
COVID-19 and Alzheimer’s Disease Share Common Neurological and Ophthalmological Manifestations: A Bidirectional Risk in the Post-Pandemic Future

Abstract
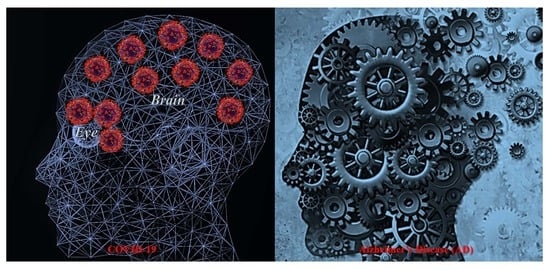
:
{kind=link}
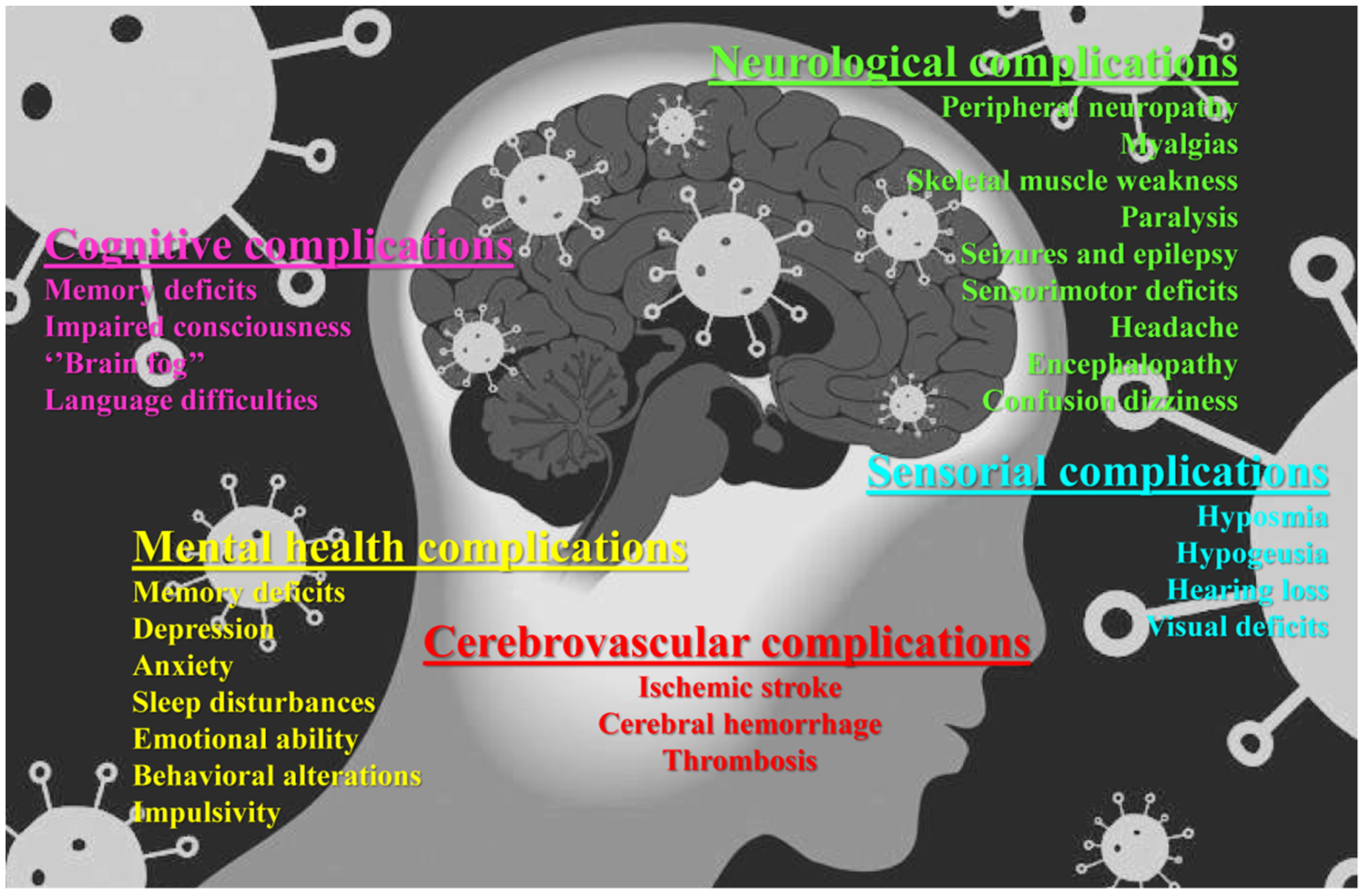{kind=link}
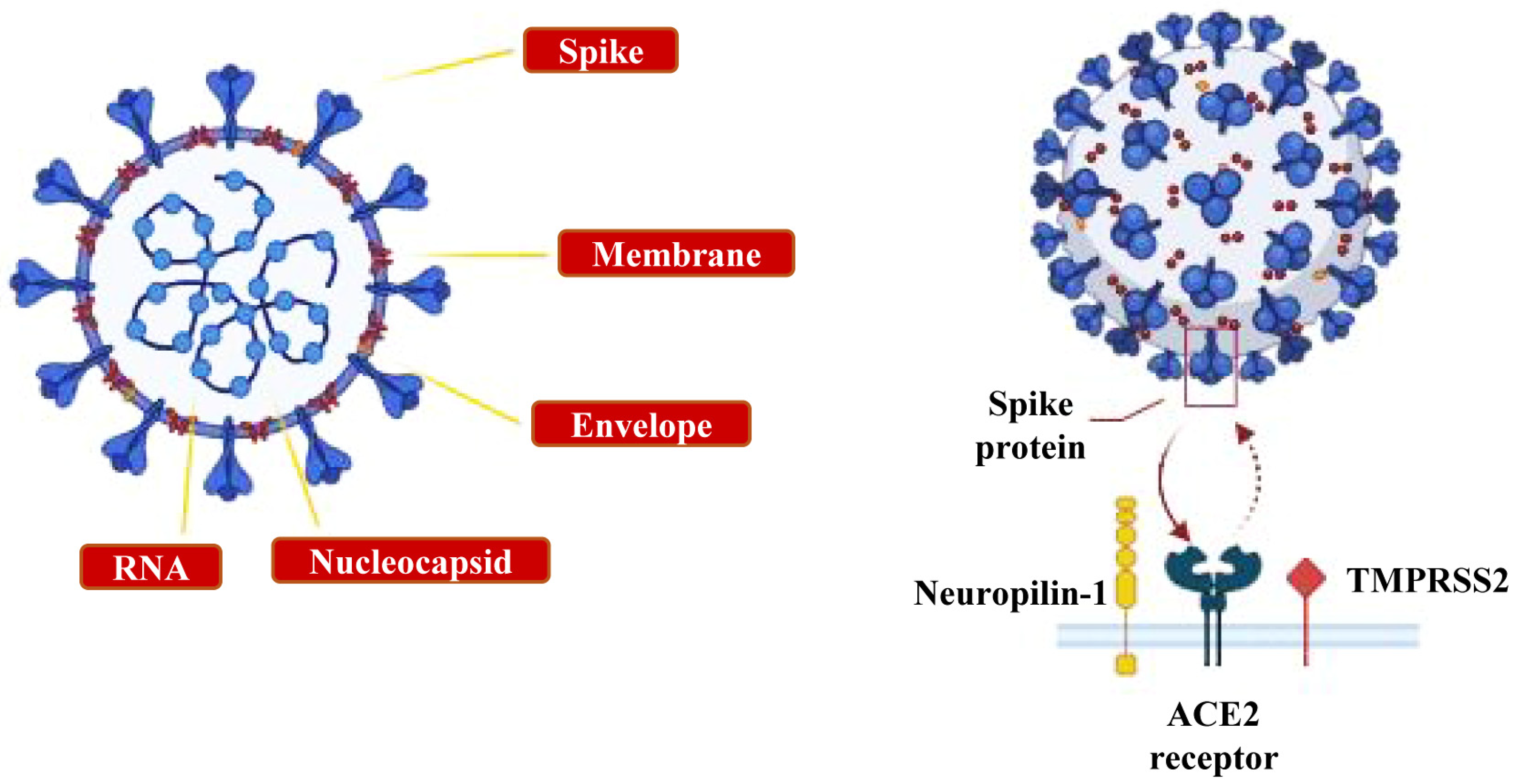{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Neuroinvasive Mechanisms of SARS-CoV-2 and Neurological Manifestations of COVID-19
- -
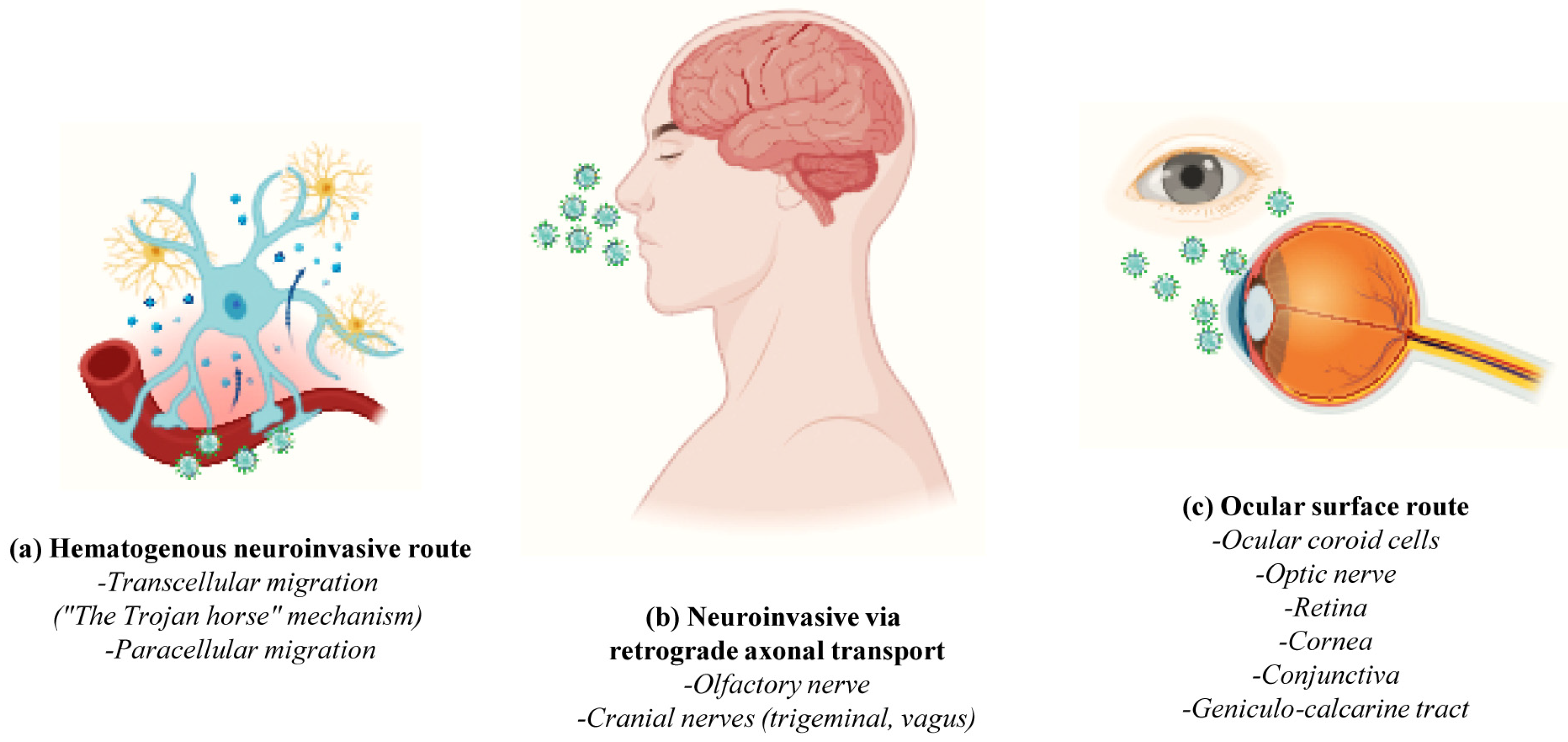
- The hematogenous pathway or “Trojan horse mechanism” wherein infected circulating immune cells serve as reservoirs for the virus that traverses from the bloodstream to the CNS (cell transmigration); SARS-CoV-2 infects, near the vessel wall, the resident peripheral immune cells of the blood circulation (phagocytic monocytes/macrophages, neutrophils and lymphocytes) which, in turn, penetrate the neurovascular unit of the Blood–Brain Barrier (BBB), becoming a pool of viral dissemination toward the CNS [54,55,56] (Figure 3a);
- -
- The Blood–CerebroSpinal Fluid (B-CSF) pathway (paracellular migration): SARS-CoV-2 binds to the ACE2 receptors of the endothelial cells and damages the integral citoarchitecture of the BBB. To get into the brain, the virus locally activates the signaling transduction pathway of Nuclear Factor kappa B (NF-kB) transcription factor, leading to an up-regulation in the basal expression level of Matrix MetalloPeptidase 9 (MMP9) which, in turn, degrades the extracellular matrix with consequent increased B-CSF permeability and alterations in immune cell trafficking (MMP8, Monocyte Chemoattractant Protein-1 (MCP-1), InterCellular Adhesion Molecule 1 (ICAM-1), a neuroinflammatory response with the release of pro-inflammatory cytokines and chemokines such as Interleukin (IL) IL-2, IL-6, IL-7 and IL-8, Tumor Necrosis Factor (TNF) TNFα, C-C Motif Chemokine Ligand (CCL) CCL2, CCL3 and CCL7 and C-X-C motif chemokine ligand (CXCL) CXCL10) [54,57] (Figure 3a);
- -
- The transneuronal spreading or “neuronal route” (via exocytosis/endocytosis or “fast axonal transport” mechanisms of vesicles along the microtubules track in order to move the virus from synaptic terminals back towards neuronal cell bodies) from systemic organs to the CNS throughout the cranial nerves: In this process, the virus first enters the nerve endings (i.e., the peripheral nerves) and then is retrogradely transported to the soma to invade the CNS; in detail, SARS-CoV-2 enters through (i) the olfactory mucosa (causing anosmia), and it spreads via the olfactory nerve to the olfactory cortex; (ii) the lacrimal and salivary glands, and it spreads via the facial VII and glossopharyngeal IX nerves to their respective brainstem nuclei; (iii) the taste buds of gustatory mucosa (triggering ageusia), and it spreads via the VII and IX nerves to the Nucleus Tractus Solitarius (NTS) located in the brainstem; or (iv) the respiratory system, and, via the vagus nerve X, it spreads both to other systemic organs (the heart, kidneys and gastrointestinal tract) innervated by this nerve and to the brainstem [58] (Figure 3b);
- -
- The circumventricular organs (CVO) lacking the BBB: SARS-CoV-2 enters the CNS through the ACE2-expressing and vascularized subfornical organ, the paraventricular nucleus, the NTS and the rostral ventrolateral medulla by triggering local neurovascular damage (Figure 3b);
- -
- The ocular system: the epithelial cells of the cornea and conjunctiva, the trabecular meshwork, choroid and retinal cells, optic nerve and geniculo-calcarin tract expressing the ACE2 receptor and neuropilin-1 are also entry points for the SARS-CoV-2 infection towards the occipital cortical areas [26,30] (Figure 3c).
3. Bidirectional Relationships between Long COVID-19 and AD
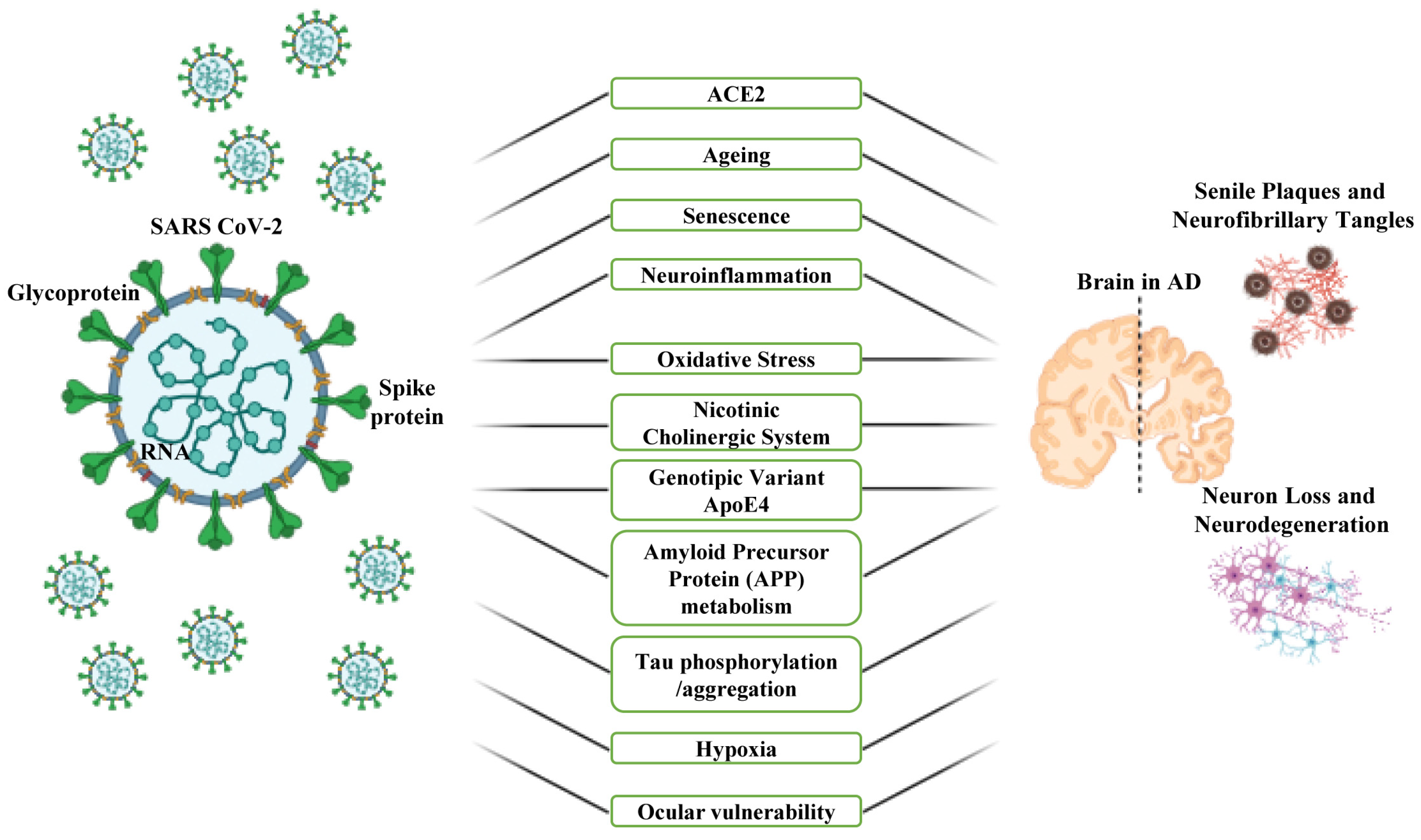
4. Common Risk Factors and Involved Mechanisms That Mediate the Association between COVID-19 and AD
4.1. ACE2 and Ageing
4.2. Neuroinflammation, Oxidative Stress and Nicotinic Cholinergic System
4.3. APOE Genetic Variant and Signal Pathways
4.4. Hypoxia
4.5. Serotonin or 5-Hydroxytryptamine (5-HT)
5. Neuro-Ophthalmic Complications Shared by COVID-19 and AD
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Breijyeh, Z.; Karaman, R. Comprehensive review on alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Bchetnia, M.; Girard, C.; Duchaine, C.; Laprise, C. The outbreak of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): A review of the current global status. J. Infect. Public Health 2020, 13, 1601–1610. [Google Scholar] [CrossRef]
- Hiscott, J.; Alexandridi, M.; Muscolini, M.; Tassone, E.; Palermo, E.; Soultsioti, M.; Zevini, A. The global impact of the coronavirus pandemic. Cytokine Growth Factor Rev. 2020, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.R.L.; Martin, M.R.; Martin, J.D.; Kuhn, P.; Hicks, J.B. Modeling the onset of symptoms of COVID-19. Front. Public Health 2020, 8, 473. [Google Scholar] [CrossRef] [PubMed]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Hampshire, A.; Trender, W.; Chamberlain, S.R.; Jolly, A.E.; Grant, J.E.; Patrick, F.; Mazibuko, N.; Williams, S.C.; Barnby, J.M.; Hellyer, P.; et al. Cognitive deficits in people who have recovered from COVID-19. eClinicalMedicine 2021, 39, 101044. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Vieites Prado, A.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef]
- Harapan, B.N.; Yoo, H.J. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J. Neurol. 2021, 268, 3059–3071. [Google Scholar] [CrossRef] [PubMed]
- Sharifkashani, S.; Bafrani, M.A.; Khaboushan, A.S.; Pirzadeh, M.; Kheirandish, A.; Bali, H.Y.; Hessami, A.; Saghazadeh, A.; Rezaei, N. Angiotensin-converting enzyme 2 (ACE2) receptor and SARS-CoV-2: Potential therapeutic targeting. Eur. J. Pharmacol. 2020, 884, 173455. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, J.F.; Yuen, T.T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.-Y.; Tsang, J.O.-L.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Sun, S.-H.; Chen, Q.; Gu, H.-J.; Yang, G.; Wang, Y.-X.; Huang, X.-Y.; Liu, S.-S.; Zhang, N.-N.; Li, X.-F.; Xiong, R.; et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbe 2020, 28, 124–133.e4. [Google Scholar] [CrossRef] [PubMed]
- Von Weyhern, C.H.; Kaufmann, I.; Neff, F.; Kremer, M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet 2020, 395, e109. [Google Scholar] [CrossRef]
- Zhang, B.-Z.; Chu, H.; Han, S.; Shuai, H.; Deng, J.; Hu, Y.-F.; Gong, H.-R.; Lee, A.C.-Y.K.; Zou, Z.; Yau, T.; et al. SARS-CoV-2 infects human neural progenitor cells and brain organoids. Cell Res. 2020, 30, 928–931. [Google Scholar] [CrossRef]
- Tan, B.-H.; Liu, J.-M.; Gui, Y.; Wu, S.; Suo, J.-L.; Li, Y.-C. Neurological involvement in the respiratory manifestations of COVID-19 patients. Aging 2021, 13, 4713–4730. [Google Scholar] [CrossRef]
- Fišar, Z. Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets. Biomolecules 2022, 12, 1676. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Bowirrat, A. Immunosenescence and Aging: Neuroinflammation Is a Prominent Feature of Alzheimer’s Disease and Is a Likely Contributor to Neurodegenerative Disease Pathogenesis. J. Pers. Med. 2022, 12, 1817. [Google Scholar] [CrossRef] [PubMed]
- Holubiec, M.I.; Gellert, M.; Hanschmann, E.M. Redox signaling and metabolism in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1003721. [Google Scholar] [CrossRef] [PubMed]
