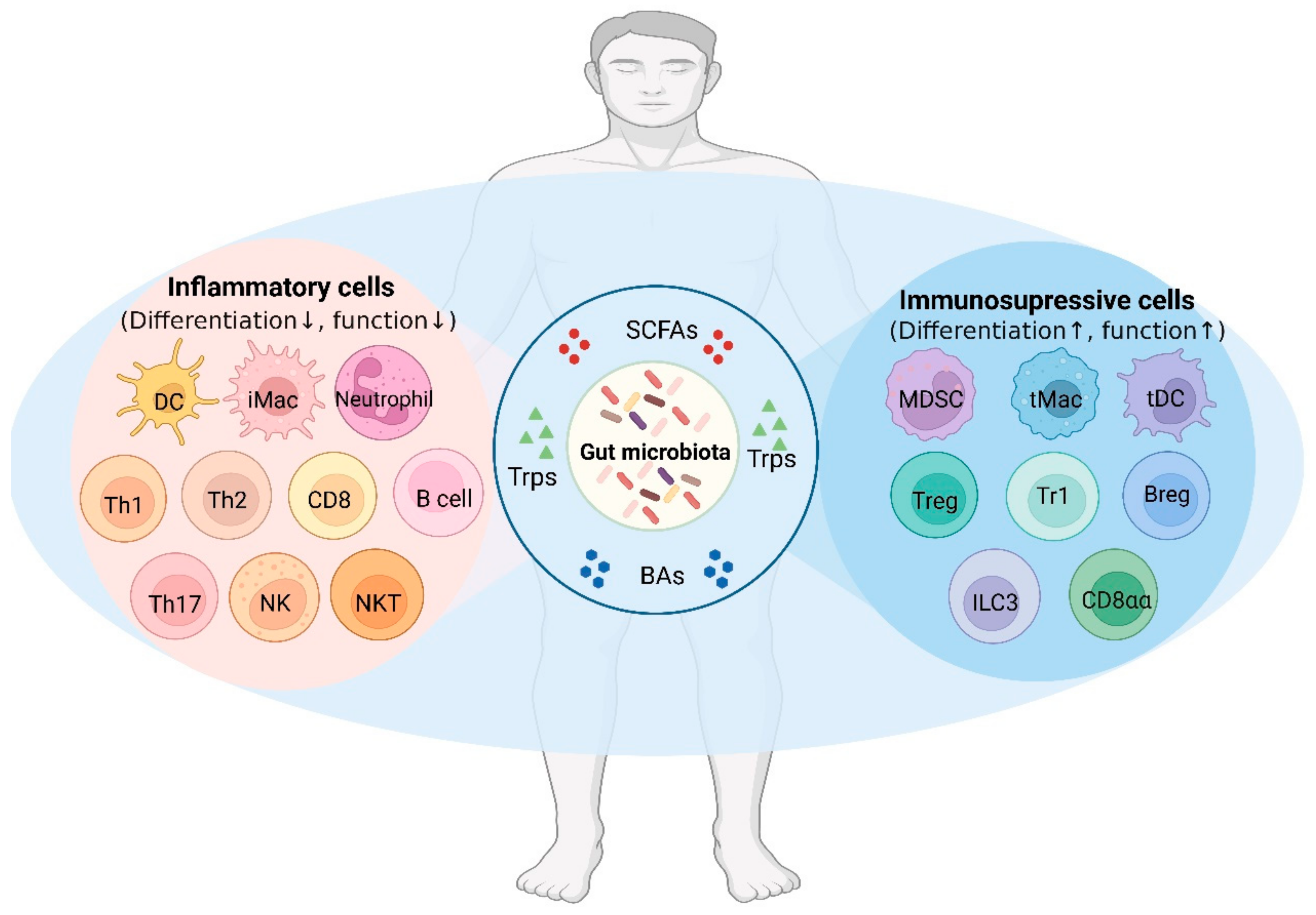
3. Receptors of Gut-Microbiota-Derived Metabolites in the Immune Cells
3.1. Receptors of Short-Chain Fatty Acids
3.2. Receptors of Tryptophan Metabolites
3.3. Receptors of Bile Acid Metabolites
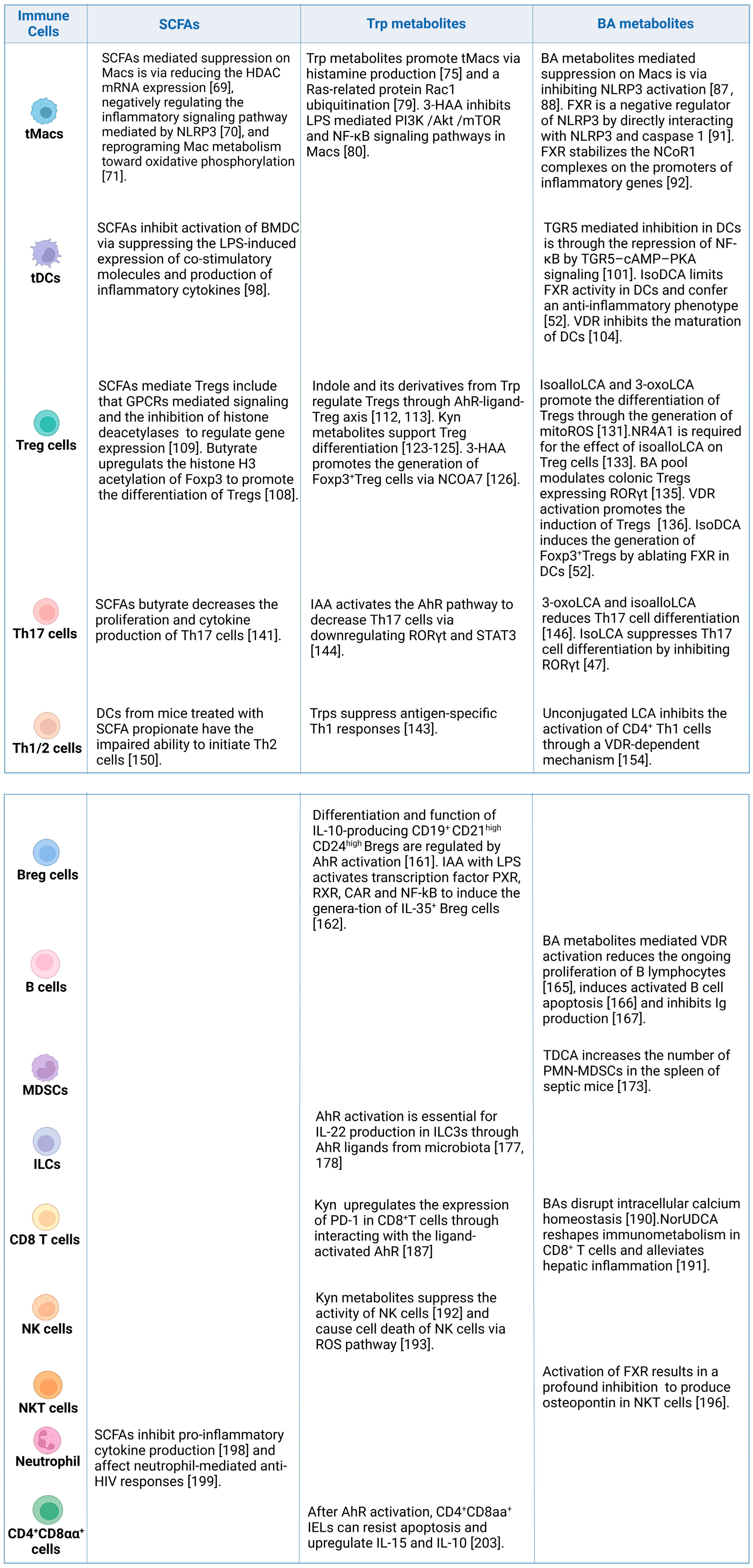
4. Effects of Gut-Microbiota-Derived Metabolites on the Immune Cells
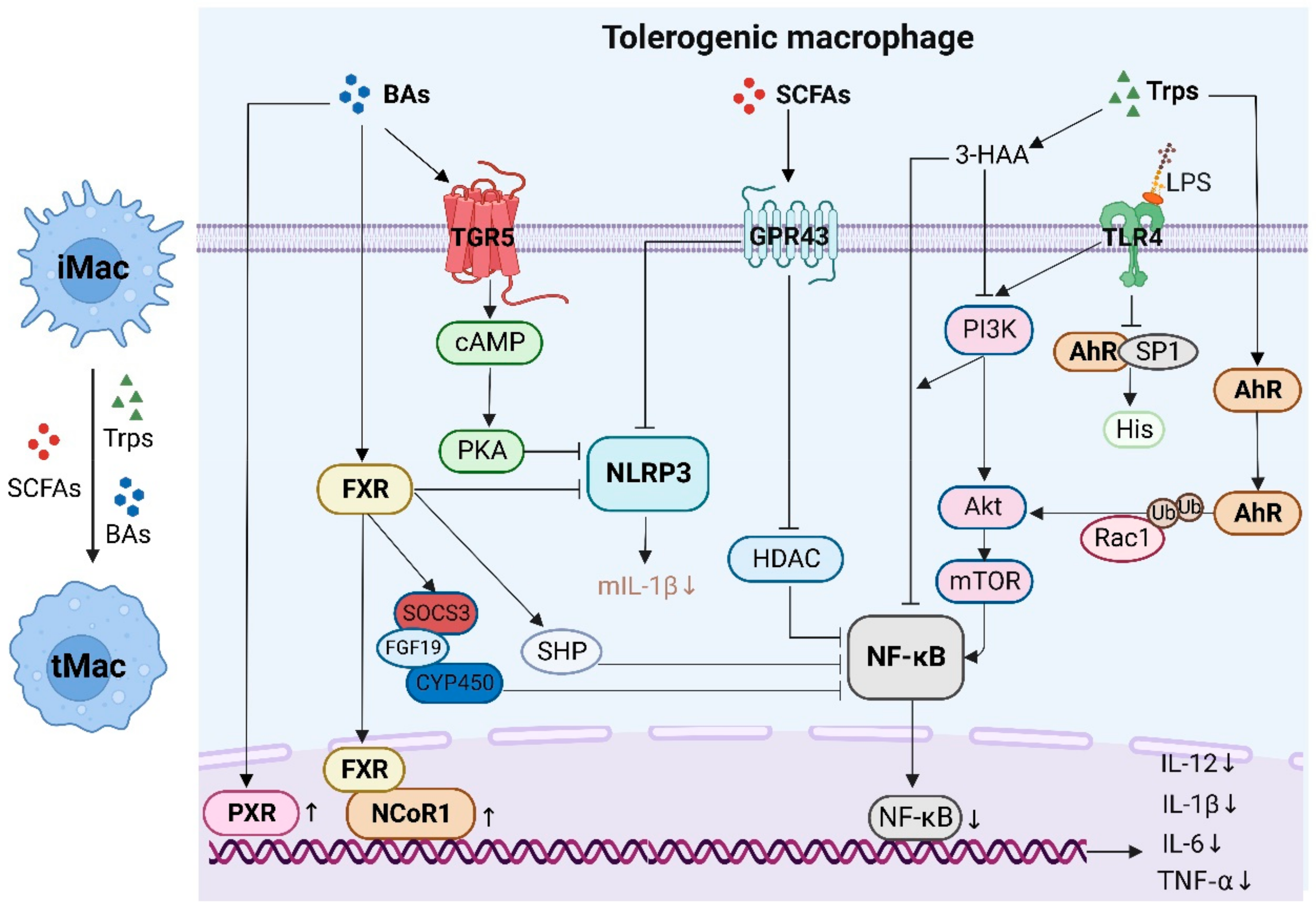
4.1. Tolerogenic and Inflammatory Macrophages
4.2. Tolerogenic Dendritic Cells
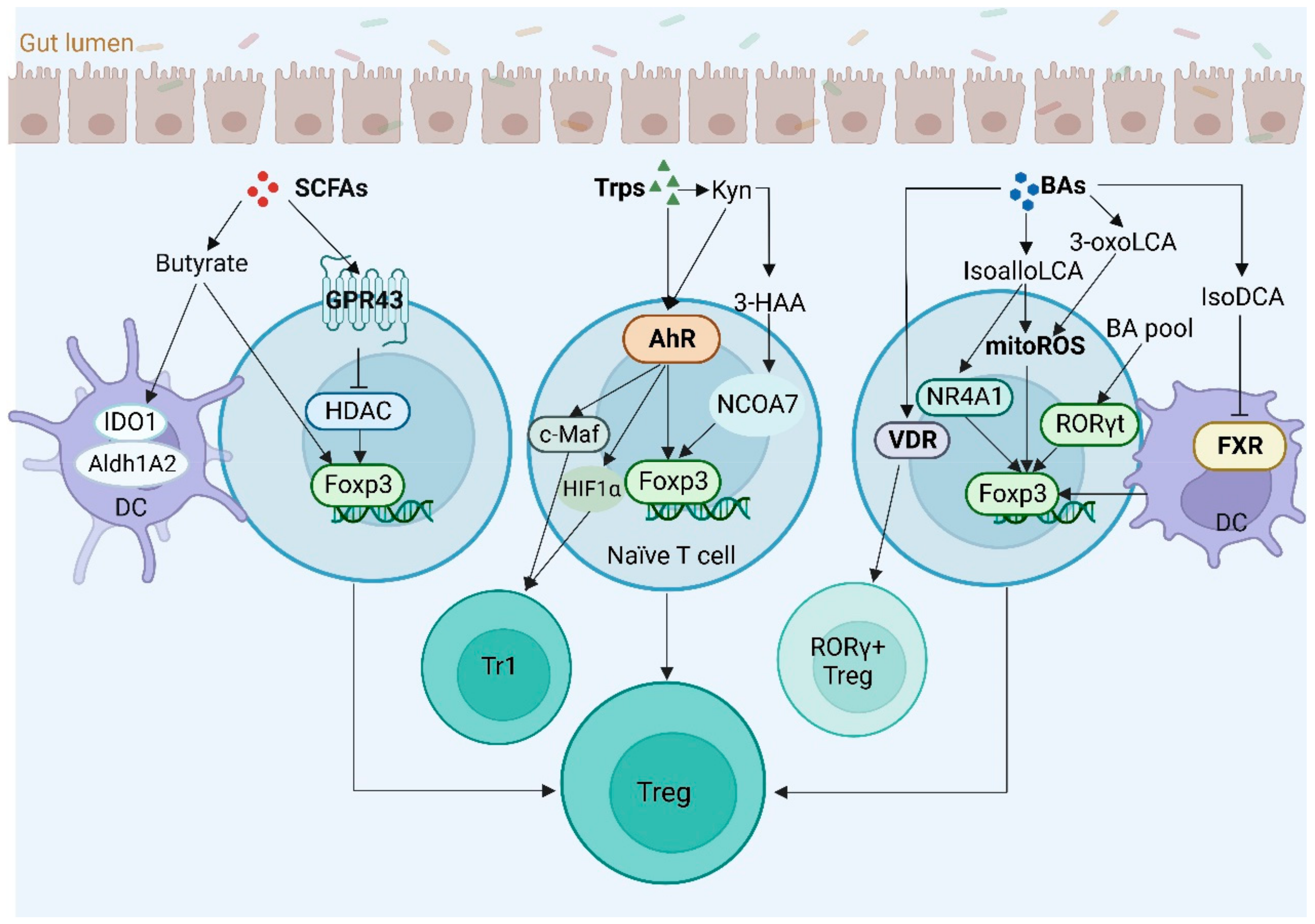
4.3. Regulatory T Cells
4.4. T Helper 17 Cells
4.5. CD4+Th1 and Th2 Cells
4.6. Regulatory B Cells
4.7. B Cells
4.8. Myeloid-Derived Suppressor Cells
4.9. Innate Lymphoid Cells
4.10. CD8+ T Cells
4.11. Natural Killer Cells
4.12. NKT Cells
4.13. Neutrophils
4.14. CD4+CD8αα+ Cells
5. Gut-Microbiota-Derived Metabolites and Immune-Associated Disorders
6. Conclusions and Perspectives
- (1)
- Discovery of new culture method(s) for gut microbiota. A key question for gut microbiota metabolites is whether gut microorganisms can be successfully cultured in vitro. The discovery of any new culture technique will be beneficial to the identification of gut microbiota metabolites.
