
Age-Related Dysfunction in Proteostasis and Cellular Quality Control in the Development of Sarcopenia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Skeletal Muscle Anabolism
2.1. Anabolic Signaling
2.2. Skeletal Muscle Anabolic Resistance
2.2.1. Amino Acid Induced MPS
2.2.2. MPS and Sarcopenic Obesity
2.2.3. Gastrointestinal–Muscle Axis
2.2.4. Microbiome and Inflammation in Sarcopenia
2.3. Aging and Skeletal Muscle Recovery from Disuse
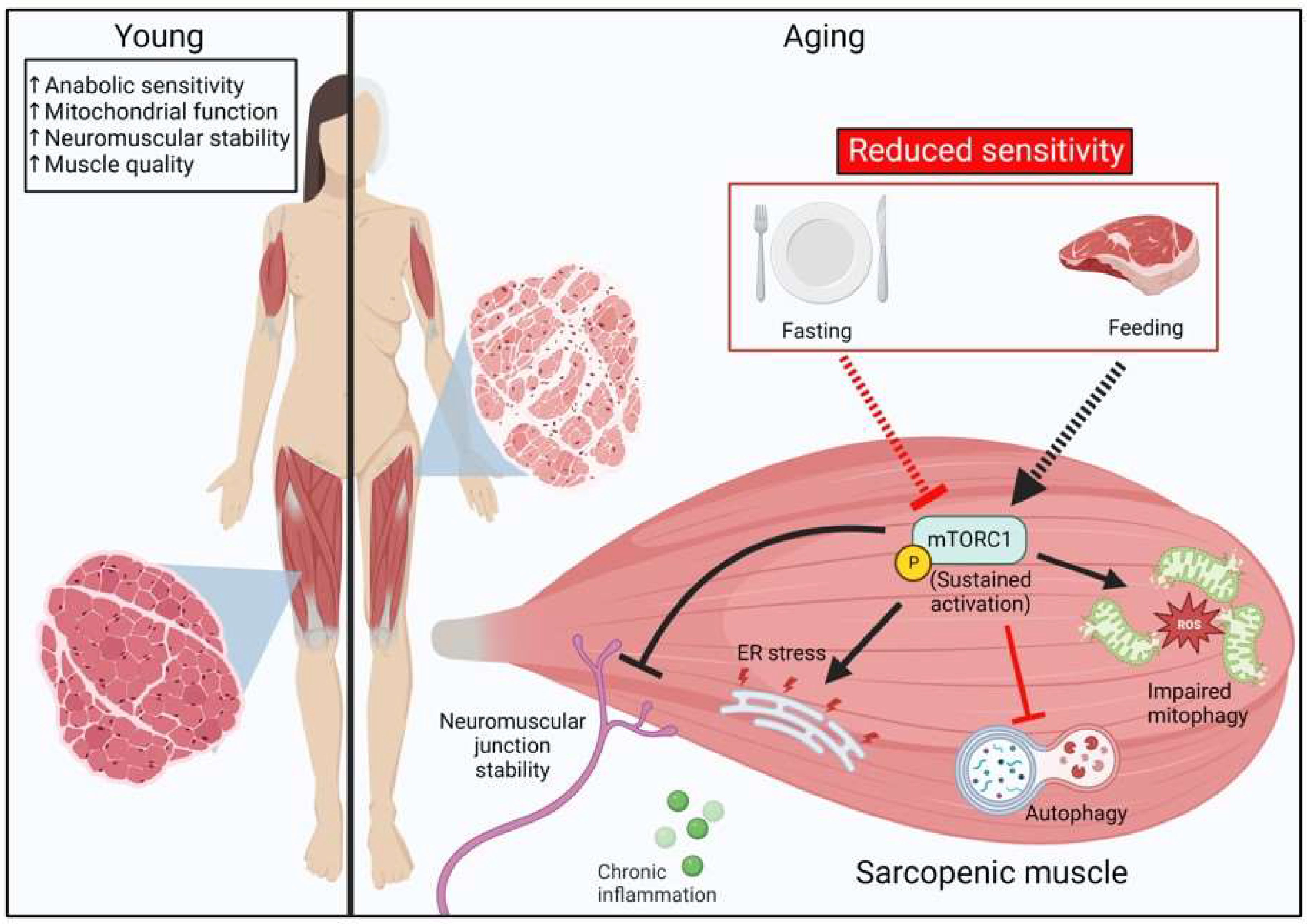
2.4. Sarcopenia and mTORC1 Signaling
3. Skeletal Muscle Catabolism
3.1. The Ubiquitin Proteasome System
3.1.1. Ubiquitin Proteasome Degradation of Cellular Proteins
3.1.2. The Ubiquitin Proteasome System in Sarcopenia
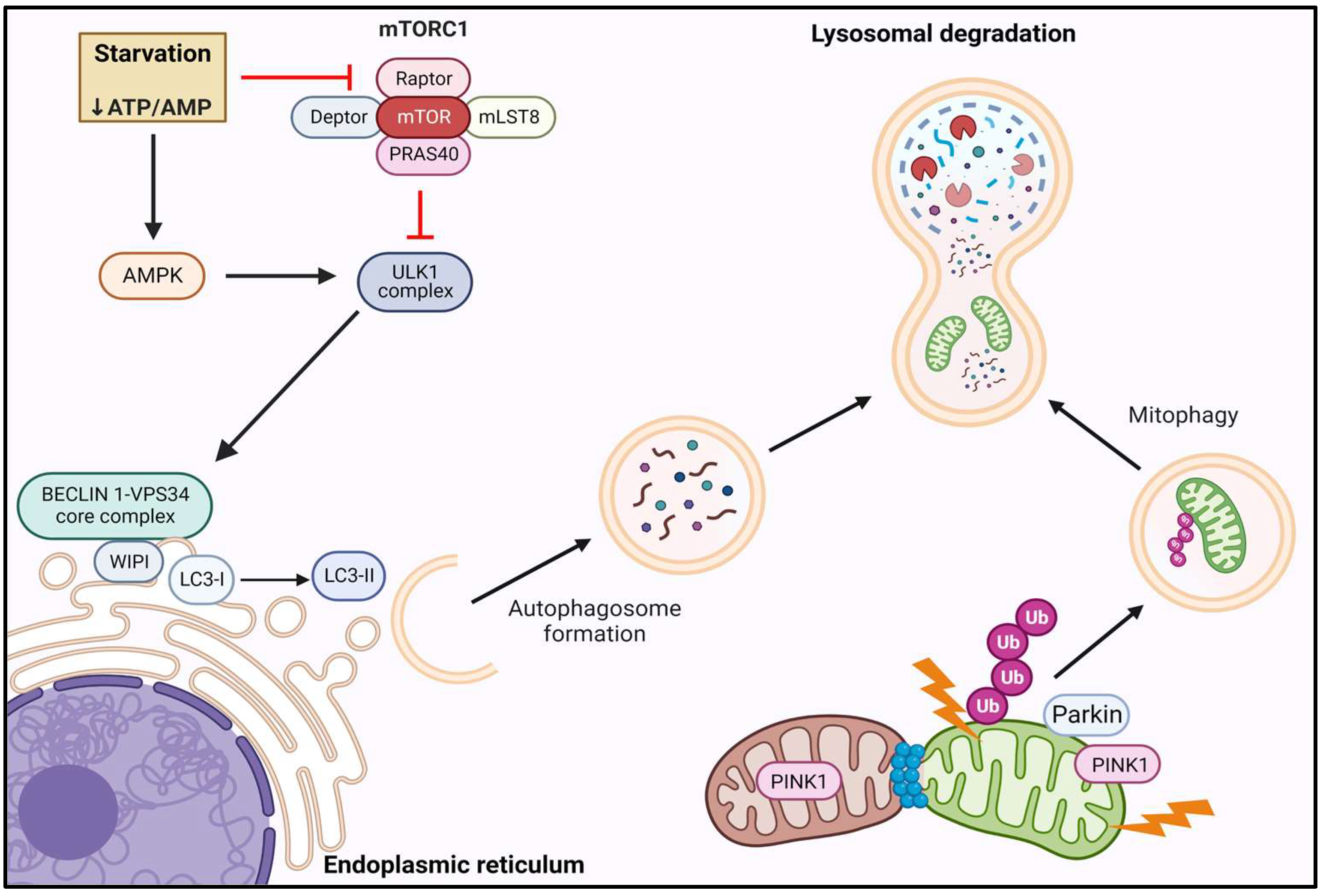
3.2. Autophagy and Mitophagy
3.2.1. Autophagy and Mitophagy Signaling
3.2.2. Defective Autophagy Impairs Skeletal Muscle Function and Mass
3.2.3. mTORC1 and Autophagy
3.2.4. Autophagy and Mitochondrial Dysfunction
3.2.5. Ca2+ Dysregulation in Sarcopenia
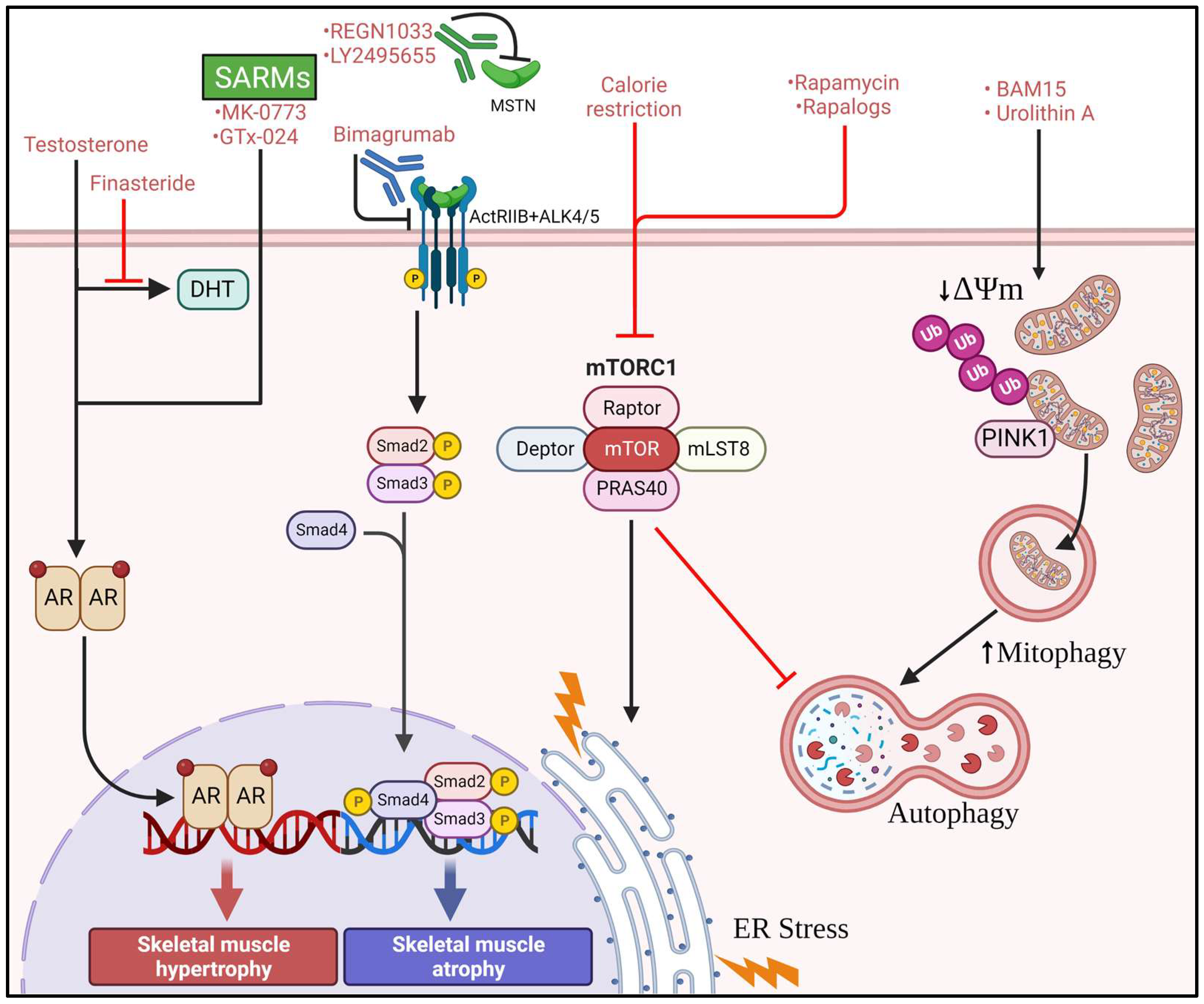
4. Therapeutics for the Treatment of Sarcopenia
4.1. Branched-Chain Amino Acid Supplementation
4.2. Rapamycin, Rapalogs, and Calorie Restriction
4.3. Mitochondrial Uncouplers
4.4. Androgens and SARMs
4.5. Myostatin Inhibitors
4.6. Present Challenges and Future Direction
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Janssen, I.; Heymsfield, S.B.; Wang, Z.; Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. 2000, 89, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Xia, J.; Zhang, X.; Gathirua-Mwangi, W.G.; Guo, J.; Li, Y.; McKenzie, S.; Song, Y. Associations of muscle mass and strength with all-cause mortality among US older adults. Med. Sci. Sport. Exerc. 2018, 50, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Szulc, P.; Munoz, F.; Marchand, F.; Chapurlat, R.; Delmas, P.D. Rapid loss of appendicular skeletal muscle mass is associated with higher all-cause mortality in older men: The prospective MINOS study. Am. J. Clin. Nutr. 2010, 91, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, F.; Liperoti, R.; Russo, A.; Giovannini, S.; Tosato, M.; Capoluongo, E.; Bernabei, R.; Onder, G. Sarcopenia as a risk factor for falls in elderly individuals: Results from the ilSIRENTE study. Clin. Nutr. 2012, 31, 652–658. [Google Scholar] [CrossRef]
- Koo, B.K.; Roh, E.; Yang, Y.S.; Moon, M.K. Difference between old and young adults in contribution of β-cell function and sarcopenia in developing diabetes mellitus. J. Diabetes Investig. 2016, 7, 233–240. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [Green Version]
- Koster, A.; Ding, J.; Stenholm, S.; Caserotti, P.; Houston, D.K.; Nicklas, B.J.; You, T.; Lee, J.S.; Visser, M.; Newman, A.B. Does the amount of fat mass predict age-related loss of lean mass, muscle strength, and muscle quality in older adults? J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2011, 66, 888–895. [Google Scholar] [CrossRef]
- Delmonico, M.J.; Harris, T.B.; Visser, M.; Park, S.W.; Conroy, M.B.; Velasquez-Mieyer, P.; Boudreau, R.; Manini, T.M.; Nevitt, M.; Newman, A.B.; et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009, 90, 1579–1585. [Google Scholar]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef] [Green Version]