- Kara, B.; Gordon, M.N.; Gifani, M.; Dorrance, A.M.; Counts, S.E. Vascular and Nonvascular Mechanisms of Cognitive Impairment and Dementia. Clin. Geriatr. Med. 2023, 39, 109–122. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, X.; Ma, L.; Wei, W.; Li, Z.; Chang, S.; Wen, J.; Sun, J.; Li, H. Role of Aβ in Alzheimer’s-related synaptic dysfunction. Front. Cell Dev. Biol. 2022, 10, 964075. [Google Scholar] [CrossRef]
- Pacheco-Herrero, M.; Soto-Rojas, L.O.; Harrington, C.R.; Flores-Martinez, Y.M.; Villegas-Rojas, M.M.; León-Aguilar, A.M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Corniel-Taveras, C.N.; et al. Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection. Front. Neurol. 2021, 12, 660087. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef]
- Karuppan, M.K.M.; Devadoss, D.; Nair, M.; Chand, H.S.; Lakshmana, M.K. SARS-CoV-2 Infection in the Central and Peripheral Nervous System-Associated Morbidities and Their Potential Mechanism. Mol. Neurobiol. 2021, 58, 2465–2480. [Google Scholar] [CrossRef]
- Jha, N.K.; Ojha, S.; Jha, S.K.; Dureja, H.; Singh, S.K.; Shukla, S.D.; Chellappan, D.K.; Gupta, G.; Bhardwaj, S.; Kumar, N.; et al. Evidence of Coronavirus (CoV) Pathogenesis and Emerging Pathogen SARS-CoV-2 in the Nervous System: A Review on Neurological Impairments and Manifestations. J. Mol. Neurosci. 2021, 71, 2192–2209. [Google Scholar] [CrossRef]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Puvvada, N.; Kandimalla, R.; Reddy, P.H. Emerging COVID-19 Neurological Manifestations: Present Outlook and Potential Neurological Challenges in COVID-19 Pandemic. Mol. Neurobiol. 2021, 58, 4694–4715. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J.; Kamel, H. Effects of COVID-19 on the Nervous System. Cell 2020, 183, 16–27.e1. [Google Scholar] [CrossRef]
- Liu, J.M.; Tan, B.H.; Wu, S.; Gui, Y.; Suo, J.L.; Li, Y.C. Evidence of central nervous system infection and neuroinvasive routes, as well as neurological involvement, in the lethality of SARS-CoV-2 infection. J. Med. Virol. 2021, 93, 1304–1313. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Duarte-Neto, A.N.; Saldiva, P.H.N.; Caldini, E.G. Using EM data to understand COVID-19 pathophysiology. Lancet 2021, 397, 196–197. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef]
- Al-Dalahmah, O.; Thakur, K.T.; Nordvig, A.S.; Prust, M.L.; Roth, W.; Lignelli, A.; Uhlemann, A.-C.; Happy Miller, E.; Kunnath-Velayudhan, S.; Del Portillo, A.; et al. Neuronophagia and microglial nodules in a SARS-CoV-2 patient with cerebellar hemorrhage. Acta Neuropathol. Commun. 2020, 8, 147. [Google Scholar] [CrossRef]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; De Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef] [PubMed]
- Matschke, J.; Lutgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schroder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of COVID-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Kremer, S.; Lersy, F.; Anheim, M.; Merdji, H.; Schenck, M.; Oesterlé, H.; Bolognini, F.; Messie, J.; Khalil, A.; Gaudemer, A.; et al. Neurologic and neuroimaging findings in patients with COVID-19: A retrospective multicenter study. Neurology 2020, 95, e1868–e1882. [Google Scholar] [CrossRef]
- Edén, A.; Kanberg, N.; Gostner, J.; Fuchs, D.; Hagberg, L.; Andersson, L.-A.; Lindh, M.; Price, R.W.; Zetterberg, H.; Gisslén, M. CSF Biomarkers in Patients with COVID-19 and Neurologic Symptoms: A Case Series. Neurology 2021, 96, e294–e300. [Google Scholar] [CrossRef]
- Destras, G.; Bal, A.; Escuret, V.; Morfin, F.; Lina, B.; Josset, L. COVID-Diagnosis HCL Study Group. Systematic SARS-CoV-2 screening in cerebrospinal fluid during the COVID-19 pandemic. Lancet Microbe 2020, 1, e149. [Google Scholar] [CrossRef] [PubMed]
- Espíndola, O.M.; Siqueira, M.; Soares, C.N.; Lima, M.A.S.D.; Leite, A.C.C.B.; Araujo, A.Q.C.; Brandão, C.O.; Silva, M.T.T. Patients with COVID-19 and neurological manifestations show undetectable SARS-CoV-2 RNA levels in the cerebrospinal fluid. Int. J. Infect. Dis. 2020, 96, 567–569. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Steiner, J.; Pasternack, N.; Li, W.; Maric, D.; Safavi, F.; Horkayne-Szakaly, I.; Jones, R.; Stram, M.N.; et al. Neurovascular injury with complement activation and inflammation in COVID-19. Brain 2022, 145, 2555–2568. [Google Scholar] [CrossRef]
- Zheng, J.; Roy Wong, L.Y.; Li, K.; Verma, A.K.; Ortiz, M.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B.; et al. K18-hACE2 Mice for Studies of COVID-19 Treatments and Pathogenesis Including Anosmia. Nature 2021, 589, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Rathnasinghe, R.; Strohmeier, S.; Amanat, F.; Gillespie, V.L.; Krammer, F.; García-Sastre, A.; Coughlan, L.; Schotsaert, M.; Uccellini, M. Comparison of Transgenic and Adenovirus hACE2 Mouse Models for SARS-CoV-2 Infection. Emerg. Microbes Infect. 2020, 9, 2433–2445. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rozen-Gagnon, K.; Miles, L.A.; Schuster, F.; Razooky, B.; Jacobson, E.; Wu, X.; Yi, S.; Rudin, C.M.; et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell 2021, 184, 133–148.e20. [Google Scholar] [CrossRef]
- Mostafavi, E.; Dubey, A.K.; Teodori, L.; Ramakrishna, S.; Kaushik, A. SARS-CoV-2 Omicron variant: A next phase of the COVID-19 pandemic and a call to arms for system sciences and precision medicine. MedComm 2022, 3, e119. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Lane, T.E.; Stohlman, S.A. Coronavirus infection of the central nervous system: Host-virus stand-off. Nat. Rev. Microbiol. 2006, 4, 121–132. [Google Scholar] [CrossRef]
- Chen, F.; Chen, Y.; Wang, Y.; Qiongwei Ke, Q.; Cui, L. The COVID-19 pandemic and Alzheimer’s disease: Mutual risks and mechanisms. Transl. Neurodegener. 2022, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Kujawska, M.; Mostafavi, E.; Kaushik, A. SARS-CoV-2 getting into the brain; neurological phenotype of COVID-19, and management by nano-biotechnology. Neural Regen. Res. 2023, 18, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.; Siokas, V.; Aloizou, A.-M.; Liampas, I.; Mentis, A.-F.A.; Tsouris, Z.; Papadimitriou, A.; Mitsias, P.D.; Tsatsakis, A.; Bogdanos, D.P.; et al. Unraveling the Possible Routes of SARS-CoV-2 Invasion into the Central Nervous System. Curr. Treat. Options Neurol. 2020, 22, 37. [Google Scholar] [CrossRef]
- Ur, A.; Verma, K. Cytokine Storm in COVID-19: A Neural Hypothesis. ACS Chem. Neurosci. 2020, 11, 1868–1870. [Google Scholar] [CrossRef] [PubMed]
- MacLean, M.A.; Kamintsky, L.; Leck, E.D.; Friedman, A. The potential role of microvascular pathology in the neurological manifestations of coronavirus infection Fluids Barriers. Fluids Barriers CNS 2020, 17, 55. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Stodola, J.K.; Meessen-Pinard, M.; Talbot, P.J. Human coronaviruses: Viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res. 2014, 194, 145–158. [Google Scholar] [CrossRef]
- Fenrich, M.; Mrdenovic, S.; Balog, M.; Tomic, S.; Zjalic, M.; Roncevic, A.; Mandic, D.; Debeljak, Z.; Heffer, M. SARS-CoV-2 Dissemination through Peripheral Nerves Explains Multiple Organ Injury. Front. Cell. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef]