- (2)
- Improvement of the metabolite analyses. For currently targeted metabolomics, the restricted standard samples have limited application, whereas for untargeted metabolomics, it is easy to produce “false positive” data.
- (3)
- Synthesis of gut microbiota metabolites. Some metabolites from the gut microbiota need to be synthesized for their functions and application.
- (4)
- Determination of immune cell subset function. With the development of single-cell sequencing techniques, more immune cell subpopulations related to the gut microbiota or metabolites will be identified. However, the functional potential of these immune cell subsets remains to be determined.
- (5)
- Establishment of new animal models. Some gut microbiota metabolites may exert their function through new mechanism(s), including receptor, signal pathway, genetic and epigenetic modification and metabolism. All of these need new animal models to explain how the metabolites exert their effects on the immune cells and/or diseases.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sommer, F.; Backhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Dixit, K.; Chaudhari, D.; Dhotre, D.; Shouche, Y.; Saroj, S. Restoration of dysbiotic human gut microbiome for homeostasis. Life Sci. 2021, 278, 119622. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Y.; Zhang, S.; Dong, L. Gut microbiota-mediated immunomodulation in tumor. J. Exp. Clin. Cancer Res. 2021, 40, 221. [Google Scholar] [CrossRef] [PubMed]
- Margolis, K.G.; Cryan, J.F.; Mayer, E.A. The Microbiota-Gut-Brain Axis: From Motility to Mood. Gastroenterology 2021, 160, 1486–1501. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Gundamaraju, R.; Shastri, M.D.; Shukla, S.D.; Kalpurath, K.; Ball, M.; Tristram, S.; Shankar, E.M.; Ahuja, K.; Eri, R. Gut Microbial Changes, Interactions, and Their Implications on Human Lifecycle: An Ageing Perspective. Biomed. Res. Int. 2018, 2018, 4178607. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Michaudel, C.; Sokol, H. The Gut Microbiota at the Service of Immunometabolism. Cell Metab. 2020, 32, 514–523. [Google Scholar] [CrossRef]
- Ratajczak, W.; Ryl, A.; Mizerski, A.; Walczakiewicz, K.; Sipak, O.; Laszczynska, M. Immunomodulatory potential of gut microbiome-derived short-chain fatty acids (SCFAs). Acta Biochim. Pol. 2019, 66, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; McWilliam Leitch, C.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.P.; Martin, J.C.; Campbell, G.; Mayer, C.D.; Flint, H.J. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans”. J. Bacteriol. 2006, 188, 4340–4349. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio. 2014, 5, e00889. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Gao, Y.; Yang, R. Gut Microbiota-Derived Tryptophan Metabolites Maintain Gut and Systemic Homeostasis. Cells 2022, 11, 2296. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Luo, C.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Vujkovic-Cvijin, I.; Dunham, R.M.; Iwai, S.; Maher, M.C.; Albright, R.G.; Broadhurst, M.J.; Hernandez, R.D.; Lederman, M.M.; Huang, Y.; Somsouk, M.; et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci. Transl. Med. 2013, 5, 193ra191. [Google Scholar] [CrossRef] [Green Version]
- Bourgin, M.; Kriaa, A.; Mkaouar, H.; Mariaule, V.; Jablaoui, A.; Maguin, E.; Rhimi, M. Bile Salt Hydrolases: At the Crossroads of Microbiota and Human Health. Microorganisms 2021, 9, 1122. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorucci, S.; Carino, A.; Baldoni, M.; Santucci, L.; Costanzi, E.; Graziosi, L.; Distrutti, E.; Biagioli, M. Bile Acid Signaling in Inflammatory Bowel Diseases. Dig. Dis. Sci. 2021, 66, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce TauEta17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Gotoh, Y.; Ogura, Y.; Hayashi, T.; Fukiya, S.; Yokota, A. Comparative Genomic and Physiological Analysis against Clostridium scindens Reveals Eubacterium sp. c-25 as an Atypical Deoxycholic Acid Producer of the Human Gut Microbiota. Microorganisms 2021, 9, 2254. [Google Scholar] [CrossRef]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef]
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- Lucas, L.N.; Barrett, K.; Kerby, R.L.; Zhang, Q.; Cattaneo, L.E.; Stevenson, D.; Rey, F.E.; Amador-Noguez, D. Dominant Bacterial Phyla from the Human Gut Show Widespread Ability To Transform and Conjugate Bile Acids. mSystems 2021, 6, e0080521. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.H.; O’Flaherty, S.; Allen, G.; Rivera, A.J.; Stewart, A.K.; Barrangou, R.; Theriot, C.M. Lactobacillus bile salt hydrolase substrate specificity governs bacterial fitness and host colonization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017709118. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.P.; Hudson, L.L. Cloning and characterization of a conjugated bile acid hydrolase gene from Clostridium perfringens. Appl. Environ. Microbiol. 1995, 61, 2514–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal-Srivastava, R.; Hylemon, P.B. Purification and characterization of bile salt hydrolase from Clostridium perfringens. J. Lipid Res. 1988, 29, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Mining genome traits that determine the different gut colonization potential of Lactobacillus and Bifidobacterium species. Microb. Genom. 2021, 7, 000581. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.; Annapure, U.S. In vitro assessment of metabolic profile of Enterococcus strains of human origin. J. Genet. Eng. Biotechnol. 2019, 17, 11. [Google Scholar] [CrossRef]
- Adhikari, A.A.; Seegar, T.C.M.; Ficarro, S.B.; McCurry, M.D.; Ramachandran, D.; Yao, L.; Chaudhari, S.N.; Ndousse-Fetter, S.; Banks, A.S.; Marto, J.A.; et al. Development of a covalent inhibitor of gut bacterial bile salt hydrolases. Nat. Chem. Biol. 2020, 16, 318–326. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.C.; Devendran, S.; Mendez-Garcia, C.; Mythen, S.M.; Wright, C.L.; Fields, C.J.; Hernandez, A.G.; Cann, I.; Hylemon, P.B.; Ridlon, J.M. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243(T). Gut Microbes. 2018, 9, 523–539. [Google Scholar]
- Eggert, T.; Bakonyi, D.; Hummel, W. Enzymatic routes for the synthesis of ursodeoxycholic acid. J Biotechnol. 2014, 191, 11–21. [Google Scholar] [CrossRef]