- McCastlain, K.; Howell, C.R.; Welsh, C.E.; Wang, Z.; Wilson, C.L.; Mulder, H.L.; Easton, J.; Mertens, A.C.; Zhang, J.; Yasui, Y. The association of mitochondrial copy number with sarcopenia in adult survivors of childhood cancer. JNCI J. Natl. Cancer Inst. 2021, 113, 1570–1580. [Google Scholar] [CrossRef]
- Ooi, P.H.; Thompson-Hodgetts, S.; Pritchard-Wiart, L.; Gilmour, S.M.; Mager, D.R. Pediatric sarcopenia: A paradigm in the overall definition of malnutrition in children? J. Parenter. Enter. Nutr. 2020, 44, 407–418. [Google Scholar] [CrossRef]
- Suzuki, D.; Kobayashi, R.; Sano, H.; Hori, D.; Kobayashi, K. Sarcopenia after induction therapy in childhood acute lymphoblastic leukemia: Its clinical significance. Int. J. Hematol. 2018, 107, 486–489. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Orav, J.; Kanis, J.A.; Rizzoli, R.; Schlögl, M.; Staehelin, H.; Willett, W.C.; Dawson-Hughes, B. Comparative performance of current definitions of sarcopenia against the prospective incidence of falls among community-dwelling seniors age 65 and older. Osteoporos. Int. 2015, 26, 2793–2802. [Google Scholar] [CrossRef]
- Schaap, L.A.; Van Schoor, N.M.; Lips, P.; Visser, M. Associations of sarcopenia definitions, and their components, with the incidence of recurrent falling and fractures: The longitudinal aging study Amsterdam. J. Gerontol. Ser. A 2018, 73, 1199–1204. [Google Scholar] [CrossRef]
- Morley, J.E.; Abbatecola, A.M.; Argiles, J.M.; Baracos, V.; Bauer, J.; Bhasin, S.; Cederholm, T.; Coats, A.J.S.; Cummings, S.R.; Evans, W.J. Sarcopenia with limited mobility: An international consensus. J. Am. Med. Dir. Assoc. 2011, 12, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Lauretani, F.; Russo, C.R.; Bandinelli, S.; Bartali, B.; Cavazzini, C.; Di Iorio, A.; Corsi, A.M.; Rantanen, T.; Guralnik, J.M.; Ferrucci, L. Age-associated changes in skeletal muscles and their effect on mobility: An operational diagnosis of sarcopenia. J. Appl. Physiol. 2003, 95, 1851–1860. [Google Scholar] [CrossRef] [Green Version]
- Janssen, I.; Heymsfield, S.B.; Ross, R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J. Am. Geriatr. Soc. 2002, 50, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, L.; Cyrino, E.S.; Antunes, M.; Santos, D.A.; Sardinha, L.B. Sarcopenia and physical independence in older adults: The independent and synergic role of muscle mass and muscle function. J. Cachexia Sarcopenia Muscle 2017, 8, 245–250. [Google Scholar] [CrossRef]
- Goates, S.; Du, K.; Arensberg, M.; Gaillard, T.; Guralnik, J.; Pereira, S.L. Economic impact of hospitalizations in US adults with sarcopenia. J. Frailty Aging 2019, 8, 93–99. [Google Scholar] [CrossRef]
- Janssen, I.; Shepard, D.S.; Katzmarzyk, P.T.; Roubenoff, R. The healthcare costs of sarcopenia in the United States. J. Am. Geriatr. Soc. 2004, 52, 80–85. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Ageing and Health; World Health Organization: Geneva, Switzerland, 2018.