- Perrottelli, A.; Sansone, N.; Giordano, G.M.; Caporusso, E.; Giuliani, L.; Melillo, A.; Pezzella, P.; Bucci, P.; Mucci, A.; Galderisi, S. Cognitive Impairment after Post-Acute COVID-19 Infection: A Systematic Review of the Literature. J. Pers. Med. 2022, 12, 2070. [Google Scholar] [CrossRef]
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive impact of COVID-19: Looking beyond the short term. Alzheimers Res. Ther. 2020, 12, 170. [Google Scholar] [CrossRef]
- Ceban, F.; Ling, S.; Lui, L.M.W.; Lee, Y.; Gill, H.; Teopiz, K.M.; Rodrigues, N.B.; Subramaniapillai, M.; Di Vincenzo, J.D.; Cao, B.; et al. Fatigue and cognitive impairment in Post-COVID-19 Syndrome: A systematic review and meta-analysis. Brain Behav. Immun. 2022, 101, 93–135. [Google Scholar] [CrossRef]
- Crivelli, L.; Palmer, K.; Calandri, I.; Guekht, A.; Beghi, E.; Carroll, W.; Frontera, J.; García-Azorín, D.; Westenberg, E.; Winkler, A.S.; et al. Changes in cognitive functioning after COVID-19: A systematic review and meta-analysis. Alzheimers Dement. 2022, 18, 1047–1066. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Li, Q.; Melino, S.; Melino, G.; Shi, Y. Can COVID-19 pandemic boost the epidemic of neurodegenerative diseases? Biol. Direct. 2020, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, M.A.; Bagnato, G.; Ioppolo, C.; Versace, A.G.; Irrera, N. Impact of the COVID-19 Pandemic on Chronic Neurological Disorders: Focus on Patients with Dementia. CNS Neurol. Disord. Drug Targets 2022, 21, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, W.; Yu, X.; Hu, D.; Bao, M.; Li, H.; Zhou, J.; Jiang, H. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on4-week follow-up. J. Infect. 2020, 80, 639–645. [Google Scholar] [CrossRef]
- de Erausquin, G.A.; Snyder, H.; Brugha, T.S.; Seshadri, S.; Carrillo, M.; Sagar, R.; Huang, Y.; Newton, C.; Tartaglia, C.; Teunissen, C.; et al. Chronic neuropsychiatric sequelae of SARS-CoV-2: Protocol and methods from the Alzheimer’s Association Global Consortium. Alzheimers Dement. 2022, 8, e12348. [Google Scholar] [CrossRef] [PubMed]
- Golzari-Sorkheh, M.; Weaver, D.F.; Reed, M.A. COVID-19 as a Risk Factor for Alzheimer’s Disease. J. Alzheimers Dis. 2023, 91, 1–23. [Google Scholar] [CrossRef]
- Olivera, E.; Sáez, A.; Carniglia, L.; Caruso, C.; Lasaga, M.; Durand, D. Alzheimer’s disease risk after COVID-19: A view from the perspective of the infectious hypothesis of neurodegeneration. Neural Regen. Res. 2023, 18, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Ferini-Strambi, L.; Salsone, M. COVID-19 and neurological disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 2021, 268, 409–419. [Google Scholar] [CrossRef]
- Daroische, R.; Hemminghyth, M.S.; Eilertsen, T.H.; Breitve, M.H.; Chwiszczuk, L.J. Cognitive Impairment after COVID-19—A Review on Objective Test Data. Front. Neurol. 2021, 12, 699582. [Google Scholar] [CrossRef]
- Numbers, K.; Brodaty, H. The effects of the COVID-19 pandemic on people with dementia. Nat. Rev. Neurol. 2021, 17, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Magusali, N.; Graham, A.C.; Piers, T.M.; Panichnantakul, P.; Yaman, U.; Shoai, M.; Reynolds, R.H.; Botia, J.A.; Brookes, K.J.; Guetta-Baranes, T.; et al. A genetic link between risk for Alzheimer’s disease and severe COVID-19 outcomes via the OAS1 gene. Brain 2021, 144, 3727–3741. [Google Scholar] [CrossRef] [PubMed]
- Hariyanto, T.I.; Putri, C.; Arisa, J.; Situmeang, R.F.V.; Kurniawan, A. Dementia and outcomes from coronavirus disease 2019 (COVID-19) pneumonia: A systematic review and meta-analysis. Arch. Gerontol. Geriatr. 2021, 93, 104299. [Google Scholar] [CrossRef] [PubMed]
- Tahira, A.C.; Verjovski-Almeida, S.; Ferreira, S.T. Dementia is an age-independent risk factor for severity and death in COVID-19 in patients. Alzheimers Dement. 2021, 17, 1818–1831. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jee, Y.; Park, S.; Ha, E.H.; Jo, I.; Lee, H.W.; Song, M.S. Mortality risk within 14 days after coronavirus disease 2019 diagnosis in dementia patients: A nation wide analysis. Dement. Geriatr. Cogn. Disord. 2021, 50, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Park, S.H.; Kim, N.Y.; Kang, D.W.; Na, H.R.; Um, Y.H.; Han, S.; Park, S.S.; Lim, H.K. Association between dementia and clinical outcome after COVID-19: A nationwide cohort study with propensity score matched control in South Korea. Psychiatry Investig. 2021, 18, 523–529. [Google Scholar] [CrossRef]
- Chung, S.J.; Chang, Y.; Jeon, J.; Shin, J.I.; Song, T.-J.; Kim, J. Association of Alzheimer’s Disease with COVID-19 Susceptibility and Severe Complications: A Nationwide Cohort Study. J. Alzheimers Dis. 2022, 87, 701–710. [Google Scholar] [CrossRef]
- Wang, F.; Kream, R.M.; Stefano, G.B. Long-Term Respiratory and Neurological Sequelae of COVID-19. Med. Sci. Monit. 2020, 26, e928996. [Google Scholar] [CrossRef]
- Wang, L.; Davis, P.B.; Volkow, N.D.; Berger, N.A.; Kaelber, D.C.; Xu, R. Association of COVID-19 with with new-onset Alzheimer’s disease. J. Alzheimers Dis. 2022, 89, 411–414. [Google Scholar] [CrossRef]
- Covino, M.; De Matteis, G.; Santoro, M.; Sabia, L.; Simeoni, B.; Candelli, M.; Ojetti, V.; Franceschi, F. Clinical characteristics and prognostic factors in COVID-19 patients aged ≥80 years. Geriatr. Gerontol. Int. 2020, 20, 704–708. [Google Scholar] [CrossRef]
- Bianchetti, A.; Rozzini, R.; Guerini, F.; Bofelli, S.; Ranieri, P.; Minelli, G.; Bianchetti, L.; Trabucchi, M. Clinical presentation of COVID-19 in dementia patients. J. Nutr. Health Aging 2020, 24, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Negrini, F.; Ferrario, I.; Mazziotti, D.; Berchicci, M.; Bonazzi, M.; de Sire, A.; Negrini, S.; Zapparoli, L. Neuropsychological features of severe hospitalized coronavirus disease 2019 patients at clinical stability and clues for postacute rehabilitation. Arch. Phys. Med. Rehabil. 2021, 102, 155–158. [Google Scholar] [CrossRef]
- De Lorenzo, R.; Conte, C.; Lanzani, C.; Benedetti, F.; Roveri, L.; Mazza, M.G.; Brioni, E.; Giacalone, G.; Canti, V.; Sofia, V.; et al. Residual clinical damage after COVID-19: A retrospective and prospective observational cohort study. PLoS ONE 2020, 15, e0239570. [Google Scholar] [CrossRef] [PubMed]
- Beaud, V.; Crottaz-Herbette, S.; Dunet, V.; Vaucher, J.; Bernard-Valnet, R.; Du Pasquier, R.; Bart, P.-A.; Clarke, S. Pattern of cognitive deficits in severe COVID-19. J. Neurol. Neurosurg. Psychiatry 2021, 92, 567–568. [Google Scholar] [CrossRef] [PubMed]
- van den Borst, B.; Peters, J.B.; Brink, M.; Schoon, Y.; Bleeker-Rovers, C.P.; Schers, H.; van Hees, H.W.H.; van Helvoort, H.; van den Boogaard, M.; van der Hoeven, H.; et al. Comprehensive health assessment three months after recovery from acute COVID-19. Clin. Infect. Dis. 2021, 73, e1089–e1098. [Google Scholar] [CrossRef]
- Almeria, M.; Cejudo, J.C.; Sotoca, J.; Deus, J.; Krupinski, J. Cognitive profile following COVID-19 infection: Clinical predictors leading to neuropsychological impairment. Brain Behav. Immun. Health 2020, 9, 100163. [Google Scholar] [CrossRef]
- Woo, M.S.; Malsy, J.; Pottgen, J.; Seddiq Zai, S.; Ufer, F.; Hadjilaou, A.; Schmiedel, S.; Addo, M.M.; Gerloff, C.; Heesen, C.; et al. Frequent neurocognitive deficits after recovery from mild COVID-19. Brain Commun. 2020, 2, fcaa205. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, S.; Chen, J.; Wei, N.; Wang, D.; Lyu, H.; Shi, C.; Hu, S. The landscape of cognitive function in recovered COVID-19 patients. J. Psychiatr. Res. 2020, 129, 98–102. [Google Scholar] [CrossRef]