- Lee, J.Y.; Arai, H.; Nakamura, Y.; Fukiya, S.; Wada, M.; Yokota, A. Contribution of the 7beta-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J. Lipid. Res. 2013, 54, 3062–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandi, E.E.; Bertolesi, G.M.; Polentini, F.; Negri, A.; Riva, S.; Monti, D. In search of sustainable chemical processes: Cloning, recombinant expression, and functional characterization of the 7alpha- and 7beta-hydroxysteroid dehydrogenases from Clostridium absonum. Appl. Microbiol. Biotechnol. 2012, 95, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Bolognini, D.; Tobin, A.B.; Milligan, G.; Moss, C.E. The Pharmacology and Function of Receptors for Short-Chain Fatty Acids. Mol. Pharmacol. 2016, 89, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluznick, J.L. Gut microbiota in renal physiology: Focus on short-chain fatty acids and their receptors. Kidney Int. 2016, 90, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug. Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Dong, F.; Hao, F.; Murray, I.A.; Smith, P.B.; Koo, I.; Tindall, A.M.; Kris-Etherton, P.M.; Gowda, K.; Amin, S.G.; Patterson, A.D.; et al. Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microbes 2020, 12, 1788899. [Google Scholar] [CrossRef]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef]
- Vyhlidalova, B.; Krasulova, K.; Pecinkova, P.; Marcalikova, A.; Vrzal, R.; Zemankova, L.; Vanco, J.; Travnicek, Z.; Vondracek, J.; Karasova, M.; et al. Gut Microbial Catabolites of Tryptophan Are Ligands and Agonists of the Aryl Hydrocarbon Receptor: A Detailed Characterization. Int. J. Mol. Sci. 2020, 21, 2614. [Google Scholar] [CrossRef] [Green Version]
- Sari, Z.; Miko, E.; Kovacs, T.; Janko, L.; Csonka, T.; Lente, G.; Sebo, E.; Toth, J.; Toth, D.; Arkosy, P.; et al. Indolepropionic Acid, a Metabolite of the Microbiome, Has Cytostatic Properties in Breast Cancer by Activating AHR and PXR Receptors and Inducing Oxidative Stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef]
- Ye, X.; Li, H.; Anjum, K.; Zhong, X.; Miao, S.; Zheng, G.; Liu, W.; Li, L. Dual Role of Indoles Derived From Intestinal Microbiota on Human Health. Front. Immunol. 2022, 13, 903526. [Google Scholar] [CrossRef]
- Biagioli, M.; Marchiano, S.; Carino, A.; Di Giorgio, C.; Santucci, L.; Distrutti, E.; Fiorucci, S. Bile Acids Activated Receptors in Inflammatory Bowel Disease. Cells 2021, 10, 1281. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhao, T.; Liu, S.; Wu, Q.; Johnson, O.; Wu, Z.; Zhuang, Z.; Shi, Y.; Peng, L.; He, R.; et al. MRGPRX4 is a bile acid receptor for human cholestatic itch. Elife 2019, 8, e48431. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415. [Google Scholar] [CrossRef] [Green Version]
- Honda, M.; Surewaard, B.G.J.; Watanabe, M.; Hedrick, C.C.; Lee, W.Y.; Brown, K.; McCoy, K.D.; Kubes, P. Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat. Commun. 2020, 11, 1329. [Google Scholar] [CrossRef] [Green Version]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Eslick, S.; Williams, E.J.; Berthon, B.S.; Wright, T.; Karihaloo, C.; Gately, M.; Wood, L.G. Weight Loss and Short-Chain Fatty Acids Reduce Systemic Inflammation in Monocytes and Adipose Tissue Macrophages from Obese Subjects. Nutrients 2022, 14, 765. [Google Scholar] [CrossRef]
- Shao, X.; Sun, S.; Zhou, Y.; Wang, H.; Yu, Y.; Hu, T.; Yao, Y.; Zhou, C. Bacteroides fragilis restricts colitis-associated cancer via negative regulation of the NLRP3 axis. Cancer Lett. 2021, 523, 170–181. [Google Scholar] [CrossRef]
- Scott, N.A.; Andrusaite, A.; Andersen, P.; Lawson, M.; Alcon-Giner, C.; Leclaire, C.; Caim, S.; Le Gall, G.; Shaw, T.; Connolly, J.P.R.; et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci. Transl. Med. 2018, 10, eaao4755. [Google Scholar] [CrossRef] [Green Version]
- Goudot, C.; Coillard, A.; Villani, A.C.; Gueguen, P.; Cros, A.; Sarkizova, S.; Tang-Huau, T.L.; Bohec, M.; Baulande, S.; Hacohen, N.; et al. Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages. Immunity 2017, 47, 582–596.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Kimura, A.; Hanieh, H.; Nguyen, N.T.; Nakahama, T.; Chinen, I.; Otoyo, Y.; Murotani, T.; Yamatodani, A.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int. Immunol. 2011, 23, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadonaga, J.T.; Courey, A.J.; Ladika, J.; Tjian, R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science 1988, 242, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.Y.; Khachigian, L.M. Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell Biol. 2009, 29, 2483–2488. [Google Scholar] [CrossRef] [Green Version]
- Mulero-Navarro, S.; Carvajal-Gonzalez, J.M.; Herranz, M.; Ballestar, E.; Fraga, M.F.; Ropero, S.; Esteller, M.; Fernandez-Salguero, P.M. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis 2006, 27, 1099–1104. [Google Scholar] [CrossRef]
- Grosskopf, H.; Walter, K.; Karkossa, I.; von Bergen, M.; Schubert, K. Non-Genomic AhR-Signaling Modulates the Immune Response in Endotoxin-Activated Macrophages After Activation by the Environmental Stressor BaP. Front. Immunol. 2021, 12, 620270. [Google Scholar] [CrossRef]
- Lee, K.; Kwak, J.H.; Pyo, S. Inhibition of LPS-induced inflammatory mediators by 3-hydroxyanthranilic acid in macrophages through suppression of PI3K/NF-kappaB signaling pathways. Food Funct. 2016, 7, 3073–3082. [Google Scholar] [CrossRef]
- Riemschneider, S.; Kohlschmidt, J.; Fueldner, C.; Esser, C.; Hauschildt, S.; Lehmann, J. Aryl hydrocarbon receptor activation by benzo(a)pyrene inhibits proliferation of myeloid precursor cells and alters the differentiation state as well as the functional phenotype of murine bone marrow-derived macrophages. Toxicol. Lett. 2018, 296, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Platzer, B.; Richter, S.; Kneidinger, D.; Waltenberger, D.; Woisetschlager, M.; Strobl, H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and Langerhans dendritic cells. J. Immunol. 2009, 183, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Haselow, K.; Bode, J.G.; Wammers, M.; Ehlting, C.; Keitel, V.; Kleinebrecht, L.; Schupp, A.K.; Haussinger, D.; Graf, D. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J. Leukoc. Biol. 2013, 94, 1253–1264. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [Green Version]