- Idoate, F.; Cadore, E.L.; Casas-Herrero, A.; Zambom-Ferraresi, F.; Marcellán, T.; de Gordoa, A.R.; Rodriguez-Mañas, L.; Bastarrika, G.; Marques, M.C.; Martínez-Velilla, N. Adipose tissue compartments, muscle mass, muscle fat infiltration, and coronary calcium in institutionalized frail nonagenarians. Eur. Radiol. 2015, 25, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Landi, F.; Calvani, R.; Cesari, M.; Tosato, M.; Martone, A.M.; Bernabei, R.; Onder, G.; Marzetti, E. Sarcopenia as the biological substrate of physical frailty. Clin. Geriatr. Med. 2015, 31, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Roby, M.A.; Eason, M.K.; Harris, M.B. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp. Gerontol. 2010, 45, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, T.; Hirata, M.; Uchiyama, Y. Qualitative alteration of peripheral motor system begins prior to appearance of typical sarcopenia syndrome in middle-aged rats. Front. Aging Neurosci. 2014, 6, 296. [Google Scholar] [CrossRef]
- Spendiff, S.; Vuda, M.; Gouspillou, G.; Aare, S.; Perez, A.; Morais, J.A.; Jagoe, R.T.; Filion, M.E.; Glicksman, R.; Kapchinsky, S. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol. 2016, 594, 7361–7379. [Google Scholar] [CrossRef] [Green Version]
- Visser, M.; Pahor, M.; Taaffe, D.R.; Goodpaster, B.H.; Simonsick, E.M.; Newman, A.B.; Nevitt, M.; Harris, T.B. Relationship of interleukin-6 and tumor necrosis factor-α with muscle mass and muscle strength in elderly men and women: The Health ABC Study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2002, 57, M326–M332. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Yu, K.; Shyh-Chang, N.; Li, G.X.; Jiang, L.J.; Yu, S.L.; Xu, L.Y.; Liu, R.J.; Guo, Z.J.; Xie, H.Y. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J. Cachexia Sarcopenia Muscle 2019, 10, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The role of inflammation in age-related sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef] [Green Version]
- Pistilli, E.E.; Jackson, J.R.; Alway, S.E. Death receptor-associated pro-apoptotic signaling in aged skeletal muscle. Apoptosis 2006, 11, 2115–2126. [Google Scholar] [CrossRef]
- Pistilli, E.E.; Siu, P.M.; Alway, S.E. Molecular regulation of apoptosis in fast plantaris muscles of aged rats. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 245–255. [Google Scholar] [CrossRef]
- Alway, S.E.; Siu, P.M. Nuclear apoptosis contributes to sarcopenia. Exerc. Sport Sci. Rev. 2008, 36, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Bian, A.; Ma, Y.; Zhou, X.; Guo, Y.; Wang, W.; Zhang, Y.; Wang, X. Association between sarcopenia and levels of growth hormone and insulin-like growth factor-1 in the elderly. BMC Musculoskelet. Disord. 2020, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yuki, A.; Otsuka, R.; Kozakai, R.; Kitamura, I.; Okura, T.; Ando, F.; Shimokata, H. Relationship between low free testosterone levels and loss of muscle mass. Sci. Rep. 2013, 3, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.H.; Kim, J.H.; Lee, J.H.; Hong, A.R.; Shin, C.S.; Cho, N.H. Dehydroepiandrosterone sulfate and free testosterone but not estradiol are related to muscle strength and bone microarchitecture in older adults. Calcif. Tissue Int. 2019, 105, 285–293. [Google Scholar] [CrossRef]
- Gianoudis, J.; Bailey, C.; Daly, R. Associations between sedentary behaviour and body composition, muscle function and sarcopenia in community-dwelling older adults. Osteoporos. Int. 2015, 26, 571–579. [Google Scholar] [CrossRef]
- Distefano, G.; Standley, R.A.; Zhang, X.; Carnero, E.A.; Yi, F.; Cornnell, H.H.; Coen, P.M. Physical activity unveils the relationship between mitochondrial energetics, muscle quality, and physical function in older adults. J. Cachexia Sarcopenia Muscle 2018, 9, 279–294. [Google Scholar] [CrossRef] [Green Version]
- Landi, F.; Liperoti, R.; Russo, A.; Giovannini, S.; Tosato, M.; Barillaro, C.; Capoluongo, E.; Bernabei, R.; Onder, G. Association of anorexia with sarcopenia in a community-dwelling elderly population: Results from the ilSIRENTE study. Eur. J. Nutr. 2013, 52, 1261–1268. [Google Scholar] [CrossRef]
- Morley, J.E. Anorexia of ageing: A key component in the pathogenesis of both sarcopenia and cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Coelho, H.J., Jr.; Calvani, R.; Azzolino, D.; Picca, A.; Tosato, M.; Landi, F.; Cesari, M.; Marzetti, E. Protein Intake and Sarcopenia in Older Adults: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 8718. [Google Scholar] [CrossRef]
- Drummond, M.J.; Fry, C.S.; Glynn, E.L.; Dreyer, H.C.; Dhanani, S.; Timmerman, K.L.; Volpi, E.; Rasmussen, B.B. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J. Physiol. 2009, 587, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.J.; Börsch, A.; Lin, S.; Thürkauf, M.; Weihrauch, M.; Reinhard, J.R.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S. The neuromuscular junction is a focal point of mTORC1 signaling in sarcopenia. Nat. Commun. 2020, 11, 4510. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.-C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [CrossRef]
- Browne, G.J.; Proud, C.G. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell. Biol. 2004, 24, 2986–2997. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef]
- Mosoni, L.; Valluy, M.; Serrurier, B.; Prugnaud, J.; Obled, C.; Guezennec, C.-Y.; Mirand, P.P. Altered response of protein synthesis to nutritional state and endurance training in old rats. Am. J. Physiol.-Endocrinol. Metab. 1995, 268, E328–E335. [Google Scholar] [CrossRef]
- Volpi, E.; Mittendorfer, B.; Rasmussen, B.B.; Wolfe, R.R. The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J. Clin. Endocrinol. Metab. 2000, 85, 4481–4490. [Google Scholar] [CrossRef] [Green Version]
- Cuthbertson, D.; Smith, K.; Babraj, J.; Leese, G.; Waddell, T.; Atherton, P.; Wackerhage, H.; Taylor, P.M.; Rennie, M.J. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. 2005, 19, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Volpi, E.; Sheffield-Moore, M.; Rasmussen, B.B.; Wolfe, R.R. Basal muscle amino acid kinetics and protein synthesis in healthy young and older men. JAMA 2001, 286, 1206–1212. [Google Scholar] [CrossRef]
- Moore, D.R.; Churchward-Venne, T.A.; Witard, O.; Breen, L.; Burd, N.A.; Tipton, K.D.; Phillips, S.M. Protein ingestion to stimulate myofibrillar protein synthesis requires greater relative protein intakes in healthy older versus younger men. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 70, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. Am. J. Clin. Nutr. 2005, 82, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Szwiega, S.; Pencharz, P.B.; Rafii, M.; Lebarron, M.; Chang, J.; Ball, R.O.; Kong, D.; Xu, L.; Elango, R.; Courtney-Martin, G. Dietary leucine requirement of older men and women is higher than current recommendations. Am. J. Clin. Nutr. 2021, 113, 410–419. [Google Scholar] [CrossRef]
- Evenson, K.R.; Buchner, D.M.; Morland, K.B. Objective measurement of physical activity and sedentary behavior among US adults aged 60 years or older. Prev. Chronic Dis. 2012, 9, E26. [Google Scholar] [CrossRef] [PubMed]