- Rass, V.; Beer, R.; Schiefecker, A.J.; Lindner, A.; Kofler, M.; Ianosi, B.A.; Mahlknecht, P.; Heim, B.; Peball, M.; Carbone, F.; et al. Neurological outcomes 1 year after COVID-19diagnosis: A prospective longitudinal cohort study. Eur. J. Neurol. 2022, 29, 1685–1696. [Google Scholar] [CrossRef]
- Søraas, A.; Bø, R.; Kalleberg, K.T.; Støer, N.C.; Ellingjord-Dale, M.; Landrø, N.I. Self-reported memory problems 8 months after COVID-19 infection. JAMA Netw. Open 2021, 4, e2118717. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.-H.; Wood, J.; O’Dea, M.R.; Dutton, S.; Shamardani, K.; Nwangwu, K.; et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell 2022, 185, 2452–2468.e16. [Google Scholar] [CrossRef] [PubMed]
- Charnley, M.; Islam, S.; Bindra, G.B.; Engwirda, J.; Ratcliffe, J.; Zhou, J.; Mezzenga, R.; Hulett, M.D.; Han, K.; Berryman, J.T.; et al. Neurotoxic amyloidogenic peptides in the proteome of SARS-CoV2: Potential implications for neurological symptoms in COVID-19. Nat. Commun. 2022, 13, 3387. [Google Scholar] [CrossRef] [PubMed]
- de Melo, G.D.; Lazarini, F.; Levallois, S.; Hautefort, C.; Michel, V.; Larrous, F.; Verillaud, B.; Aparicio, C.; Wagner, S.; Gheusi, G.; et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021, 13, eabf8396. [Google Scholar] [CrossRef] [PubMed]
- Sodagar, A.; Javed, R.; Tahir, H.; Razak, S.I.A.; Shakir, M.; Naeem, M.; Yusof, A.H.A.; Sagadevan, S.; Hazafa, A.; Uddin, J.; et al. Pathological Features and Neuroinflammatory Mechanisms of SARS-CoV-2 in the Brain and Potential Therapeutic Approaches. Biomolecules 2022, 12, 971. [Google Scholar] [CrossRef]
- Mohammadi, S.; Gouravani, M.; Salehi, M.A.; Harandi, H.; Moosaie, F.; Firouzabadi, F.D.; Yousem, D.M. Olfactory system measurements in COVID-19: A systematic review and meta-analysis. Neuroradiology 2023, 65, 25–39. [Google Scholar] [CrossRef]
- Ho, C.-Y.; Salimian, M.; Hegert, J.; O’Brien, J.; Choi, S.G.; Ames, H.; Morris, M.; Papadimitriou, J.C.; Mininni, J.; Niehaus, P.; et al. Postmortem Assessment of Olfactory Tissue Degeneration and Microvasculopathy in Patients with COVID-19. JAMA Neurol. 2022, 79, 544–553. [Google Scholar] [CrossRef]
- Kay, L.M. COVID-19 and olfactory dysfunction: A looming wave of dementia? J. Neurophysiol. 2022, 128, 436–444. [Google Scholar] [CrossRef]
- Ziuzia-Januszewska, L.; Januszewski, M. Pathogenesis of Olfactory Disorders in COVID-19. Brain Sci. 2022, 12, 449. [Google Scholar] [CrossRef]
- Tu, L.; Lv, X.; Fan, Z.; Zhang, M.; Wang, H.; Yu, X. Association of odor identification ability with amyloid-β and tau burden: A systematic review and meta-analysis. Front. Neurosci. 2020, 14, 586330. [Google Scholar] [CrossRef]
- Guedj, E.; Campion, J.Y.; Horowitz, T.; Barthelemy, F.; Cammilleri, S.; Ceccaldi, M. The impact of COVID-19 lockdown on brain metabolism. Hum. Brain Mapp. 2022, 43, 593–597. [Google Scholar] [CrossRef]
- Hugon, J.; Msika, E.F.; Queneau, M.; Farid, K.; Paquet, C. Long COVID: Cognitive complaints (brain fog) and dysfunction of the cingulate cortex. J. Neurol. 2022, 269, 44–46. [Google Scholar] [CrossRef]
- Plangár, I.; Zádori, D.; Klivényi, P.; Toldi, J.; Vécsei, L. Targeting the kynurenine pathway-related alterations in Alzheimer’s disease: A future therapeutic strategy. J. Alzheimers Dis. 2011, 24, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Jakabek, D.; Bracken, S.G.; Allen-Davidian, Y.; Heng, B.; Chow, S.; Dehhaghi, M.; Staats Pires, A.; Darley, D.R.; Byrne, A.; et al. The kynurenine pathway relates to post-acute COVID-19 objective cognitive impairment and PASC. Ann. Clin. Transl. Neurol. 2023, 10, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Deeks, S.G.; Mustapic, M.; Kapogiannis, D.; Henrich, T.J.; Lu, S.; Goldberg, S.A.; Hoh, R.; Chen, J.Y.; Martinez, E.O.; et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Ann. Neurol. 2022, 91, 772–781. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J. COVID-19 and Alzheimer’s disease: How one crisis worsens the other. Transl. Neurodegener. 2021, 10, 15. [Google Scholar] [CrossRef]
- Barrantes, F.J. Central Nervous System Targets and Routes for SARS-CoV-2: Current Views and New Hypotheses. ACS Chem. Neurosci. 2020, 11, 2793–2803. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Pogue, A.; Hill, J.M. SARS-CoV-2 Infectivity and Neurological Targets in the Brain. Cell Mol. Neurobiol. 2022, 42, 217–224. [Google Scholar] [CrossRef]
- Seaks, C.E.; Wilcock, D.M. Infectious hypothesis of Alzheimer disease. PLoS Pathog. 2020, 16, e1008596. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka-Drożak, E.; Drożak, P.; Mizerski, G.; Zaborowski, T.; Ślusarska, B.; Nowicki, G.; Drożak, M. Links between COVID-19 and Alzheimer’s Disease-What Do We Already Know? Int. J. Environ. Res. Public Health 2023, 20, 2146. [Google Scholar] [CrossRef]
- Lim, K.-H.; Yang, S.; Kim, S.H.; Joo, J.Y. Elevation of ACE2 as a SARS-CoV-2 entry receptor gene expression in Alzheimer’s disease. J. Infect. 2020, 81, e33–e34. [Google Scholar] [CrossRef]
- Ding, Q.; Shults, N.V.; Gychka, S.G.; Harris, B.T.; Suzuki, Y.J. Protein Expression of Angiotensin-Converting Enzyme 2 (ACE2) is Upregulated in Brains with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1687. [Google Scholar] [CrossRef]
- Louise, R.; Manon, L.; Vincent, E.; Cyntia, T.; Andréanne, L.; Philippe, B.; David, A.B.; Hébert, S.S.; Frédéric, C. Higher angiotensin-converting enzyme 2 (ACE2) levels in the brain of individuals with Alzheimer’s disease. Acta Neuropathol. Commun. 2023, 11, 159. [Google Scholar] [CrossRef]
- Cao, S.; Song, Z.; Rong, J.; Andrikopoulos, N.; Liang, X.; Wang, Y.; Peng, G.; Ding, F.; Ke, P.C. Spike Protein Fragments Promote Alzheimer’s Amyloidogenesis. ACS Appl. Mater. Interfaces 2023, 15, 40317–40329. [Google Scholar] [CrossRef] [PubMed]
- Motaghinejad, M.; Gholami, M. Possible Neurological and Mental Outcomes of COVID-19 Infection: A Hypothetical Role of ACE-2\Mas\BDNF Signaling Pathway. Int. J. Prev. Med. 2020, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Haque, M.E.; Balakrishnan, R.; Kim, I.S.; Choi, D.K. The Ageing Brain: Molecular and Cellular Basis of Neurodegeneration. Front. Cell Dev. Biol. 2021, 9, 683459. [Google Scholar] [CrossRef]
- Behfar, Q.; Ramirez Zuniga, A.; Martino-Adami, P.V. Aging, Senescence, and Dementia. J. Prev. Alzheimers Dis. 2022, 9, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.R.; Rani, L.; Subba, R.; Mondal, A.C. Cellular senescence in the aging brain: A promising target for neurodegenerative diseases. Mech. Ageing Dev. 2022, 204, 111675. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Tchkonia, T.; Niedernhofer, L.J.; Robbins, P.D.; Kirkland, J.L.; Lee, S. COVID-19 and cellular senescence. Nat. Rev. Immunol. 2023, 23, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Nehme, J.; Borghesan, M.; Mackedenski, S.; Bird, T.G.; Demaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell. 2020, 19, e13237. [Google Scholar] [CrossRef]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2021, 11, 571416. [Google Scholar] [CrossRef]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “inflame-aging”. Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Lynch, S.M.; Guo, G.; Gibson, D.S.; Bjourson, A.J.; Rai, T.S. Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease. Cells 2021, 10, 3367. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Tian, D.S. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Ulhaq, Z.S.; Soraya, G.V. Interleukin-6 as a potential biomarker of COVID-19 progression. Med. Mal. Infect. 2020, 50, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-J.; Koh, S.-H. Is Telomere Length Shortening a Risk Factor for Neurodegenerative Disorders? Dement. Neurocogn. Disord. 2022, 21, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vazquez, R.; Guío-Carrión, A.; Zapatero-Gaviria, A.; Martínez, P.; Blasco, M.A. Shorter telomere lengths in patients with severe COVID-19 disease. Aging 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults. JAMA 2020, 323, 2427–2429. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2023, 24, 1869. [Google Scholar] [CrossRef]