- Pols, T.W.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 944. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Su, W.; Zhang, L.; Shi, C.; Zhou, J.; Wang, P.; Wang, H.; Shi, X.; Wei, S.; Wang, Q.; et al. TGR5 Regulates Macrophage Inflammation in Nonalcoholic Steatohepatitis by Modulating NLRP3 Inflammasome Activation. Front. Immunol. 2020, 11, 609060. [Google Scholar] [CrossRef]
- Di Vincenzo, F.; Puca, P.; Lopetuso, L.R.; Petito, V.; Masi, L.; Bartocci, B.; Murgiano, M.; De Felice, M.; Petronio, L.; Gasbarrini, A.; et al. Bile Acid-Related Regulation of Mucosal Inflammation and Intestinal Motility: From Pathogenesis to Therapeutic Application in IBD and Microscopic Colitis. Nutrients 2022, 14, 2664. [Google Scholar] [CrossRef]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, A.; Fiorotto, R.; Strazzabosco, M. Bile acids and their receptors: Modulators and therapeutic targets in liver inflammation. Semin. Immunopathol. 2022, 44, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Park, J.H.; Choi, H.S. Molecular basis of endocrine regulation by orphan nuclear receptor Small Heterodimer Partner. Endocr. J. 2008, 55, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Yang, C.S.; Kim, J.J.; Kim, T.S.; Lee, P.Y.; Kim, S.Y.; Lee, H.M.; Shin, D.M.; Nguyen, L.T.; Lee, M.S.; Jin, H.S.; et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [Green Version]
- Nastasi, C.; Candela, M.; Bonefeld, C.M.; Geisler, C.; Hansen, M.; Krejsgaard, T.; Biagi, E.; Andersen, M.H.; Brigidi, P.; Odum, N.; et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci. Rep. 2015, 5, 16148. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Ruby, C.E.; Leid, M.; Kerkvliet, N.I. 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses tumor necrosis factor-alpha and anti-CD40-induced activation of NF-kappaB/Rel in dendritic cells: p50 homodimer activation is not affected. Mol. Pharmacol. 2002, 62, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, C.; Huang, X.; Yi, S.; Pan, S.; Zhang, Y.; Yuan, G.; Cao, Q.; Ye, X.; Li, H. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 2021, 36, 109726. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, R.; Takayama, T.; Yoneno, K.; Kamada, N.; Kitazume, M.T.; Higuchi, H.; Matsuoka, K.; Watanabe, M.; Itoh, H.; Kanai, T.; et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012, 136, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Szeles, L.; Keresztes, G.; Torocsik, D.; Balajthy, Z.; Krenacs, L.; Poliska, S.; Steinmeyer, A.; Zuegel, U.; Pruenster, M.; Rot, A.; et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J. Immunol. 2009, 182, 2074–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, S.; Haghikia, A.; Wilck, N.; Muller, D.N.; Linker, R.A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2016, 44, 951–953. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.J.; Rossi, M.; Romano, E.; Ghith, J.; Yuan, J.; Munn, D.H.; Young, J.W. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood 2009, 114, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal. Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Jiang, Y.; Yang, Y.; Shao, J.; Sun, X.; Chen, J.; Dong, L.; Zhang, J. 3,3′-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Mol. Immunol. 2013, 53, 335–344. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Wang, X.; Rohlman, D.; Punj, S.; Lohr, C.V.; Newman, M.T.; Kolluri, S.K.; Kerkvliet, N.I. Activation of the Aryl Hydrocarbon Receptor by 10-Cl-BBQ Prevents Insulitis and Effector T Cell Development Independently of Foxp3+ Regulatory T Cells in Nonobese Diabetic Mice. J. Immunol. 2016, 196, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhs, A.; Haarmann-Stemmann, T.; Frauenstein, K.; Krutmann, J.; Schwarz, T.; Schwarz, A. Activation of the arylhydrocarbon receptor causes immunosuppression primarily by modulating dendritic cells. J. Investig. Dermatol. 2015, 135, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Abron, J.D.; Singh, N.P.; Mishra, M.K.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P. An endogenous aryl hydrocarbon receptor ligand, ITE, induces regulatory T cells and ameliorates experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G220–G230. [Google Scholar] [CrossRef]
- Hao, N.; Whitelaw, M.L. The emerging roles of AhR in physiology and immunity. Biochem. Pharmacol. 2013, 86, 561–570. [Google Scholar] [CrossRef]
- Ye, J.; Qiu, J.; Bostick, J.W.; Ueda, A.; Schjerven, H.; Li, S.; Jobin, C.; Chen, Z.E.; Zhou, L. The Aryl Hydrocarbon Receptor Preferentially Marks and Promotes Gut Regulatory T Cells. Cell Rep. 2017, 21, 2277–2290. [Google Scholar] [CrossRef] [Green Version]
- de Araujo, E.F.; Feriotti, C.; Galdino, N.A.L.; Preite, N.W.; Calich, V.L.G.; Loures, F.V. The IDO-AhR Axis Controls Th17/Treg Immunity in a Pulmonary Model of Fungal Infection. Front. Immunol. 2017, 8, 880. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettel, J.A.; Gandhi, R.; Kenison, J.E.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval, D.S.; Weiner, H.L.; et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashita, C.; Hoshi, M.; Hirata, A.; Nakamoto, K.; Ando, T.; Hattori, T.; Yamamoto, Y.; Tezuka, H.; Tomita, H.; Hara, A.; et al. Kynurenine plays an immunosuppressive role in 2,4,6-trinitrobenzene sulfate-induced colitis in mice. World J. Gastroenterol. 2020, 26, 918–932. [Google Scholar] [CrossRef]
- Gargaro, M.; Vacca, C.; Massari, S.; Scalisi, G.; Manni, G.; Mondanelli, G.; Mazza, E.M.C.; Bicciato, S.; Pallotta, M.T.; Orabona, C.; et al. Engagement of Nuclear Coactivator 7 by 3-Hydroxyanthranilic Acid Enhances Activation of Aryl Hydrocarbon Receptor in Immunoregulatory Dendritic Cells. Front. Immunol. 2019, 10, 1973. [Google Scholar] [CrossRef] [Green Version]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, A.K.; Pennington, J.M.; Tilton, S.; Wang, X.; Marshall, N.B.; Rohlman, D.; Funatake, C.; Punj, S.; O’Donnell, E.; Yu, Z.; et al. AhR activation increases IL-2 production by alloreactive CD4(+) T cells initiating the differentiation of mucosal-homing Tim3(+) Lag3(+) Tr1 cells. Eur. J. Immunol. 2017, 47, 1989–2001. [Google Scholar] [CrossRef] [Green Version]
- Giovannini, M.; Lodi, L.; Ricci, S. T-cell immunomodulation by bile acid metabolites. Allergy 2020, 75, 1833–1834. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of T(H)17 and induced T(reg) cell balance by an epigenetic mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef]