- Breen, L.; Stokes, K.A.; Churchward-Venne, T.A.; Moore, D.R.; Baker, S.K.; Smith, K.; Atherton, P.J.; Phillips, S.M. Two weeks of reduced activity decreases leg lean mass and induces “anabolic resistance” of myofibrillar protein synthesis in healthy elderly. J. Clin. Endocrinol. Metab. 2013, 98, 2604–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardevet, D.; Sornet, C.; Balage, M.; Grizard, J. Stimulation of in vitro rat muscle protein synthesis by leucine decreases with age. J. Nutr. 2000, 130, 2630–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. Am. J. Physiol.-Endocrinol. Metab. 2006, 291, E381–E387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, T.; Brightwell, C.R.; Deer, R.R.; Graber, T.G.; Galvan, E.; Fry, C.S.; Volpi, E.; Rasmussen, B.B. Muscle protein anabolic resistance to essential amino acids does not occur in healthy older adults before or after resistance exercise training. J. Nutr. 2018, 148, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.N.; Park, M.S.; Lim, K.I.; Choi, H.Y.; Yang, S.J.; Yoo, H.J.; Kang, H.J.; Song, W.; Choi, H.; Baik, S.H. Relationships between sarcopenic obesity and insulin resistance, inflammation, and vitamin D status: The K orean S arcopenic O besity S tudy. Clin. Endocrinol. 2013, 78, 525–532. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Vieira-Lara, M.A.; Dommerholt, M.B.; Zhang, W.; Blankestijn, M.; Wolters, J.C.; Abegaz, F.; Gerding, A.; van der Veen, Y.T.; Thomas, R.; van Os, R.P. Age-related susceptibility to insulin resistance arises from a combination of CPT1B decline and lipid overload. BMC Biol. 2021, 19, 154. [Google Scholar] [CrossRef]
- Messa, G.A.; Piasecki, M.; Hurst, J.; Hill, C.; Tallis, J.; Degens, H. The impact of a high-fat diet in mice is dependent on duration and age, and differs between muscles. J. Exp. Biol. 2020, 223, jeb217117. [Google Scholar] [CrossRef] [Green Version]
- Stephens, F.B.; Chee, C.; Wall, B.T.; Murton, A.J.; Shannon, C.E.; Van Loon, L.J.; Tsintzas, K. Lipid-induced insulin resistance is associated with an impaired skeletal muscle protein synthetic response to amino acid ingestion in healthy young men. Diabetes 2015, 64, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Beals, J.W.; Sukiennik, R.A.; Nallabelli, J.; Emmons, R.S.; Van Vliet, S.; Young, J.R.; Ulanov, A.V.; Li, Z.; Paluska, S.A.; De Lisio, M. Anabolic sensitivity of postprandial muscle protein synthesis to the ingestion of a protein-dense food is reduced in overweight and obese young adults. Am. J. Clin. Nutr. 2016, 104, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Murton, A.J.; Marimuthu, K.; Mallinson, J.E.; Selby, A.L.; Smith, K.; Rennie, M.J.; Greenhaff, P.L. Obesity appears to be associated with altered muscle protein synthetic and breakdown responses to increased nutrient delivery in older men, but not reduced muscle mass or contractile function. Diabetes 2015, 64, 3160–3171. [Google Scholar] [CrossRef] [Green Version]
- Smeuninx, B.; Mckendry, J.; Wilson, D.; Martin, U.; Breen, L. Age-related anabolic resistance of myofibrillar protein synthesis is exacerbated in obese inactive individuals. J. Clin. Endocrinol. Metab. 2017, 102, 3535–3545. [Google Scholar] [CrossRef] [Green Version]
- Purves-Smith, F.M.; Solbak, N.M.; Rowan, S.L.; Hepple, R.T. Severe atrophy of slow myofibers in aging muscle is concealed by myosin heavy chain co-expression. Exp. Gerontol. 2012, 47, 913–918. [Google Scholar] [CrossRef]
- Siddharth, J.; Chakrabarti, A.; Pannerec, A.; Karaz, S.; Morin-Rivron, D.; Masoodi, M.; Feige, J.N.; Parkinson, S.J. Aging and sarcopenia associate with specific interactions between gut microbes, serum biomarkers and host physiology in rats. Aging 2017, 9, 1698–1720. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Yang, X.; Zheng, L.; Wang, Z.; Wu, L.; Jiang, J.; Yang, T.; Ma, L.; Fu, Z. Lactobacillus and Bifidobacterium improves physiological function and cognitive ability in aged mice by the regulation of gut microbiota. Mol. Nutr. Food Res. 2019, 63, 1900603. [Google Scholar] [CrossRef]
- Lahiri, S.; Kim, H.; Garcia-Perez, I.; Reza, M.M.; Martin, K.A.; Kundu, P.; Cox, L.M.; Selkrig, J.; Posma, J.M.; Zhang, H. The gut microbiota influences skeletal muscle mass and function in mice. Sci. Transl. Med. 2019, 11, eaan5662. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-C.; Chen, Y.-H.; Chuang, H.-L.; Chiu, C.-C.; Huang, C.-C. Investigation of the effects of microbiota on exercise physiological adaption, performance, and energy utilization using a gnotobiotic animal model. Front. Microbiol. 2019, 10, 1906. [Google Scholar] [CrossRef] [Green Version]
- Stanley, D.; Denman, S.E.; Hughes, R.J.; Geier, M.S.; Crowley, T.M.; Chen, H.; Haring, V.R.; Moore, R.J. Intestinal microbiota associated with differential feed conversion efficiency in chickens. Appl. Microbiol. Biotechnol. 2012, 96, 1361–1369. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 2017, 21, 455–466.e4. [Google Scholar] [CrossRef] [Green Version]
- Sovran, B.; Hugenholtz, F.; Elderman, M.; Van Beek, A.A.; Graversen, K.; Huijskes, M.; Boekschoten, M.V.; Savelkoul, H.F.; De Vos, P.; Dekker, J. Age-associated impairment of the mucus barrier function is associated with profound changes in microbiota and immunity. Sci. Rep. 2019, 9, 1437. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lertwattanarak, R.; Garduño, J.d.J.; Galeana, J.J.; Li, J.; Zamarripa, F.; Lancaster, J.L.; Mohan, S.; Hussey, S.; Musi, N. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 70, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’connor, E.M.; Cusack, S.; Harris, H.; Coakley, M.; Lakshminarayanan, B.; O’sullivan, O. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Kilroe, S.P.; Fulford, J.; Jackman, S.R.; Van Loon, L.J.; Wall, B.T. Temporal muscle-specific disuse atrophy during one week of leg immobilization. Med. Sci. Sport. Exerc. 2020, 52, 944–954. [Google Scholar] [CrossRef]
- Lang, F.; Aravamudhan, S.; Nolte, H.; Tuerk, C.; Hölper, S.; Müller, S.; Günther, S.; Blaauw, B.; Braun, T.; Krüger, M. Dynamic changes in the mouse skeletal muscle proteome during denervation-induced atrophy. Dis. Model. Mech. 2017, 10, 881–896. [Google Scholar]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565, 309–323. [Google Scholar] [CrossRef]
- Chen, K.D.; Alway, S.E. Clenbuterol reduces soleus muscle fatigue during disuse in aged rats. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2001, 24, 211–222. [Google Scholar] [CrossRef]
- Myers, M.J.; Shepherd, D.L.; Durr, A.J.; Stanton, D.S.; Mohamed, J.S.; Hollander, J.M.; Alway, S.E. The role of SIRT1 in skeletal muscle function and repair of older mice. J. Cachexia Sarcopenia Muscle 2019, 10, 929–949. [Google Scholar] [CrossRef] [Green Version]
- Suetta, C.; Frandsen, U.; Mackey, A.L.; Jensen, L.; Hvid, L.G.; Bayer, M.; Petersson, S.J.; Schrøder, H.D.; Andersen, J.L.; Aagaard, P. Ageing is associated with diminished muscle re-growth and myogenic precursor cell expansion early after immobility-induced atrophy in human skeletal muscle. J. Physiol. 2013, 591, 3789–3804. [Google Scholar] [CrossRef]