- Wang, H.; Shen, Y.; Chuang, H.; Chiu, C.; Ye, Y.; Zhao, L. Neuroinflammation in Alzheimer’s Disease: Microglia, Molecular Participants and Therapeutic Choices. Curr. Alzheimer Res. 2019, 16, 659–674. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Navarrete, L.P.; González, A.; González-Canacer, A.; Guzmán-Martínez, L.; Cortés, N. Inflammation: A Major Target for Compounds to Control Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1199–1213. [Google Scholar] [CrossRef]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Boubour, A.; Bruce, S.S.; Chong, A.M.; Claassen, J.; et al. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 2021, 144, 2696–2708. [Google Scholar] [CrossRef]
- Schwabenland, M.; Salie, H.; Tanevski, J.; Killmer, S.; Lago, M.S.; Schlaak, A.E.; Mayer, L.; Matschke, J.; Püschel, K.; Fitzek, A.; et al. Deep spatial profiling of human COVID-19 brains reveals neuroinflammation with distinct microanatomical microglia-T-cell interactions. Immunity 2021, 54, 1594–1610.e11. [Google Scholar] [CrossRef]
- Rahman, M.A.; Islam, K.; Rahman, S.; Alamin, M. Neurobiochemical Cross-Talk between COVID-19 and Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Feldman, E.L.; Stino, E.L. Neurological sequela and disruption of neuron-glia homeostasis in SARS-CoV-2 infection. Neurobiol. Dis. 2022, 68, 105715. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, E.A.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Aguado, J.; Pliego Zamora, A.; McMillan, C.L.D.; Liang, B.; et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psychiatry 2023, 28, 2878–2893. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. [Google Scholar] [CrossRef]
- Kozlov, E.M.; Ivanova, E.; Grechko, A.V.; Wu, W.K.; Starodubova, A.V.; Orekhov, A.N. Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection. Diseases 2021, 9, 17. [Google Scholar] [CrossRef]
- Choe, K.; Park, H.Y.; Ikram, M.; Lee, H.J.; Park, T.J.; Ullah, R.; Kim, M.O. Systematic Review of the Common Pathophysiological Mechanisms in COVID-19 and Neurodegeneration: The Role of Bioactive Compounds and Natural Antioxidants. Cells 2022, 11, 1298. [Google Scholar] [CrossRef]
- Golonka, R.M.; Saha, P.; Yeoh, B.S.; Chattopadhyay, S.; Gewirtz, A.T.; Joe, B.; Vijay-Kumar, M. Harnessing innate immunity to eliminate SARS-CoV-2 and ameliorate COVID-19 disease. Physiol. Genom. 2020, 52, 217–221. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaud, C.; Nuss, P.; Benoliel, J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol.-Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Villaume, W.A. Marginal BH4 deficiencies, iNOS, and self-perpetuating oxidative stress in post-acute sequelae of COVID-19. Med. Hypotheses 2022, 163, 110842. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Expression and function of the cholinergic system in immune cells. Front. Immunol. 2017, 8, 1085. [Google Scholar] [CrossRef] [PubMed]
- Kopańska, M.; Batoryna, M.; Bartman, P.; Szczygielski, J.; Banaś-Ząbczyk, A. Disorders of the Cholinergic System in COVID-19 Era—A Review of the Latest Research. Int. J. Mol. Sci. 2022, 23, 672. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Copeland, R.L.; Aschner, M. Nicotine and the nicotinic cholinergic system in COVID-19. FEBS J. 2020, 287, 3656–3663. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Farsalinos, K.; Eliopoulos, E.; Leonidas, D.; Papadopoulos, G.; Tzartos, S.; Poulas, K. Nicotinic cholinergic system and COVID-19: In silico identification of an interaction between SARS-CoV-2 and nicotinic receptors with potential therapeutic targeting implications. Int. J. Mol. Sci. 2020, 21, 5807. [Google Scholar] [CrossRef] [PubMed]
- Lagoumintzis, G.; Chasapis, C.T.; Alexandris, N.; Kouretas, D.; Tzartos, S.; Eliopoulos, E.; Farsalinos, K.; Poulas, K. Nicotinic cholinergic system and COVID-19: In silico identification of interactions between α7 nicotinic acetylcholine receptor and the cryptic epitopes of SARS-Co-V and SARS-CoV-2 Spike glycoproteins. Food Chem. Toxicol. 2021, 149, 112009. [Google Scholar] [CrossRef]
- Alexandris, N.; Lagoumintzis, G.; Chasapis, C.T.; Leonidas, D.D.; Papadopoulos, G.E.; Tzartos, S.J.; Tsatsakis, A.; Eliopoulos, E.; Poulas, K.; Farsalinos, K. Nicotinic cholinergic system and COVID-19: In silico evaluation of nicotinic acetylcholine receptor agonists as potential therapeutic interventions. Toxicol. Rep. 2020, 8, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Leitzke, M. Is the post-COVID-19 syndrome a severe impairment of acetylcholine-orchestrated neuromodulation that responds to nicotine administration? Bioelectron. Med. 2023, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Hauser, P.S.; Ryan, R.O. Impact of Apolipoprotein E on Alzheimer’s Disease. Curr. Alzheimer Res. 2013, 10, 809–817. [Google Scholar] [CrossRef]
- Del Ser, T.; Fernandez-Blazquez, M.A.; Valenti, M.; Zea-Sevilla, M.A.; Frades, B.; Alfayate, E.; Saiz, L.; Calero, O.; García-López, F.J.; Rábano, A.; et al. Residence, clinical features, and genetic risk factors associated with symptoms of COVID-19 in a cohort of older people in Madrid. Gerontology 2021, 67, 281–289. [Google Scholar] [CrossRef]
- Al-Jaf, S.M.A.; Niranji, S.S.; Ali, H.N.; Mohammed, O.A. Association of apolipoprotein e polymorphism with SARS-CoV-2 infection. Infect. Genet. Evol. 2021, 95, 105043. [Google Scholar] [CrossRef]
- Kurki, S.N.; Kantonen, J.; Kaivola, K.; Hokkanen, L.; Mayranpaa, M.I.; Puttonen, H.; Gen, F.; Martola, J.; Pöyhönen, M.; Kero, M.; et al. APOE epsilon4 associates with increased risk of severe COVID-19, cerebral microhaemorrhages and post-COVID mental fatigue: A finnish biobank, autopsy and clinical study. Acta Neuropathol. Commun. 2021, 9, 199. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Dlouha, L.; Dusek, L.; Majek, O.; Adamkova, V. Apolipoprotein E4 allele in subjects with COVID-19. Gerontology 2021, 67, 320–322. [Google Scholar] [CrossRef]
- Abondio, P.; Sazzini, M.; Garagnani, P.; Boattini, A.; Monti, D.; Franceschi, C.; Luiselli, D.; Giuliani, C. The genetic variability of APOE in different human populations and its implications for longevity. Genes 2019, 10, 222. [Google Scholar] [CrossRef]