- Li, W.; Hang, S.; Fang, Y.; Bae, S.; Zhang, Y.; Zhang, M.; Wang, G.; McCurry, M.D.; Bae, M.; Paik, D.; et al. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe 2021, 29, 1366–1377. [Google Scholar] [CrossRef]
- Fassett, M.S.; Jiang, W.; D’Alise, A.M.; Mathis, D.; Benoist, C. Nuclear receptor Nr4a1 modulates both regulatory T-cell (Treg) differentiation and clonal deletion. Proc. Natl. Acad. Sci. USA 2012, 109, 3891–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Gregori, S.; Casorati, M.; Amuchastegui, S.; Smiroldo, S.; Davalli, A.M.; Adorini, L. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. J. Immunol. 2001, 167, 1945–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Zhou, R.; Luger, D.; Zhu, W.; Silver, P.B.; Grajewski, R.S.; Su, S.B.; Chan, C.C.; Adorini, L.; Caspi, R.R. Calcitriol suppresses antiretinal autoimmunity through inhibitory effects on the Th17 effector response. J. Immunol. 2009, 182, 4624–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.Y.; Yeung, S.T.; Silva, H.M.; Li, D.; et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2020, 180, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Kenny, D.J.; Xavier, R.J. Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu. Rev. Immunol. 2019, 37, 599–624. [Google Scholar] [CrossRef]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat(+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Kibbie, J.J.; Dillon, S.M.; Thompson, T.A.; Purba, C.M.; McCarter, M.D.; Wilson, C.C. Butyrate directly decreases human gut lamina propria CD4 T cell function through histone deacetylase (HDAC) inhibition and GPR43 signaling. Immunobiology 2021, 226, 152126. [Google Scholar] [CrossRef]
- Wen, S.; He, L.; Zhong, Z.; Zhao, R.; Weng, S.; Mi, H.; Liu, F. Stigmasterol Restores the Balance of Treg/Th17 Cells by Activating the Butyrate-PPARgamma Axis in Colitis. Front. Immunol. 2021, 12, 741934. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Becker, S.; Mohapatra, S.R.; Opitz, C.A.; Wick, W.; Platten, M. Suppression of Th1 differentiation by tryptophan supplementation in vivo. Amino. Acids. 2017, 49, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Yang, L.; You, K.; Chen, T.; Su, Z.; Cui, Z.; Wang, M.; Zhang, W.; Liu, B.; Zhou, K.; et al. Indole-3-Acetic Acid Alters Intestinal Microbiota and Alleviates Ankylosing Spondylitis in Mice. Front. Immunol. 2022, 13, 762580. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Christensen, J.; O’Garra, A.; Stockinger, B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 2009, 206, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Withers, D.R.; Hepworth, M.R.; Wang, X.; Mackley, E.C.; Halford, E.E.; Dutton, E.E.; Marriott, C.L.; Brucklacher-Waldert, V.; Veldhoen, M.; Kelsen, J.; et al. Transient inhibition of ROR-gammat therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat. Med. 2016, 22, 319–323. [Google Scholar] [CrossRef] [Green Version]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Park, J.; Goergen, C.J.; HogenEsch, H.; Kim, C.H. Chronically Elevated Levels of Short-Chain Fatty Acids Induce T Cell-Mediated Ureteritis and Hydronephrosis. J. Immunol. 2016, 196, 2388–2400. [Google Scholar] [CrossRef] [Green Version]
- Tykocinski, L.O.; Lauffer, A.M.; Bohnen, A.; Kaul, N.C.; Krienke, S.; Tretter, T.; Adam, I.; Mohapatra, S.R.; Saikali, P.; Lohning, M.; et al. Synovial Fibroblasts Selectively Suppress Th1 Cell Responses through IDO1-Mediated Tryptophan Catabolism. J. Immunol. 2017, 198, 3109–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Kayama, H.; Chen, M.L.; Delmas, A.; Sun, A.; Kim, S.Y.; Rangarajan, E.S.; McKevitt, K.; Beck, A.P.; Jackson, C.B.; et al. The Xenobiotic Transporter Mdr1 Enforces T Cell Homeostasis in the Presence of Intestinal Bile Acids. Immunity 2017, 47, 1182–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; de Vries, C.J.M. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE 2017, 12, e0176715. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Zhou, L.; Li, Z.; Shen, S.; Zhao, Y.; Liu, C.; Zhong, X.; Chang, Y.; Kermode, A.G.; Qiu, W. Gut Microbiome and Bile Acid Metabolism Induced the Activation of CXCR5+ CD4+ T Follicular Helper Cells to Participate in Neuromyelitis Optica Spectrum Disorder Recurrence. Front. Immunol. 2022, 13, 827865. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Piper, C.J.M.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Zheng, Y.; Zhang, M.; Fei, W.; Sun, D.; Zhao, M.; Ye, Y.; Zheng, C. Short-chain fatty acids regulate B cells differentiation via the FFA2 receptor to alleviate rheumatoid arthritis. Br. J. Pharmacol. 2022, 179, 4315–4329. [Google Scholar] [CrossRef]
- Fagarasan, S.; Muramatsu, M.; Suzuki, K.; Nagaoka, H.; Hiai, H.; Honjo, T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 2002, 298, 1424–1427. [Google Scholar] [CrossRef]
- Lundell, A.C.; Bjornsson, V.; Ljung, A.; Ceder, M.; Johansen, S.; Lindhagen, G.; Tornhage, C.J.; Adlerberth, I.; Wold, A.E.; Rudin, A. Infant B cell memory differentiation and early gut bacterial colonization. J. Immunol. 2012, 188, 4315–4322. [Google Scholar] [CrossRef] [Green Version]
- Piper, C.J.M.; Rosser, E.C.; Oleinika, K.; Nistala, K.; Krausgruber, T.; Rendeiro, A.F.; Banos, A.; Drozdov, I.; Villa, M.; Thomson, S.; et al. Aryl Hydrocarbon Receptor Contributes to the Transcriptional Program of IL-10-Producing Regulatory B Cells. Cell Rep. 2019, 29, 1878–1892. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Zhang, M.; Qi, H.; Gao, Y.; Yang, Y.; Yun, H.; Zhang, Q.; Yang, X.; Zhang, Y.; He, J.; et al. Gut microbiota-derived metabolite 3-idoleacetic acid together with LPS induces IL-35(+) B cell generation. Microbiome 2022, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhu, S.; Xu, G.; Feng, J.; Han, T.; Zhao, F.; She, Y.L.; Liu, S.; Ye, L.; Zhu, Y. Gene Expression and Antiviral Activity of Interleukin-35 in Response to Influenza A Virus Infection. J. Biol. Chem. 2016, 291, 16863–16876. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, A.K.; Amento, E.P.; Krane, S.M. Differential effects of 1,25-dihydroxyvitamin D3 on human lymphocytes and monocyte/macrophages: Inhibition of interleukin-2 and augmentation of interleukin-1 production. Cell Immunol. 