- Ahmadi, M.; Karlsen, A.; Mehling, J.; Soendenbroe, C.; Mackey, A.L.; Hyldahl, R.D. Aging is associated with an altered macrophage response during human skeletal muscle regeneration. Exp. Gerontol. 2022, 169, 111974. [Google Scholar] [CrossRef]
- Phillips, S.M.; McGlory, C. CrossTalk proposal: The dominant mechanism causing disuse muscle atrophy is decreased protein synthesis. J. Physiol. 2014, 592, 5341. [Google Scholar] [CrossRef]
- Alway, S.E.; Bennett, B.T.; Wilson, J.C.; Sperringer, J.; Mohamed, J.S.; Edens, N.K.; Pereira, S.L. Green tea extract attenuates muscle loss and improves muscle function during disuse, but fails to improve muscle recovery following unloading in aged rats. J. Appl. Physiol. 2015, 118, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Siu, P.M.; Pistilli, E.E.; Alway, S.E. Age-dependent increase in oxidative stress in gastrocnemius muscle with unloading. J. Appl. Physiol. 2008, 105, 1695–1705. [Google Scholar] [CrossRef] [Green Version]
- Siu, P.M.; Pistilli, E.E.; Murlasits, Z.; Alway, S.E. Hindlimb unloading increases muscle content of cytosolic but not nuclear Id2 and p53 proteins in young adult and aged rats. J. Appl. Physiol. 2006, 100, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, P.M.; Pistilli, E.E.; Butler, D.C.; Alway, S.E. Aging influences cellular and molecular responses of apoptosis to skeletal muscle unloading. Am. J. Physiol.-Cell Physiol. 2005, 288, C338–C349. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Pistilli, E.E.; Alway, S.E. Apoptotic responses to hindlimb suspension in gastrocnemius muscles from young adult and aged rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2005, 289, R1015–R1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.R.S.; Mohamed, J.S.; Myers, M.J.; Brooks, M.J.; Alway, S.E. Effects of hindlimb suspension and reloading on gastrocnemius and soleus muscle mass and function in geriatric mice. Exp. Gerontol. 2019, 115, 19–31. [Google Scholar] [CrossRef]
- Bennett, B.T.; Mohamed, J.S.; Alway, S.E. Effects of resveratrol on the recovery of muscle mass following disuse in the plantaris muscle of aged rats. PLoS ONE 2013, 8, e83518. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Jackson, J.R.; Wang, Y.; Edens, N.; Pereira, S.L.; Alway, S.E. β-Hydroxy-β-methylbutyrate reduces myonuclear apoptosis during recovery from hind limb suspension-induced muscle fiber atrophy in aged rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R701–R715. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Trevino, M.B.; Wang, M.; Gardell, S.J.; Ayala, J.E.; Han, X.; Kelly, D.P.; Goodpaster, B.H.; Vega, R.B.; Coen, P.M. Impaired mitochondrial energetics characterize poor early recovery of muscle mass following hind limb unloading in old mice. J. Gerontol. Ser. A 2018, 73, 1313–1322. [Google Scholar] [CrossRef] [Green Version]
- Kirby, T.J.; Lee, J.D.; England, J.H.; Chaillou, T.; Esser, K.A.; McCarthy, J.J. Blunted hypertrophic response in aged skeletal muscle is associated with decreased ribosome biogenesis. J. Appl. Physiol. 2015, 119, 321–327. [Google Scholar] [CrossRef]
- Trevino, M.B.; Zhang, X.; Standley, R.A.; Wang, M.; Han, X.; Reis, F.C.; Periasamy, M.; Yu, G.; Kelly, D.P.; Goodpaster, B.H. Loss of mitochondrial energetics is associated with poor recovery of muscle function but not mass following disuse atrophy. Am. J. Physiol.-Endocrinol. Metab. 2019, 317, E899–E910. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; West, D.W.; Marcotte, G.; Marshall, A.G.; De Sousa, L.G.; Baar, K.; Bodine, S.C. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 2016, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; West, D.W.; Marshall, A.G.; Marcotte, G.R.; Baar, K.; Bodine, S.C. Muscle-specific and age-related changes in protein synthesis and protein degradation in response to hindlimb unloading in rats. J. Appl. Physiol. 2017, 122, 1336–1350. [Google Scholar] [CrossRef] [Green Version]
- Waterlow, J. Protein turnover with special reference to man. Q. J. Exp. Physiol. Transl. Integr. 1984, 69, 409–438. [Google Scholar] [CrossRef]
- Rolfe, D.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Peng, Y.; Feng, Z.; Shi, W.; Qu, L.; Li, Y.; Liu, J.; Long, J. Reloading functionally ameliorates disuse-induced muscle atrophy by reversing mitochondrial dysfunction, and similar benefits are gained by administering a combination of mitochondrial nutrients. Free Radic. Biol. Med. 2014, 69, 116–128. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Brooks, M.J.; Hajira, A.; Mohamed, J.S.; Alway, S.E. Voluntary wheel running increases satellite cell abundance and improves recovery from disuse in gastrocnemius muscles from mice. J. Appl. Physiol. 2018, 124, 1616–1628. [Google Scholar] [CrossRef]
- Hwee, D.T.; Bodine, S.C. Age-related deficit in load-induced skeletal muscle growth. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2009, 64, 618–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetta, C.; Hvid, L.G.; Justesen, L.; Christensen, U.; Neergaard, K.; Simonsen, L.; Ortenblad, N.; Magnusson, S.P.; Kjaer, M.; Aagaard, P. Effects of aging on human skeletal muscle after immobilization and retraining. J. Appl. Physiol. 2009, 107, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Tanner, R.E.; Brunker, L.B.; Agergaard, J.; Barrows, K.M.; Briggs, R.A.; Kwon, O.S.; Young, L.M.; Hopkins, P.N.; Volpi, E.; Marcus, R.L. Age-related differences in lean mass, protein synthesis and skeletal muscle markers of proteolysis after bed rest and exercise rehabilitation. J. Physiol. 2015, 593, 4259–4273. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Barberi, L.; Bijlsma, A.; Blaauw, B.; Dyar, K.; Milan, G.; Mammucari, C.; Meskers, C.; Pallafacchina, G.; Paoli, A. Signalling pathways regulating muscle mass in ageing skeletal muscle. The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef] [PubMed]
- White, Z.; White, R.B.; McMahon, C.; Grounds, M.D.; Shavlakadze, T. High mTORC1 signaling is maintained, while protein degradation pathways are perturbed in old murine skeletal muscles in the fasted state. Int. J. Biochem. Cell Biol. 2016, 78, 10–21. [Google Scholar] [CrossRef]
- Joseph, G.A.; Wang, S.X.; Jacobs, C.E.; Zhou, W.; Kimble, G.C.; Tse, H.W.; Eash, J.K.; Shavlakadze, T.; Glass, D.J. Partial inhibition of mTORC1 in aged rats counteracts the decline in muscle mass and reverses molecular signaling associated with sarcopenia. Mol. Cell. Biol. 2019, 39, e00141-19. [Google Scholar] [CrossRef] [Green Version]
- Markofski, M.M.; Dickinson, J.M.; Drummond, M.J.; Fry, C.S.; Fujita, S.; Gundermann, D.M.; Glynn, E.L.; Jennings, K.; Paddon-Jones, D.; Reidy, P.T. Effect of age on basal muscle protein synthesis and mTORC1 signaling in a large cohort of young and older men and women. Exp. Gerontol. 2015, 65, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Baar, E.L.; Carbajal, K.A.; Ong, I.M.; Lamming, D.W. Sex-and tissue-specific changes in mTOR signaling with age in C57 BL/6J mice. Aging Cell 2016, 15, 155–166. [Google Scholar] [CrossRef]
