
Genetic Inhibition of Mitochondrial Permeability Transition Pore Exacerbates Ryanodine Receptor 2 Dysfunction in Arrhythmic Disease
 ,
,  and
and
Abstract
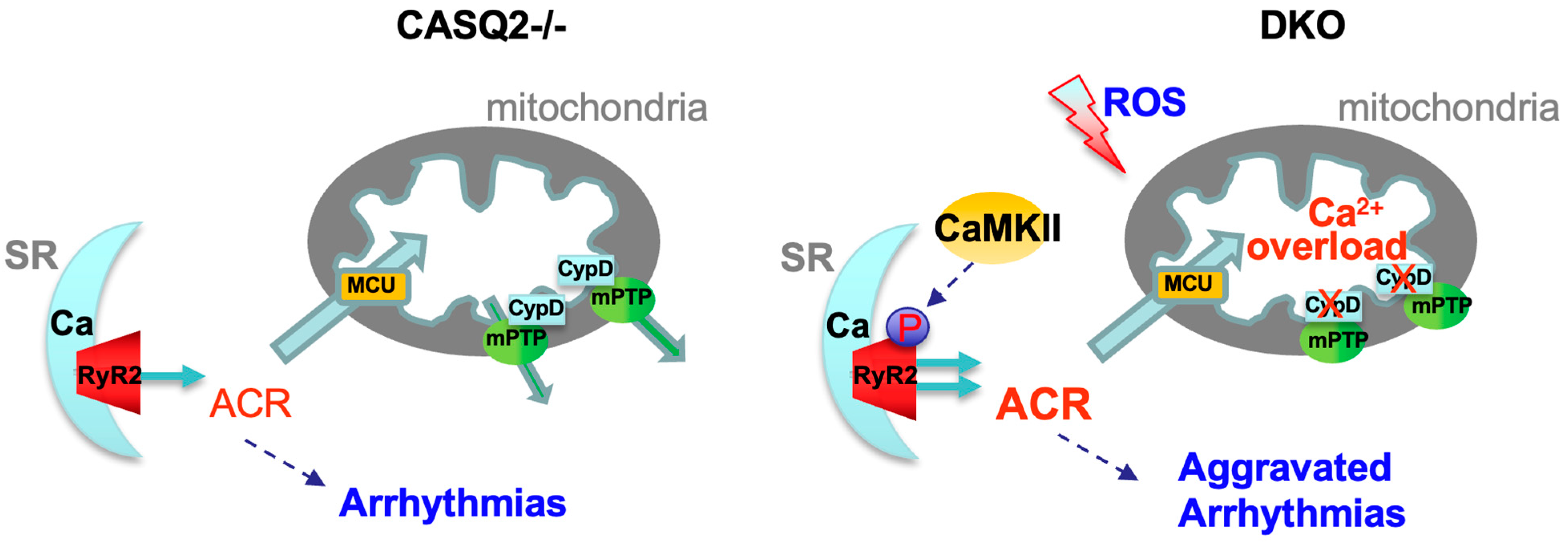
:1. Introduction
2. Materials and Methods
2.1. Generation of Mouse Models
2.2. Echocardiography
2.3. Cardiomyocyte Isolation
2.4. Fluorescent Imaging
2.4.1. Ca2+ Imaging
2.4.2. ROS Imaging
2.4.3. Mitochondrial Membrane Potential (MitoWinks Recordings)
2.5. Western Blot
2.6. Staining of Cardiac Tissue Sections
2.7. RNA Extraction, Library Construction, and Sequencing
2.8. mRNA Sequence Processing
2.9. Statistical Analysis