- Dhangadamajhi, G.; Mishra, S.; Mukherjee, P. Association of ApoE isoforms with COVID-19 outcomes: A world-wide epidemiological study. Hum. Cell 2021, 34, 1932–1933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shao, L.; Lin, Z.; Long, Q.-X.; Yuan, H.; Cai, L.; Jiang, G.; Guo, X.; Yang, R.; Zhang, Z.; et al. APOE interacts with ACE2 inhibiting SARS-CoV-2 cellular entry and inflammation in COVID-19 patients. Signal Transduct. Target Ther. 2022, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Moore, J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, M.; Garcia, G., Jr.; Tian, E.; Cui, Q.; Chen, X.; Sun, G.; Wang, J.; Arumugaswami, V.; Shi, Y. ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 2021, 28, 331–342.e5. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Jablonski, S.M.; Jablonski, J.; Hobson, R.; Valente, S.; Reddy, C.B.; Hansen, S.B. The role of high cholesterol in age-related COVID-19 lethality. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xiong, N.; Schiller, M.R.; Li, J.; Chen, X.; Lin, Z. Severe COVID-19 in Alzheimer’s disease: APOE4′s fault again? Alzheimers Res. Ther. 2021, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Kushwah, P.S.; Agrawal, N.; Pathak, S. APOE4: A Culprit for the Vulnerability of COVID-19 in Alzheimer’s Patients. Curr. Neurovasc. Res. 2023, 20, 162–169. [Google Scholar] [CrossRef]
- Ramachandran, A.K.; Das, S.; Shenoy, G.G.; Mudgal, J.; Joseph, A. Relation between apolipoprotein e in alzheimer’s disease and Sars-CoV-2 and their treatment strategy: A review. CNS Neurol. Disord. Drug Targets 2022. [Google Scholar] [CrossRef]
- Chen, F.; Chen, Y.; Ke, Q.; Wang, Y.; Gong, Z.; Chen, X.; Cai, Y.; Li, S.; Sun, Y.; Peng, X.; et al. ApoE4 associated with severe COVID-19 outcomes via downregulation of ACE2 and imbalanced RAS pathway. J. Transl. Med. 2023, 21, 103. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, M.B.; Sanusi, K.O.; Ugusman, A.; Mohamed, W.; Kamal, H.; Ibrahim, N.H.; Khoo, C.S.; Kumar, J. Alzheimer’s Disease: An Update and Insights Into Pathophysiology. Front. Aging Neurosci. 2022, 14, 742408. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40.e7. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Ziff, O.J.; Ashton, N.J.; Mehta, P.R.; Brown, R.; Athauda, D.; Heaney, J.; Heslegrave, A.J.; Lessa Benedet, A.; Blennow, K.; Checkley, A.M.; et al. Amyloid processing in COVID-19-associated neurological syndromes. J. Neurochem. 2022, 161, 146–157. [Google Scholar] [CrossRef]
- Chiricosta, L.; Gugliandolo, A.; Mazzon, E. SARS-CoV-2 Exacerbates Beta-Amyloid Neurotoxicity, Inflammation and Oxidative Stress in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2021, 22, 13603. [Google Scholar] [CrossRef]
- Hsu, J.T.-A.; Tien, C.F.; Yu, G.-Y.; Shen, S.; Lee, Y.-H.; Hsu, P.-C.; Wang, Y.; Chao, P.-K.; Tsay, H.-J.; Shie, F.-S. The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2021, 22, 8226. [Google Scholar] [CrossRef]
- Priemer, D.S.; Rhodes, C.H.; Karlovich, E.; Perl, D.P.; Goldman, J.E. 5β Deposits in the Neocortex of Adult and Infant Hypoxic Brains, Including in Cases of COVID-19. J. Neuropathol. Exp. Neurol. 2022, 81, 988–995. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, D.F.; Zou, Q.C.; Xie, X.; Xu, L.; Feng, X.L.; Li, X.; Han, J.B.; Yu, D.; Deng, Z.H.; et al. SARS-CoV-2 Spike protein S2 subunit modulates γ-secretase and enhances amyloid-β production in COVID-19 neuropathy. Cell Discov. 2022, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, C.; Schreiber, C.S.; Hartke, A.S.; Denden, I.; Stanelle-Bertram, S.; Beck, S.; Kouassi, N.M.; Beythien, G.; Becker, K.; Schreiner, T.; et al. Microgliosis and neuronal proteinopathy in brain persist beyond viral clearance in SARS-CoV-2 hamster model. EBioMedicine 2022, 79, 103999. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.J.; Ye, L.B.; Timani, K.A.; Zeng, Y.C.; She, Y.L.; Ye, L.; Wu, Z.H. Activation of NF-kappa B by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim. Biophys. Sin. 2005, 37, 607–612. [Google Scholar] [CrossRef] [PubMed]
- De Diego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeno, J.M.; Fernandez-Delgado, R.; Fett, C.; Castano-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Reiken, S.; Sittenfeld, L.; Dridi, H.; Liu, Y.; Liu, X.; Marks, A.R. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimers Dement. 2022, 18, 955–965. [Google Scholar] [CrossRef]
- Virhammar, J.; Nääs, A.; Fällmar, D.; Cunningham, J.L.; Klang, A.; Ashton, N.J.; Jackmann, S.; Westman, G.; Frithiof, R.; Blennow, K.; et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and diseaseseverity. Eur. J. Neurol. 2021, 28, 3324–3331. [Google Scholar] [CrossRef]
- Needham, E.; Ren, A.L.; Digby, R.J.; Norton, E.J.; Ebrahimi, S.; Outtrim, J.G.; Chatfield, D.A.; Manktelow, A.E.; Leibowitz, M.M.; Newcombe, V.F.J.; et al. Brain injury in COVID-19 is associated with dysregulated innate and adaptive immune responses. Brain 2022, 145, 4097–4107. [Google Scholar] [CrossRef]
- Ramani, A.; Müller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Müller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef]
- Di Primio, C.; Quaranta, P.; Mignanelli, M.; Siano, G.; Bimbati, M.; Scarlatti, A.; Piazza, C.R.; Spezia, P.G.; Perrera, P.; Basolo, F.; et al. Severe acute respiratory syndrome coronavirus 2 infection leads to Tau pathological signature in neurons. PNAS Nexus 2023, 2, pgad282. [Google Scholar] [CrossRef]
- Eberle, R.J.; Coronado, M.A.; Gering, I.; Sommerhage, S.; Korostov, K.; Stefanski, A.; Stühler, K.; Kraemer-Schulien, V.; Blömeke, L.; Bannach, O.; et al. Tau protein aggregation associated with SARS-CoV-2 main protease. PLoS ONE 2023, 18, e0288138. [Google Scholar] [CrossRef]
- Paidas, M.J.; Cosio, D.S.; Ali, S.; Kenyon, N.S.; Jayakumar, A.R. Long-Term Sequelae of COVID-19 in Experimental Mice. Mol. Neurobiol. 2022, 59, 5970–5986. [Google Scholar] [CrossRef] [PubMed]
- Sivagurunathan, N.; Calivarathan, L. SARS-CoV-2 Infection to Premature Neuronal Aging and Neurodegenerative Diseases: Is there any Connection with Hypoxia? CNS Neurol. Disord. Drug Targets 2023, 23, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Appelberg, S.; Gupta, S.; Akusjärvi, S.S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, A.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Liu, W.; Li, X.; Zhao, P.; Shereen, M.A.; Zhu, C.; Huang, S.; Liu, S.; Yu, X.; Yue, M.; et al. HIF-1α promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct. Target Ther. 2021, 6, 308. [Google Scholar] [CrossRef]
- Jana, S.; Heaven, M.R.; Stauft, C.B.; Wang, T.T.; Williams, M.C.; D’Agnillo, F.; Alayash, A.I. 1 HIF-1α-Dependent Metabolic Reprogramming, Oxidative Stress, and Bioenergetic Dysfunction in SARS-CoV-2-Infected Hamsters. Int. J. Mol. Sci. 2022, 24, 558. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of AmyloidPrecursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Quing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef]
- Kerridge, C.; Kozlova, D.I.; Nalivaeva, N.N.; Turner, A.J. Hypoxia Affects Neprilysin Expression through Caspase Activation and an APP Intracellular Domain-dependent Mechanism. Front. Neurosci. 2015, 9, 426. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Huang, Z.-T.; Yuan, M.-H.; Jing, F.; Cai, R.-L.; Zou, Q.; Pu, Y.-S.; Wang, S.Y.; Chen, F.; Yi, W.-M.; et al. Role of Hypoxia Inducible Factor-1α in Alzheimer’s Disease2. J. Alzheimers Dis. 2021, 80, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.H.; Priemer, D.S.; Karlovich, E.; Perl, D.P.; Goldman, J. Β-Amyloid Deposits in Young COVID Patients. Lancet 2023. [Google Scholar] [CrossRef]