- Blaauw, B.; Canato, M.; Agatea, L.; Toniolo, L.; Mammucari, C.; Masiero, E.; Abraham, R.; Sandri, M.; Schiaffino, S.; Reggiani, C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009, 23, 3896–3905. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S.; Handschin, C.; Tintignac, L.A.; Hall, M.N.; Rüegg, M.A. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet. Muscle 2013, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.; Ji, X. mTORC1 underlies age-related muscle fiber damage and loss by inducing oxidative stress and catabolism. Aging Cell 2019, 18, e12943. [Google Scholar] [CrossRef] [Green Version]
- Guridi, M.; Tintignac, L.A.; Lin, S.; Kupr, B.; Castets, P.; Rüegg, M.A. Activation of mTORC1 in skeletal muscle regulates whole-body metabolism through FGF21. Sci. Signal. 2015, 8, ra113. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Ham, D.J.; Börsch, A.; Chojnowska, K.; Lin, S.; Leuchtmann, A.B.; Ham, A.S.; Thürkauf, M.; Delezie, J.; Furrer, R.; Burri, D. Distinct and additive effects of calorie restriction and rapamycin in aging skeletal muscle. Nat. Commun. 2022, 13, 2025. [Google Scholar] [CrossRef]
- Alway, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria initiate and regulate sarcopenia. Exerc. Sport. Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Triolo, M.; Oliveira, A.N.; Kumari, R.; Hood, D.A. The influence of age, sex, and exercise on autophagy, mitophagy, and lysosome biogenesis in skeletal muscle. Skelet. Muscle 2022, 12, 13. [Google Scholar] [CrossRef]
- Kang, C.; Yeo, D.; Ji, L. Muscle immobilization activates mitophagy and disrupts mitochondrial dynamics in mice. Acta Physiol. 2016, 218, 188–197. [Google Scholar] [CrossRef]
- Bard, J.A.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018, 87, 697. [Google Scholar] [CrossRef]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Hughes, D.C.; Baehr, L.M.; Waddell, D.S.; Sharples, A.P.; Bodine, S.C. Ubiquitin Ligases in Longevity and Aging Skeletal Muscle. Int. J. Mol. Sci. 2022, 23, 7602. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.T.; Bhardwaj, G.; Penniman, C.M.; Krumpoch, M.T.; Suarez Beltran, P.A.; Klaus, K.; Poro, K.; Li, M.; Pan, H.; Dreyfuss, J.M. FoxO transcription factors are critical regulators of diabetes-related muscle atrophy. Diabetes 2019, 68, 556–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, K.A.; Chen, Y.-W.; Thompson, P.D.; Hoffman, E.P.; Clarkson, P.M. Forty-eight hours of unloading and 24 h of reloading lead to changes in global gene expression patterns related to ubiquitination and oxidative stress in humans. J. Appl. Physiol. 2010, 109, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.Z.; Haroun, S.; Leblanc, É.; Trensz, F.; Guindi, C.; Amrani, A.; Grenier, G. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet. Disord. 2011, 12, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.-M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [Green Version]
- Brocca, L.; Toniolo, L.; Reggiani, C.; Bottinelli, R.; Sandri, M.; Pellegrino, M.A. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 2017, 595, 1143–1158. [Google Scholar] [CrossRef] [Green Version]
- Jagoe, R.T.; Lecker, S.H.; Gomes, M.; Goldberg, A.L. Patterns of gene expression in atrophying skeletal muscles: Response to food deprivation. FASEB J. 2002, 16, 1697–1712. [Google Scholar] [CrossRef] [Green Version]
- Lagirand-Cantaloube, J.; Cornille, K.; Csibi, A.; Batonnet-Pichon, S.; Leibovitch, M.P.; Leibovitch, S.A. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 2009, 4, e4973. [Google Scholar] [CrossRef] [Green Version]
- Tintignac, L.A.; Lagirand, J.; Batonnet, S.; Sirri, V.; Leibovitch, M.P.; Leibovitch, S.A. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 2005, 280, 2847–2856. [Google Scholar] [CrossRef] [Green Version]
- Jogo, M.; Shiraishi, S.; Tamura, T.-A. Identification of MAFbx as a myogenin-engaged F-box protein in SCF ubiquitin ligase. FEBS Lett. 2009, 583, 2715–2719. [Google Scholar] [CrossRef]
- Csibi, A.; Leibovitch, M.P.; Cornille, K.; Tintignac, L.A.; Leibovitch, S.A. MAFbx/Atrogin-1 controls the activity of the initiation factor eIF3-f in skeletal muscle atrophy by targeting multiple C-terminal lysines. J. Biol. Chem. 2009, 284, 4413–4421. [Google Scholar] [CrossRef] [Green Version]
- Lokireddy, S.; Wijesoma, I.W.; Sze, S.K.; McFarlane, C.; Kambadur, R.; Sharma, M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am. J. Physiol.-Cell Physiol. 2012, 303, C512–C529. [Google Scholar] [CrossRef] [Green Version]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Baehr, L.M.; Hughes, D.C.; Lynch, S.A.; Van Haver, D.; Maia, T.M.; Marshall, A.G.; Radoshevich, L.; Impens, F.; Waddell, D.S.; Bodine, S.C. Identification of the MuRF1 skeletal muscle ubiquitylome through quantitative proteomics. Function 2021, 2, zqab029. [Google Scholar] [CrossRef]
- Rudolf, R.; Bogomolovas, J.; Strack, S.; Choi, K.-R.; Khan, M.M.; Wagner, A.; Brohm, K.; Hanashima, A.; Gasch, A.; Labeit, D. Regulation of nicotinic acetylcholine receptor turnover by MuRF1 connects muscle activity to endo/lysosomal and atrophy pathways. Age 2013, 35, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.M.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar] [PubMed] [Green Version]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Gomes, A.V.; Waddell, D.S.; Siu, R.; Stein, M.; Dewey, S.; Furlow, J.D.; Bodine, S.C. Upregulation of proteasome activity in muscle RING finger 1-null mice following denervation. FASEB J. 2012, 26, 2986–2999. [Google Scholar] [CrossRef] [Green Version]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef]
- Altun, M.; Besche, H.C.; Overkleeft, H.S.; Piccirillo, R.; Edelmann, M.J.; Kessler, B.M.; Goldberg, A.L.; Ulfhake, B. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J. Biol. Chem. 2010, 285, 39597–39608. [Google Scholar] [CrossRef] [Green Version]
- Raue, U.; Slivka, D.; Jemiolo, B.; Hollon, C.; Trappe, S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Clavel, S.; Coldefy, A.-S.; Kurkdjian, E.; Salles, J.; Margaritis, I.; Derijard, B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech. Ageing Dev. 2006, 127, 794–801. [Google Scholar] [CrossRef]
- Strucksberg, K.-H.; Tangavelou, K.; Schröder, R.; Clemen, C.S. Proteasomal activity in skeletal muscle: A matter of assay design, muscle type, and age. Anal. Biochem. 2010, 399, 225–229. [Google Scholar] [CrossRef]
- Edström, E.; Altun, M.; Hägglund, M.; Ulfhake, B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 663–674. [Google Scholar] [CrossRef]
- Kaiser, M.S.; Milan, G.; Ham, D.J.; Lin, S.; Oliveri, F.; Chojnowska, K.; Tintignac, L.A.; Mittal, N.; Zimmerli, C.E.; Glass, D.J.; et al. Dual roles of mTORC1-dependent activation of the ubiquitin-proteasome system in muscle proteostasis. Commun. Biol. 2022, 5, 1141. [Google Scholar] [CrossRef]
- Kido, K.; Sase, K.; Yokokawa, T.; Fujita, S. Enhanced skeletal muscle insulin sensitivity after acute resistance-type exercise is upregulated by rapamycin-sensitive mTOR complex 1 inhibition. Sci. Rep. 2020, 10, 8509. [Google Scholar] [CrossRef]
- Ogata, T.; Oishi, Y.; Higuchi, M.; Muraoka, I. Fasting-related autophagic response in slow-and fast-twitch skeletal muscle. Biochem. Biophys. Res. Commun. 2010, 394, 136–140. [Google Scholar] [CrossRef]