3. Results
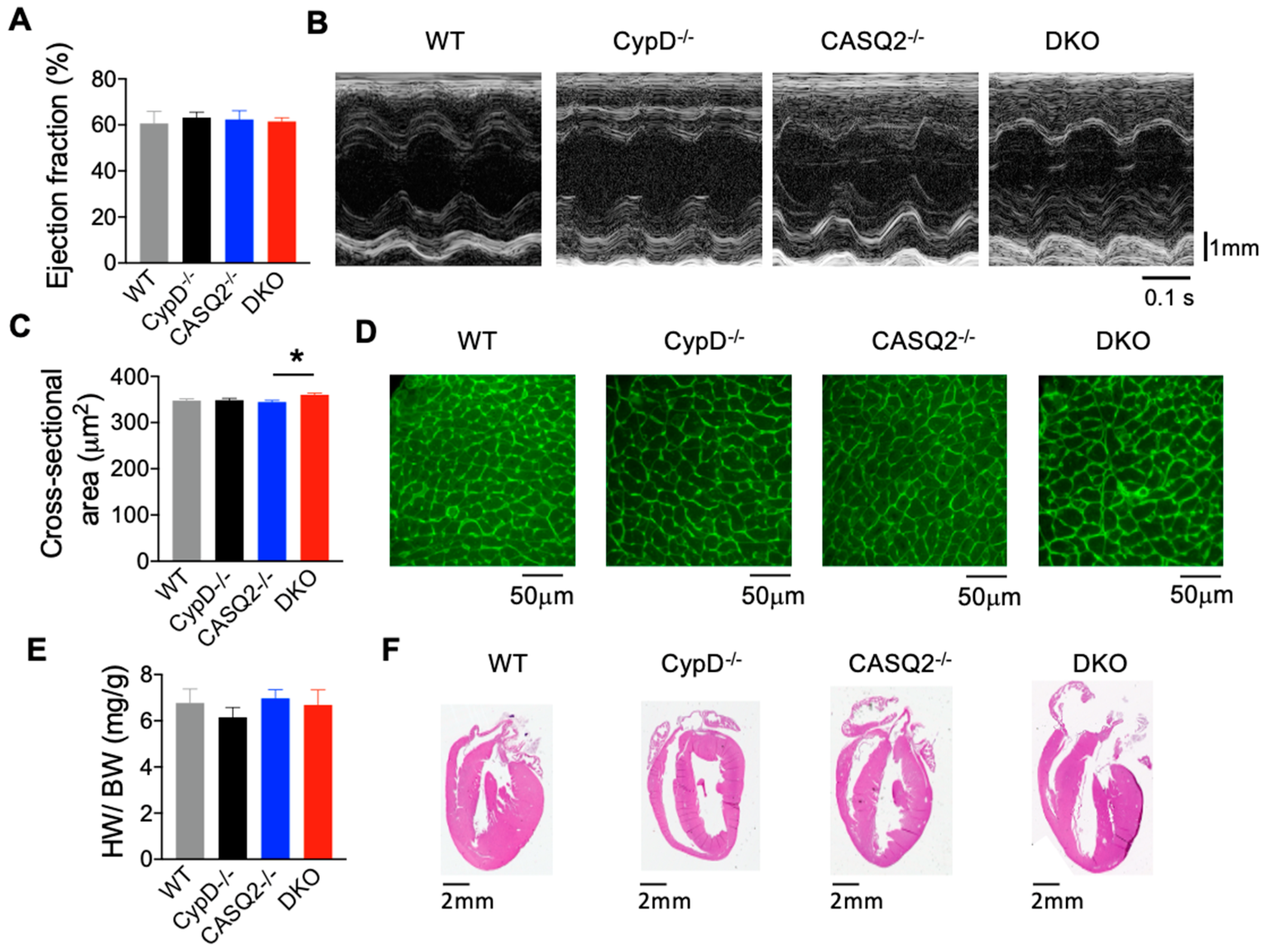
3.1. DKO Mice Did Not Display Pathological Cardiac Remodeling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Echocardiography Parameters | CypD−/− | CASQ2−/− | DKO | WT |
|---|---|---|---|---|
| E/A | 1.60 ± 0.12 | 1.76 ± 0.18 | 1.34 ± 0.05 | 1.37 ± 0.13 |
| IVS;d mm | 1.12 ± 0.10 | 1.18 ± 0.07 | 1.44 ± 0.16 | 1.03 ± 0.06 |
| IVS;s mm | 1.57 ± 0.12 | 1.64 ± 0.09 | 1.76 ± 0.12 | 1.53 ± 0.10 |
| LVID;d mm | 3.17 ± 0.14 | 3.46 ± 0.14 | 3.05 ± 0.10 | 3.50 ± 0.15 |
| LVID;s mm | 2.19 ± 0.11 | 2.30 ± 0.10 | 2.21 ± 0.39 | 2.38 ± 0.21 |