- Dondaine, T.; Ruthmann, F.; Vuotto, F.; Carton, L.; Gelé, P.; Faure, K.; Deplanque, D.; Bordet, R. Long-term cognitive impairments following COVID-19: A possible impact of hypoxia. J. Neurol. 2022, 269, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Shapira, R.; Solomon, B.; Efrati, S.; Frenkel, D.; Ashery, U. Hyperbaric oxygen therapy ameliorates pathophysiology of 3xTg-AD mouse model by attenuating neuroinflammation. Neurobiol. Aging 2018, 62, 105–119. [Google Scholar] [CrossRef]
- Harch, P.G.; Fogarty, E.F. Hyperbaric oxygen therapy for Alzheimer’s dementia with positron emission tomography imaging: A case report. Med. Gas Res. 2019, 8, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.; Devason, A.S.; Umana, I.C.; Cox, T.O.; Dohnalová, L.; Litichevskiy, L.; Perla, J.; Lundgren, P.; Etwebi, Z.; Izzo, L.T.; et al. Serotonin reduction in post-acute sequelae of viral infection. Cell 2023, 186, 4851–4867.e20. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Wilson, R.S.; Yu, L.; Wang, T.; Schneider, J.A.; Honer, W.G.; Bennett, D.A. Selective serotonin reuptake inhibitor use, age-related neuropathology and cognition in late-life. Psychiatry Res. 2023, 328, 115471. [Google Scholar] [CrossRef]
- Gottfries, C.G.; Bartfai, T.; Carlsson, A.; Eckernas, S.; Svennerholm, L. Multiple biochemical deficits in both gray and white matter of Alzheimer brains Prog. Neuropsychopharmacol. Biol. Psychiatry 1986, 10, 405–413. [Google Scholar] [CrossRef]
- Dong, J.; Chen, R.; Zhao, H.; Zhu, Y. COVID-19 and ocular complications: A review of ocular manifestations, diagnostic tools, and prevention strategies. Adv. Ophthalmol. Pract. Res. 2023, 3, 33–38. [Google Scholar] [CrossRef]
- Szczęśniak, M.; Brydak-Godowska, J. SARS-CoV-2 and the eyes: A review of the literature on transmission, detection, and ocular manifestations. Med. Sci. Monit. 2021, 27, e931863-1–e931863-10. [Google Scholar] [CrossRef]
- de Figueiredo, C.S.; Raony, Í.; Giestal-de-Araujo, E. SARS-CoV-2targeting the retina: Host-virus interaction and possible mechanisms of viral tropism. Ocul. Immunol. Inflamm. 2020, 28, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, E.; Kawasaki, A.; Eandi, C.M. Pathogenesis of Vascular Retinal Manifestations in COVID-19 Patients: A Review. Biomedicines 2022, 10, 2710. [Google Scholar] [CrossRef] [PubMed]
- Barnett, B.P.; Wahlin, K.; Krawczyk, M.; Spencer, D.; Welsbie, D.; Afshari, N.; Chao, D. Potential of Ocular Transmission of SARS-CoV-2: A Review. Vision 2020, 4, 40. [Google Scholar] [CrossRef]
- Coroneo, M.T. The eye as the discrete but defensible portal of coronavirus infection. Ocul. Surf. 2021, 19, 176–182. [Google Scholar] [CrossRef]
- deS Senanayake, P.; Drazba, J.; Shadrach, K.; Milsted, A.; Rungger-Brandle, E.; Nishiyama, K.; Miura, S.-I.; Karnik, S.; Sears, J.E.; Hollyfield, J.G. Angiotensin II and its receptor subtypes in the human retina. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3301–3311. [Google Scholar] [CrossRef]
- Choudhary, R.; Kapoor, M.S.; Singh, A.; Bodakhe, S.H. Therapeutic targets of renin-angiotensin system in ocular disorders. J. Curr. Ophthalmol. 2016, 29, 7–16. [Google Scholar] [CrossRef]
- Qing, H.; Li, Z.; Yang, Z.; Shi, M.; Huang, Z.; Song, J.; Song, Z. The possibility of COVID-19 transmission from eye to nose. Acta Ophthalmol. 2020, 98, e388. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, A.; Rosani, U.; Brun, P. Ocular Surface Expression of SARS-CoV-2 Receptors. Ocul. Immunol. Inflamm. 2020, 28, 735–738. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, Z.; Castiglione, G.M.; Soiberman, U.S.; Eberhart, C.G.; Duh, E.J. ACE2 and TMPRSS2 are expressed on the human ocular surface, suggesting susceptibility to SARS-CoV-2 infection. Ocul. Surf. 2020, 18, 537–544. [Google Scholar] [CrossRef]
- Casagrande, M.; Fitzek, A.; Spitzer, M.S.; Püschel, K.; Glatzel, M.; Krasemann, S.; Nörz, D.; Lütgehetmann, M.; Pfefferle, S.; Schultheiss, M. Presence of SARS-CoV-2 RNA in the Cornea of Viremic Patients with COVID-19. JAMA Ophthalmol. 2021, 139, 383–388. [Google Scholar] [CrossRef]
- Casagrande, M.; Fitzek, A.; Püschel, K.; Aleshcheva, G.; Schultheiss, H.-P.; Berneking, L.; Spitze, M.S.; Schultheiss, M. Detection of SARS-CoV-2 in Human Retinal Biopsies of Deceased COVID-19 Patients. Ocul. Immunol. Inflamm. 2020, 28, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, Y.; He, T.; Wei, R.; Shen, Y.; Qi, T.; Han, T.; Song, Z.; Zhu, Z.; Ma, X.; et al. Detection of SARS-CoV-2 in the ocular surface in different phases of COVID-19 patients in Shanghai, China. Ann. Transl. Med. 2021, 9, 100. [Google Scholar] [CrossRef]
- Aiello, F.; Gallo Afflitto, G.; Mancino, R.; Li, J.O.; Cesareo, M.; Giannini, C.; Nucci, C. Coronavirus disease 2019 (SARS-CoV-2) and colonization of ocular tissues and secretions: A systematic review. Eye 2020, 34, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Sawant, O.B.; Singh, S.; Wright, R.E., 3rd; Jones, K.M.; Titus, M.S.; Dennis, E.; Hicks, E.; Majmudar, P.A.; Kumar, A.; Mian, S.I. Prevalence of SARS-CoV-2 in human post-mortem ocular tissues. Ocul. Surf. 2021, 19, 322–329. [Google Scholar] [CrossRef]
- Yeo, S.; Kim, H.; Lee, J.; Yi, J.; Chung, Y.R. Retinal vascular occlusions in COVID-19 infection and vaccination: A literature review. Graefes Arch. Clin. Exp. Ophthalmol. 2023, 261, 1793–1808. [Google Scholar] [CrossRef]
- Abdul-Salam State, S.E.; Sfredel, V.; Mocanu, C.L.; Albu, C.V.; Bălășoiu, A.T. Optic neuropathies post-COVID-19-review. Rom. J. Ophthalmol. 2022, 66, 289–298. [Google Scholar] [CrossRef]
- Eissa, M.; Abdelrazek, N.A.; Saady, M. COVID-19 and its relation to the human eye: Transmission, infection, and ocular manifestations. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 261, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Dourandeesh, M. Update on overview of ocular manifestations of COVID-19. Front. Med. 2022, 9, 877023. [Google Scholar] [CrossRef] [PubMed]
- Sutandi, N.; Lee, F. Vitreoretinal abnormalities in corona virus disease 2019 patients: What we know so far Taiwan. J. Ophthalmol. 2021, 11, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, N.; Sharifi, H.; Bazrafshan, A.; Noori, A.; Karamouzian, M.; Sharifi, A. Ocular Manifestations of COVID-19: A Systematic Review and Meta-analysis. J. Ophthalmic Vis. Res. 2021, 16, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kudchadkar, U.S.; Shirodkar, R.; Usgaonkar, U.P.S.; Naik, A. Unilateral inferior altitudinal visual field defect related to COVID-19. Indian J. Ophthalmol. 2021, 69, 989–991. [Google Scholar] [CrossRef]
- Selvaraj, V.; Sacchetti, D.; Finn, A.; Dapaah-Afriyie, K. Acute vision loss in a patient with COVID-19. R. I. Med. J. 2020, 103, 37–38. [Google Scholar]
- Kaya, Y.; Kara, S.; Akinci, C.; Kocaman, A.S. Transient cortical blindness in COVID-19 pneumonia; a PRES-like syndrome: Case report. J. Neurol. Sci. 2020, 413, 116858. [Google Scholar] [CrossRef]
- Coco-Martín, M.B.; Leal-Vega, L.; Alcoceba-Herrero, I.; Molina-Martín, A.; de-Fez, D.; Luque, M.J.; Dueñas-Gutiérrez, C.; Arenillas-Lara, J.F.; Piñero, D.P. Visual perception alterations in COVID-19: A preliminary study. Int. J. Ophthalmol. 2023, 16, 1–9. [Google Scholar] [CrossRef]
- Zhu, R.; Yu, Z.Y.; Lin Han, L. Insights on the possibility of SARS-CoV-2 transmission through the eyes. Int. J. Ophthalmol. 2022, 15, 1857–1863. [Google Scholar] [CrossRef]