- Mofarrahi, M.; Guo, Y.; Haspel, J.A.; Choi, A.M.; Davis, E.C.; Gouspillou, G.; Hepple, R.T.; Godin, R.; Burelle, Y.; Hussain, S.N. Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy 2013, 9, 1604–1620. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Hood, D.A. The regulation of autophagy during exercise in skeletal muscle. J. Appl. Physiol. 2016, 120, 664–673. [Google Scholar] [CrossRef] [Green Version]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol.-Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [Green Version]
- Madaro, L.; Marrocco, V.; Carnio, S.; Sandri, M.; Bouché, M. Intracellular signaling in ER stress-induced autophagy in skeletal muscle cells. FASEB J. 2013, 27, 1990–2000. [Google Scholar] [CrossRef] [Green Version]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.-I.; Natsume, T.; Guan, J.-L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Mercer, C.A.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Proikas-Cezanne, T.; Takacs, Z.; Dönnes, P.; Kohlbacher, O. WIPI proteins: Essential PtdIns3 P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Sou, Y.-S.; Waguri, S.; Iwata, J.-I.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [Green Version]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef] [Green Version]
- Fallaize, D.; Chin, L.-S.; Li, L. Differential submitochondrial localization of PINK1 as a molecular switch for mediating distinct mitochondrial signaling pathways. Cell. Signal. 2015, 27, 2543–2554. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef] [Green Version]
- McMullen, C.A.; Ferry, A.L.; Gamboa, J.L.; Andrade, F.H.; Dupont-Versteegden, E.E. Age-related changes of cell death pathways in rat extraocular muscle. Exp. Gerontol. 2009, 44, 420–425. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, K.; Kinoshita, M.; Ito, Y.; Aizawa, M.; Aoi, W.; Yamaguchi, A. p62/SQSTM1 but not LC3 is accumulated in sarcopenic muscle of mice. J. Cachexia Sarcopenia Muscle 2016, 7, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Drummond, M.J.; Glynn, E.L.; Dickinson, J.M.; Gundermann, D.M.; Timmerman, K.L.; Walker, D.K.; Volpi, E.; Rasmussen, B.B. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2013, 68, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-S.; Varadhachary, A.S.; Miller, S.E.; Weihl, C.C. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 2010, 6, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.-D.; Yan, X.-L.; Fan, S.-D.; Chen, X.-Y.; Yan, J.-Y.; Dong, Q.-T.; Chen, W.-Z.; Liu, N.-X.; Chen, X.-L.; Yu, Z. Nrf2 deficiency promotes the increasing trend of autophagy during aging in skeletal muscle: A potential mechanism for the development of sarcopenia. Aging (Albany NY) 2020, 12, 5977. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- White, Z.; Terrill, J.; White, R.B.; McMahon, C.; Sheard, P.; Grounds, M.D.; Shavlakadze, T. Voluntary resistance wheel exercise from mid-life prevents sarcopenia and increases markers of mitochondrial function and autophagy in muscles of old male and female C57BL/6J mice. Skelet. Muscle 2016, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Bujak, A.L.; Crane, J.D.; Lally, J.S.; Ford, R.J.; Kang, S.J.; Rebalka, I.A.; Green, A.E.; Kemp, B.E.; Hawke, T.J.; Schertzer, J.D. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015, 21, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zhu, X.; Chen, K.; Lang, H.; Zhang, Y.; Hou, P.; Ran, L.; Zhou, M.; Zheng, J.; Yi, L. Resveratrol prevents sarcopenic obesity by reversing mitochondrial dysfunction and oxidative stress via the PKA/LKB1/AMPK pathway. Aging 2019, 11, 2217. [Google Scholar] [CrossRef]
- Demontis, F.; Perrimon, N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 2010, 143, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Penniman, C.M.; Bhardwaj, G.; Nowers, C.J.; Brown, C.U.; Junck, T.L.; Boyer, C.K.; Jena, J.; Fuqua, J.D.; Lira, V.A.; O’Neill, B.T. Loss of FoxOs in muscle increases strength and mitochondrial function during aging. J. Cachexia Sarcopenia Muscle 2022. [Google Scholar] [CrossRef]
- Castets, P.; Lin, S.; Rion, N.; Di Fulvio, S.; Romanino, K.; Guridi, M.; Frank, S.; Tintignac, L.A.; Sinnreich, M.; Rüegg, M.A. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013, 17, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Crombie, E.M.; Kim, S.; Adamson, S.; Dong, H.; Lu, T.C.; Wu, Y.; Wu, Y.; Levy, Y.; Stimple, N.; Lam, W.M.R. Activation of eIF4E-binding-protein-1 rescues mTORC1-induced sarcopenia by expanding lysosomal degradation capacity. J. Cachexia Sarcopenia Muscle 2022. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Wright, K.J.; Romestaing, C.; Thomas, M.M.; Rowan, S.L.; Taivassalo, T.; Hepple, R.T. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell 2010, 9, 1032–1046. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial–lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Holloway, G.P.; Holwerda, A.M.; Miotto, P.M.; Dirks, M.L.; Verdijk, L.B.; van Loon, L.J. Age-associated impairments in mitochondrial ADP sensitivity contribute to redox stress in senescent human skeletal muscle. Cell Rep. 2018, 22, 2837–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, K.E.; Hwang, J.; Zhou, C.; Veluthakal, R.; McCown, E.M.; Hamilton, A.; Oh, E.; Dai, W.; Fueger, P.T.; Jiang, L. Enrichment of the exocytosis protein STX4 in skeletal muscle remediates peripheral insulin resistance and alters mitochondrial dynamics via Drp1. Nat. Commun. 2022, 13, 424. [Google Scholar] [CrossRef] [PubMed]
- Miotto, P.M.; LeBlanc, P.J.; Holloway, G.P. High-fat diet causes mitochondrial dysfunction as a result of impaired ADP sensitivity. Diabetes 2018, 67, 2199–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, W.S.; Zunica, E.R.; Heintz, E.C.; Vandanmagsar, B.; Floyd, Z.E.; Yu, Y.; Fujioka, H.; Hoppel, C.L.; Belmont, K.P.; Axelrod, C.L. Mitochondrial uncoupling attenuates sarcopenic obesity by enhancing skeletal muscle mitophagy and quality control. J. Cachexia Sarcopenia Muscle 2022, 13, 1821–1836. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Delrio-Lorenzo, A.; Rojo-Ruiz, J.; Alonso, M.T.; García-Sancho, J. Sarcoplasmic reticulum Ca2+ decreases with age and correlates with the decline in muscle function in Drosophila. J. Cell Sci. 2020, 133, jcs240879. [Google Scholar] [CrossRef]
- Qaisar, R.; Pharaoh, G.; Bhaskaran, S.; Xu, H.; Ranjit, R.; Bian, J.; Ahn, B.; Georgescu, C.; Wren, J.D.; Van Remmen, H. Restoration of sarcoplasmic reticulum Ca2+ ATPase (SERCA) activity prevents age-related muscle atrophy and weakness in mice. Int. J. Mol. Sci. 2020, 22, 37. [Google Scholar] [CrossRef]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Sataranatarajan, K.; Premkumar, P.; Huseman, K.; Van Remmen, H. Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol. 2019, 20, 68–74. [Google Scholar] [CrossRef]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef]
- Navarro-Borelly, L.; Somasundaram, A.; Yamashita, M.; Ren, D.; Miller, R.J.; Prakriya, M. STIM1–Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J. Physiol. 2008, 586, 5383–5401. [Google Scholar] [CrossRef]
- Stiber, J.; Hawkins, A.; Zhang, Z.-S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Wei-LaPierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805. [Google Scholar] [CrossRef]