1986, 98, 311–322. [Google Scholar] [CrossRef]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemire, J.M.; Adams, J.S.; Sakai, R.; Jordan, S.C. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J. Clin. Investig. 1984, 74, 657–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, T.; Nanjo, F. Dietary cycloinulooligosaccharides enhance intestinal immunoglobulin A production in mice. Biosci. Biotechnol. Biochem. 2009, 73, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Bonezi, V.; Cataneo, A.H.D.; Branquinho, M.S.F.; Silva, M.B.B.; Gonzalez-Dias, P.; Pereira, S.S.; Ferreira, L.C.S.; Nakaya, H.I.; Campa, A.; Wowk, P.F.; et al. Flavivirus-Mediating B Cell Differentiation Into Antibody-Secreting Cells in Humans Is Associated With the Activation of the Tryptophan Metabolism. Front. Immunol. 2020, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.F.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology 2018, 7, e1494113. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Kim, Y.H.; Kim, Y.J.; Kim, Y.W.; Moon, S.; Lee, Y.Y.; Jung, J.S.; Kim, Y.; Jung, H.E.; Kim, T.J.; et al. Taurodeoxycholate Increases the Number of Myeloid-Derived Suppressor Cells That Ameliorate Sepsis in Mice. Front. Immunol. 2018, 9, 1984. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Gutierrez-Vazquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Gomez de Aguero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef]
- Ebihara, T.; Song, C.; Ryu, S.H.; Plougastel-Douglas, B.; Yang, L.; Levanon, D.; Groner, Y.; Bern, M.D.; Stappenbeck, T.S.; Colonna, M.; et al. Runx3 specifies lineage commitment of innate lymphoid cells. Nat. Immunol. 2015, 16, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J. Dietary Fiber Confers Protection against Flu by Shaping Ly6c(-) Patrolling Monocyte Hematopoiesis and CD8(+) T Cell Metabolism. Immunity 2018, 48, 992–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Zhao, C.; Wang, C.J.; Xu, W.; Zhao, J.Y.; Lin, Y.; Yuan, Y.Y.; Lin, P.C.; Li, Y.; Zhao, S.; et al. Tryptophan potentiates CD8(+) T cells against cancer cells by TRIP12 tryptophanylation and surface PD-1 downregulation. J. Immunother. Cancer. 2021, 9, e002840. [Google Scholar] [CrossRef]
- Ding, C.; Hong, Y.; Che, Y.; He, T.; Wang, Y.; Zhang, S.; Wu, J.; Xu, W.; Hou, J.; Hao, H.; et al. Bile acid restrained T cell activation explains cholestasis aggravated hepatitis B virus infection. FASEB J. 2022, 36, e22468. [Google Scholar] [CrossRef]
- Zhu, C.; Boucheron, N.; Muller, A.C.; Majek, P.; Claudel, T.; Halilbasic, E.; Baazim, H.; Lercher, A.; Viczenczova, C.; Hainberger, D.; et al. 24-Norursodeoxycholic acid reshapes immunometabolism in CD8(+) T cells and alleviates hepatic inflammation. J. Hepatol. 2021, 75, 1164–1176. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Park, H.; Kim, Y.S.; Kim, K.D.; Lee, H.K.; Cho, D.H.; Yang, J.W.; Hur, D.Y. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int. Immunopharmacol. 2011, 11, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, 6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mencarelli, A.; Renga, B.; Migliorati, M.; Cipriani, S.; Distrutti, E.; Santucci, L.; Fiorucci, S. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J. Immunol. 2009, 183, 6657–6666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinolo, M.A.; Hatanaka, E.; Lambertucci, R.H.; Newsholme, P.; Curi, R. Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem. Funct. 2009, 27, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Hatanaka, E.; Sato, F.T.; Sampaio, S.C.; Curi, R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J. Nutr. Biochem. 2011, 22, 849–855. [Google Scholar] [CrossRef]
- Carrillo-Salinas, F.J.; Parthasarathy, S.; Moreno de Lara, L.; Borchers, A.; Ochsenbauer, C.; Panda, A.; Rodriguez-Garcia, M. Short-Chain Fatty Acids Impair Neutrophil Antiviral Function in an Age-Dependent Manner. Cells 2022, 11, 2515. [Google Scholar] [CrossRef]
- Balazs, I.; Horvath, A.; Leber, B.; Feldbacher, N.; Sattler, W.; Rainer, F.; Fauler, G.; Vermeren, S.; Stadlbauer, V. Serum bile acids in liver cirrhosis promote neutrophil dysfunction. Clin. Transl. Med. 2022, 12, e735. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Y.; Ji, H.; Li, Y. Anti-inflammatory, anti-oxidative stress and novel therapeutic targets for cholestatic liver injury. Biosci. Trends. 2019, 13, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, E.E.; Dowdell, A.S.; Henen, M.A.; Lanis, J.M.; Lee, J.S.; Cartwright, I.M.; Schaefer, R.E.M.; Ornelas, A.; Onyiah, J.C.; Vogeli, B.; et al. Microbial-derived indoles inhibit neutrophil myeloperoxidase to diminish bystander tissue damage. FASEB J. 2021, 35, e21552. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pu, A.; Sheng, B.; Zhang, Z.; Li, L.; Liu, Z.; Wang, Q.; Li, X.; Ma, Y.; Yu, M.; et al. Aryl hydrocarbon receptor activation modulates CD8alphaalpha(+)TCRalphabeta(+) IELs and suppression of colitis manifestations in mice. Biomed. Pharmacother. 2017, 87, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M.; Lefort, C.; Depommier, C.; Rastelli, M.; Everard, A. Microbial regulation of organismal energy homeostasis. Nat. Metab. 2019, 1, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Leonard, M.M.; Valitutti, F.; Karathia, H.; Pujolassos, M.; Kenyon, V.; Fanelli, B.; Troisi, J.; Subramanian, P.; Camhi, S.; Colucci, A.; et al. Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proc. Natl. Acad. Sci. USA 2021, 118, e2020322118. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. [Google Scholar] [CrossRef] [Green Version]
- Hezaveh, K.; Shinde, R.S.; Klotgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e8. [Google Scholar] [CrossRef] [PubMed]
- Antushevich, H. Fecal microbiota transplantation in disease therapy. Clin. Chim. Acta 2020, 503, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Leshem, A.; Horesh, N.; Elinav, E. Fecal Microbial Transplantation and Its Potential Application in Cardiometabolic Syndrome. Front. Immunol. 2019, 10, 1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wiesnoski, D.H.; Helmink, B.A.; Gopalakrishnan, V.; Choi, K.; DuPont, H.L.; Jiang, Z.D.; Abu-Sbeih, H.; Sanchez, C.A.; Chang, C.C.; et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat. Med. 2018, 24, 1804–1808. [Google Scholar] [CrossRef] [PubMed]