- Sen, S.; Kannan, N.B.; Kumar, J.; Rajan, R.P.; Kumar, K.; Baliga, G.; Reddy, H.; Upadhyay, A.; Ramasamy, K. Retinal manifestations in patients with SARS-CoV-2 infection and pathogenetic implications: A systematic review. Int. Ophthalmol. 2022, 42, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Romaus-Sanjurjo, D.; Regueiro, U.; López-López, M.; Vázquez-Vázquez, L.; Ouro, A.; Lema, I.; Sobrino, T. Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 2486. [Google Scholar] [CrossRef] [PubMed]
- Hart, N.J.; Koronyo, Y.; Black, K.L.; Koronyo-Hamaoui, M. Ocular indicators of Alzheimer’s: Exploring disease in the retina. Acta Neuropathol. 2016, 132, 767–787. [Google Scholar] [CrossRef]
- den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain: From eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef]
- Chiquita, S.; Rodrigues-Neves, A.C.; Baptista, F.I.; Carecho, R.; Moreira, P.I.; Castelo-Branco, M.; Ambrósio, A.F. The Retina as a Window or Mirror of the Brain Changes Detected in Alzheimer’s Disease: Critical Aspects to Unravel. Mol. Neurobiol. 2019, 56, 5416–5435. [Google Scholar] [CrossRef]
- Latina, V.; Giacovazzo, G.; Cordella, F.; Balzamino, B.O.; Micera, A.; Varano, M.; Marchetti, C.; Malerba, F.; Florio, R.; Ercole, B.B.; et al. Systemic delivery of a specific antibody targeting the pathological N-terminal truncated tau peptide reduces retinal degeneration in a mouse model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 38. [Google Scholar] [CrossRef]
- Tippett, W.J.; Black, S.E. Regional Cerebral Blood Flow Correlates of Visuospatial Tasks in Alzheimer’s Disease. J. Int. Neuropsychol. Soc. 2008, 14, 1034–1045. [Google Scholar] [CrossRef]
- Rizzo, M.; Anderson, S.W.; Dawson, J.; Nawrot, M. Vision and Cognition in Alzheimer’s Disease. Neuropsychologia 2000, 38, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, H.; Siéroff, É. Visual Perceptual Disorders in Alzheimer’s Disease. Geriatr. Psychol. Neuropsychiatr. Vieil. 2019, 17, 307–316. [Google Scholar] [CrossRef]
- Kaeser, P.F.; Ghika, J.; Borruat, F.X. Visual Signs and Symptoms in Patients with the Visual Variant of Alzheimer Disease. BMC Ophthalmol. 2015, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.; Kergoat, H. Oculo-visual changes and clinical considerations affecting older patients with dementia. Ophthalmic Physiol. Opt. 2015, 35, 352–376. [Google Scholar] [CrossRef]
- Javaid, F.Z.; Brenton, J.; Guo, L.; Cordeiro, M.F. Visual and Ocular Manifestations of Alzheimer’s Disease and Their Use as Biomarkers for Diagnosis and Progression. Front. Neurol. 2016, 7, 55. [Google Scholar] [CrossRef]
- Kusne, Y.; Wolf, A.B.; Townley, K.; Conway, M.; Peyman, G.A. Visual system manifestations of Alzheimer’s disease. Acta Ophthalmol. 2017, 95, e668–e676. [Google Scholar] [CrossRef]
- McKee, A.C.; Au, R.; Cabral, H.J.; Kowall, N.W.; Seshadri, S.; Kubilus, C.A.; Drake, J.; Wolf, P.A. Visual association pathology in preclinical Alzheimer disease. Comparative Study. J. Neuropathol. Exp. Neurol. 2006, 65, 621–630. [Google Scholar] [CrossRef]
- Shah, T.M.; Gupta, S.M.; Chatterjee, P.; Campbell, M.; Martins, R.N. Beta-amyloid sequelae in the eye: A critical review on its diagnostic significance and clinical relevance in Alzheimer’s disease. Mol. Psychiatry 2017, 22, 353–363. [Google Scholar] [CrossRef]
- Mirzaei, N.; Shi, H.; Oviatt, M.; Doustar, J.; Rentsendorj, A.; Fuchs, D.-T.; Sheyn, J.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Alzheimer’s Retinopathy: Seeing Disease in the Eyes. Front. Neurosci. 2020, 14, 921. [Google Scholar] [CrossRef]
- Furman, S.; Green, K.; Lane, T.E. COVID-19 and the impact on Alzheimer’s disease pathology. J. Neurochem. 2023; ahead of print. [Google Scholar] [CrossRef]
- Bauer, L.; Laksono, B.M.; de Vrij, F.M.S.; Kushner, S.A.; Harschnitz, O.; van Riel, D. The neuroinvasiveness, neurotropism, and neurovirulence of SARS-CoV-2. Trends Neurosci. 2022, 45, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Veleri, S. Neurotropism of SARS-CoV-2 and neurological diseases of the central nervous system in COVID-19 patients. Exp. Brain Res. 2022, 240, 9–25. [Google Scholar] [CrossRef]
- Jensen, M.P.; Le Quesne, J.; Officer-Jones, L.; Teodosio, A.; Thaventhiran, J.; Ficken, C.; Goddard, M.; Smith, C.; Menon, D.; Allinson, K.S.J. Neuropathological findings in two patients with fatal COVID-19. Neuropathol. Appl. Neurobiol. 2021, 47, 17–25. [Google Scholar] [CrossRef]
- Agrawal, U.; Bedston, S.; McCowan, C.; Oke, J.; Patterson, L.; Robertson, C.; Akbari, A.; Azcoaga-Lorenzo, A.; Bradley, D.T.; Fagbamigbe, A.F.; et al. Severe COVID-19 outcomes after full vaccination of primary schedule and initial boosters: Pooled analysis of national prospective cohort studies of 30 million individuals in England, Northern Ireland, Scotland, and Wales. Lancet 2022, 400, 1305–1320. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, E.; Huang, J. Cognitive Aspects of COVID-19. Curr. Neurol. Neurosci. Rep. 2023, 23, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.L.; Richard, D.; Khachik, A.; Mistry, M.; Singh, G.; Mostaghni, N.; Yazdanmehr, S. Ophthalmic Presentations and Manifestations of COVID-19: A Systematic Review of Global Observations. Cureus 2023, 15, e40695. [Google Scholar] [CrossRef]
- Zhao, Y.; Lukiw, W.J. SARS-CoV-2 Neuroinvasion, Inflammatory Neurodegeneration and Alzheimer’s Disease. Front. Cell. Neurosci. 2022, 16, 937961. [Google Scholar] [CrossRef]
- Asghari, F.; Asghary, A.; Zolbanin, N.M.; Faraji, F.; Jafari, R. Immunosenescence and Inflammaging in COVID-19. Viral Immunol. 2023; ahead of print. [Google Scholar] [CrossRef]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G. COVID-19 Commission of the Accademia Nazionale dei Lincei Long COVID: Where we stand and challenges ahead. Cell. Death Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhang, L.Y.; Tan, Y.Y.; Chen, S.D. Links between COVID-19 and Parkinson’s disease/Alzheimer’s disease: Reciprocal impacts, medical care strategies and underlying mechanisms. Transl. Neurodegener. 2023, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Vandersteen, C.; Plonka, A.; Manera, V.; Sawchuk, K.; Lafontaine, C.; Galery, K.; Rouaud, O.; Bengaied, N.; Launay, C.; Guérin, O.; et al. Alzheimer’s early detection in post-acute COVID-19 syndrome: A systematic review and expert consensus on preclinical assessments. Front. Aging Neurosci. 2023, 15, 1206123. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amadoro, G.; Latina, V.; Stigliano, E.; Micera, A. COVID-19 and Alzheimer’s Disease Share Common Neurological and Ophthalmological Manifestations: A Bidirectional Risk in the Post-Pandemic Future. Cells 2023, 12, 2601. https://doi.org/10.3390/cells12222601
Amadoro G, Latina V, Stigliano E, Micera A. COVID-19 and Alzheimer’s Disease Share Common Neurological and Ophthalmological Manifestations: A Bidirectional Risk in the Post-Pandemic Future. Cells. 2023; 12(22):2601. https://doi.org/10.3390/cells12222601
Chicago/Turabian StyleAmadoro, Giuseppina, Valentina Latina, Egidio Stigliano, and Alessandra Micera. 2023. "COVID-19 and Alzheimer’s Disease Share Common Neurological and Ophthalmological Manifestations: A Bidirectional Risk in the Post-Pandemic Future" Cells 12, no. 22: 2601. https://doi.org/10.3390/cells12222601