- Zhao, X.; Weisleder, N.; Thornton, A.; Oppong, Y.; Campbell, R.; Ma, J.; Brotto, M. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell 2008, 7, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Thornton, A.M.; Zhao, X.; Weisleder, N.; Brotto, L.S.; Bougoin, S.; Nosek, T.M.; Reid, M.; Hardin, B.; Pan, Z.; Ma, J. Store-operated Ca2+ entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging (Albany NY) 2011, 3, 621. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.N.; Blackmore, D.G.; Gilbert, D.F.; Murphy, R.M.; Launikonis, B.S. Store-operated calcium entry remains fully functional in aged mouse skeletal muscle despite a decline in STIM1 protein expression. Aging Cell 2011, 10, 675–685. [Google Scholar] [CrossRef]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef]
- Nagaraj, R.Y.; Nosek, C.M.; Brotto, M.A.; Nishi, M.; Takeshima, H.; Nosek, T.M.; Ma, J. Increased susceptibility to fatigue of slow-and fast-twitch muscles from mice lacking the MG29 gene. Physiol. Genom. 2000, 4, 43–49. [Google Scholar] [CrossRef]
- Peterson, M.D.; Sen, A.; Gordon, P.M. Influence of resistance exercise on lean body mass in aging adults: A meta-analysis. Med. Sci. Sport. Exerc. 2011, 43, 249. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Liang, J.; Wu, L.; Zhang, H.; Lv, J.; Chen, N. Exercise-induced autophagy suppresses sarcopenia through Akt/mTOR and Akt/FoxO3a signal pathways and AMPK-mediated mitochondrial quality control. Front. Physiol. 2020, 11, 583478. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Beasley, T.M.; Allison, D.B.; Weindruch, R. Attenuation of sarcopenia by dietary restriction in rhesus monkeys. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 556–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez, G.; Tapia, J.C.; Kang, H.; Clemenson, G.D., Jr.; Gage, F.; Lichtman, J.W.; Sanes, J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA 2010, 107, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Chen, J.; Zhou, J.; Martin, C.K.; Ravussin, E.; Redman, L.M. Effect of 2-year caloric restriction on organ and tissue size in nonobese 21-to 50-year-old adults in a randomized clinical trial: The CALERIE study. Am. J. Clin. Nutr. 2021, 114, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Wolfe, R.R. The danger of weight loss in the elderly. J. Nutr. Health Aging 2008, 12, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Ter Borg, S.; Luiking, Y.; van Helvoort, A.; Boirie, Y.; Schols, J.; de Groot, C. Low levels of branched chain amino acids, eicosapentaenoic acid and micronutrients are associated with low muscle mass, strength and function in community-dwelling older adults. J. Nutr. Health Aging 2019, 23, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.J.; Hermans, W.J.; Holwerda, A.M.; Smeets, J.S.; Senden, J.M.; van Kranenburg, J.; Gijsen, A.P.; Wodzig, W.K.; Schierbeek, H.; Verdijk, L.B. Branched-chain amino acid and branched-chain ketoacid ingestion increases muscle protein synthesis rates in vivo in older adults: A double-blind, randomized trial. Am. J. Clin. Nutr. 2019, 110, 862–872. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.K.; Ankarfeldt, M.Z.; Capra, S.; Bauer, J.; Raymond, K.; Heitmann, B.L. Lean body mass change over 6 years is associated with dietary leucine intake in an older Danish population. Br. J. Nutr. 2016, 115, 1556–1562. [Google Scholar] [CrossRef] [Green Version]
- Devries, M.C.; McGlory, C.; Bolster, D.R.; Kamil, A.; Rahn, M.; Harkness, L.; Baker, S.K.; Phillips, S.M. Leucine, not total protein, content of a supplement is the primary determinant of muscle protein anabolic responses in healthy older women. J. Nutr. 2018, 148, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, R.R. Branched-chain amino acids and muscle protein synthesis in humans: Myth or reality? J. Int. Soc. Sport. Nutr. 2017, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.-H.; Wu, S.-J.; Wang, S.-T.; Chang, Y.-F.; Chang, C.-S.; Kuan, T.-S.; Chuang, H.-Y.; Chang, C.-M.; Chou, W.; Wu, C.-H. Effects of enriched branched-chain amino acid supplementation on sarcopenia. Aging (Albany NY) 2020, 12, 15091. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Bhasin, S.; Woodhouse, L.; Casaburi, R.; Singh, A.B.; Mac, R.P.; Lee, M.; Yarasheski, K.E.; Sinha-Hikim, I.; Dzekov, C.; Dzekov, J. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J. Clin. Endocrinol. Metab. 2005, 90, 678–688. [Google Scholar] [CrossRef]
- Calof, O.M.; Singh, A.B.; Lee, M.L.; Kenny, A.M.; Urban, R.J.; Tenover, J.L.; Bhasin, S. Adverse events associated with testosterone replacement in middle-aged and older men: A meta-analysis of randomized, placebo-controlled trials. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 1451–1457. [Google Scholar] [CrossRef] [Green Version]
- Borst, S.E.; Yarrow, J.F.; Conover, C.F.; Nseyo, U.; Meuleman, J.R.; Lipinska, J.A.; Braith, R.W.; Beck, D.T.; Martin, J.S.; Morrow, M. Musculoskeletal and prostate effects of combined testosterone and finasteride administration in older hypogonadal men: A randomized, controlled trial. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, E433–E442. [Google Scholar] [CrossRef] [Green Version]
- Papanicolaou, D.A.; Ather, S.; Zhu, H.; Zhou, Y.; Lutkiewicz, J.; Scott, B.; Chandler, J. A phase IIA randomized, placebo-controlled clinical trial to study the efficacy and safety of the selective androgen receptor modulator (SARM), MK-0773 in female participants with sarcopenia. J. Nutr. Health Aging 2013, 17, 533–543. [Google Scholar] [CrossRef]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J. Myostatin blockade with a fully human monoclonal antibody induces muscle hypertrophy and reverses muscle atrophy in young and aged mice. Skelet. Muscle 2015, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- Rooks, D.; Praestgaard, J.; Hariry, S.; Laurent, D.; Petricoul, O.; Perry, R.G.; Lach-Trifilieff, E.; Roubenoff, R. Treatment of sarcopenia with bimagrumab: Results from a phase II, randomized, controlled, proof-of-concept study. J. Am. Geriatr. Soc. 2017, 65, 1988–1995. [Google Scholar] [CrossRef]
- Alway, S.E.; McCrory, J.L.; Kearcher, K.; Vickers, A.; Frear, B.; Gilleland, D.L.; Bonner, D.E.; Thomas, J.M.; Donley, D.A.; Lively, M.W. Resveratrol enhances exercise-induced cellular and functional adaptations of skeletal muscle in older men and women. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2017, 72, 1595–1606. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paez, H.G.; Pitzer, C.R.; Alway, S.E. Age-Related Dysfunction in Proteostasis and Cellular Quality Control in the Development of Sarcopenia. Cells 2023, 12, 249. https://doi.org/10.3390/cells12020249
Paez HG, Pitzer CR, Alway SE. Age-Related Dysfunction in Proteostasis and Cellular Quality Control in the Development of Sarcopenia. Cells. 2023; 12(2):249. https://doi.org/10.3390/cells12020249
Chicago/Turabian StylePaez, Hector G., Christopher R. Pitzer, and Stephen E. Alway. 2023. "Age-Related Dysfunction in Proteostasis and Cellular Quality Control in the Development of Sarcopenia" Cells 12, no. 2: 249. https://doi.org/10.3390/cells12020249