- Peled, J.U.; Gomes, A.L.C.; Devlin, S.M.; Littmann, E.R.; Taur, Y.; Sung, A.D.; Weber, D.; Hashimoto, D.; Slingerland, A.E.; Slingerland, J.B.; et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2020, 382, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; van den Brink, M.R.M. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat. Rev. Cancer. 2018, 18, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Wardill, H.R.; Secombe, K.R.; Bryant, R.V.; Hazenberg, M.D.; Costello, S.P. Adjunctive fecal microbiota transplantation in supportive oncology: Emerging indications and considerations in immunocompromised patients. EBioMedicine. 2019, 44, 730–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.; Duerksen, D.R. Enteral Nutrition in the Management of Pediatric and Adult Crohn’s Disease. Nutrients 2018, 10, 537. [Google Scholar] [CrossRef] [Green Version]
- Verburgt, C.M.; Ghiboub, M.; Benninga, M.A.; de Jonge, W.J.; Van Limbergen, J.E. Nutritional Therapy Strategies in Pediatric Crohn’s Disease. Nutrients 2021, 13, 212. [Google Scholar] [CrossRef]
- Kedia, S.; Virmani, S.; Vuyyuru, S.K.; Kumar, P.; Kante, B.; Sahu, P.; Kaushal, K.; Farooqui, M.; Singh, M.; Verma, M.; et al. Faecal microbiota transplantation with anti-inflammatory diet (FMT-AID) followed by anti-inflammatory diet alone is effective in inducing and maintaining remission over 1 year in mild to moderate ulcerative colitis: A randomised controlled trial. Gut 2022, 71, 2401–2413. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SCFAs | Biosynthesis | Bacterial Species | References |
|---|---|---|---|
| Acetate (C2) | Via acetyl-CoA pathway Via Wood–Ljungdahl pathway | Akkermansia muciniphila, Bacteroides spp., Bifidobacterium spp., Prevotella spp., Ruminococcus spp Blautia hydrogenotrophica, Clostridium spp., Streptococcus spp. | [8,12,13] |
| Popionate (C3) | From succinate pathway From acrylate pathway From propanediol pathway | Bacteroides spp., Phascolarctobacterium succinatutens, Dialister spp., Veillonella spp., Roseburia spp., Firmicutes, Roseburia inulinivorans, Ruminucocus spp., Clostridium spp., Eubacterium spp., Coprococcus spp., and Akkermansia muciniphila, Megasphaera elsdenii, Coprococcus catus, Clostridiales bacterium. Coproccus catus and Clostridium spp. Salmonella spp., Roseburia inulinivorans, Ruminococcus obeum, Eubacterium halli | [8,14,15] |
| Butyrate (C4) | From butyryl-CoA acetate Co-A transferase pathway From butyrate kinase pathwayFrom lactate and acetate | Anaerostipes spp., Coprococcus catus, Eubacterium rectale, Eubacterium hallii, Faecalibacterium prausnitzii, Roseburia spp., Roseburia intestinalis, Roseburia insulinivorans, Clostridiales bacterium, Anaerostripes spp, Coprococcu spp., Costridium symbiosum and Faecalibacterium prasnitzii. Coprococcus comes and Coprococcus eutactus. Eubacterium hallii and Anaerostipes spp | [8,14,16,17] |
| Metabolite | Biosynthesis | Bacterial Species | References |
|---|---|---|---|
| Indole | Form Trp metabolism by tryptophanase | Clostridium limosum, Bacteroides ovatus, Enterococcus faecalis and Escheichia coli | [20] |
| IAA | From Trp metabolism through the oxidative and reductive pathways by tryptophan 2-monooxygenase or acyl-CoA dehydrogenase | Clostridium sporogenes Clostridium bartlettii and Bifidobacterium spp. | [22,23,24] |
| IPA | From Trp metabolism through the oxidative and reductive pathways by tryptophan 2-monooxygenase or acyl-CoA dehydrogenase and via phenyllactate dehydratae and acyl-CoA dehydrogenase | Clostridium sporogenes Clostridium bartlettii and Bifidobacterium spp. and Peptostreptococcus spp | [22,23,24,25] |
| IA | From Trp metabolism via phenyllactate dehydratae and acyl-CoA dehydrogenase | Peptostreptococcus spp. | [25] |
| Skatole | From Trp metabolism by decarboxylation of IAA | Bacteroides spp. and Clostridium spp. | [24,26] |
| IA1d | From Trp metabolism via an aromatic amino acid aminotransferase (ArAT) and indolelactic acid dehydrogenase (ILDH) | Lactobacillus johnsonii, L. reuteri, L. acidophilus and L. murinus | [27] |
| Tryptamine | From Trp metabolism via a Trp decarboxylase enzyme | Ruminococcus gnavus and Clostridium sporogenes. | [28] |
| 3-hydroxyanthranilic acid (3-HAA) | From Trp metabolism via eukaryotic Kyn pathway | Pseudomonas, Burkholderia, Stenotrophomonas, Xanthomonas, Shewanella, and Bacillus | [29] |
| Bile Acids (BAs) | Biosynthesis | Bacterial Species | References |
|---|---|---|---|
| Conjugated BAs | From primary BAs to conjugate with other amino acids | Clostridium bolteae, Bacteriodetes Bacteroides vulgatus, Firmicutes Lactobcillus rumini, Actinobacteria Hungatella hathewayi, Bacterorides vulgatus, Lactobacillus ruminis, Holdemania filiformis, Clostridium scindens | [39,40,41] |
| Deconjugated BAs | Via deconjugating by bile salt hydrolases (BSHs) | Lactobacillus spp., Clostridium spp., Bifidobacterium spp., Enterococcus spp., and Bacteroides spp. | [41,42,43,44,45,46,47,48] |
| Secondary BAs (DCA, LCA) | From deconjugated BAs through deconjugation, dehydroxylation, oxidation and epimerization | Clostridium clusters XIVa, IV, XI, C. scindens, C. hylemonae and C. perfringens, Blautia producta, Eggerthella lenta, Clostridium scindens. | [49,50] |
| 3-oxoLCA and isoLCA | Convert LCA to 3-oxoLCA and isoLCA | Adlercreutzia, Bifidobacterium, Enterocloster, Clostridium, Collinsella, Eggerthella, Gordonibacter, Monoglobus, Peptoniphilus, Phocea, Raoultibacter, and Mediterraneibacte | [35] |
| Ursodeoxycholic acid (UDCA) | Conversion of 7-oxo-LCA | Clostridium absonum, Stenotrophomonas maltophilia, Ruminococcus gnavus and Collinsella aerofaciens | [51,52] |
| UDCA | Conversion of 7α-epimerization | Clostridium baratii | [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhu, N.; Su, X.; Gao, Y.; Yang, R. Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis. Cells 2023, 12, 793. https://doi.org/10.3390/cells12050793
Wang J, Zhu N, Su X, Gao Y, Yang R. Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis. Cells. 2023; 12(5):793. https://doi.org/10.3390/cells12050793
Chicago/Turabian StyleWang, Juanjuan, Ningning Zhu, Xiaomin Su, Yunhuan Gao, and Rongcun Yang. 2023. "Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis" Cells 12, no. 5: 793. https://doi.org/10.3390/cells12050793