| LVPW;d mm | 1.31 ± 0.10 * | 1.09 ± 0.10 | 1.38 ± 0.10 * | 0.83 ± 0.10 |
| LVPW;s mm | 1.52 ± 0.14 | 1.49 ± 0.11 | 1.63 ± 0.09 * | 1.10 ± 0.12 |
| LV Vol;s µL | 16.50 ± 2.21 | 18.43 ± 1.98 | 17.19 ± 3.10 | 24.29 ± 2.08 |
| LV Vol;d µL | 41.11 ± 4.25 | 50.33 ± 4.95 | 36.93 ± 2.92 | 51.41 ± 5.05 |
| Ejection Fraction % | 63.15 ± 2.43 | 62.38 ± 3.76 | 61.48 ± 1.63 | 60.66 ± 5.18 |
| Fractional Shortening % | 33.40 ± 1.72 | 33.31 ± 2.65 | 32.02 ± 2.03 | 32.46 ± 4.05 |
| Heart Rate | 446 ± 31 | 400 ± 20 | 424 ± 20 | 431 ± 19 |
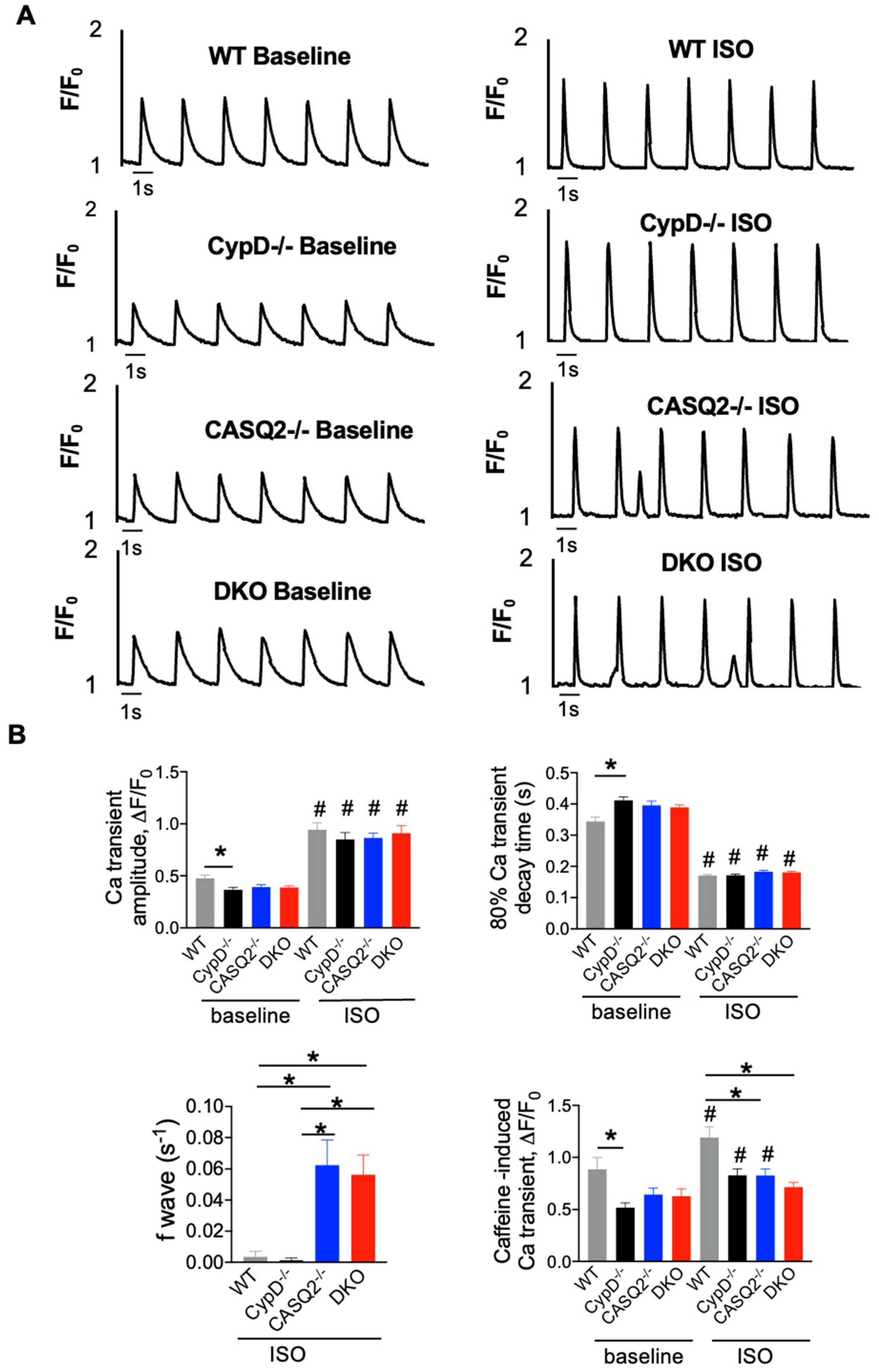
3.2. DKO Mice Displayed Abnormal Myocyte Ca2+ Handling
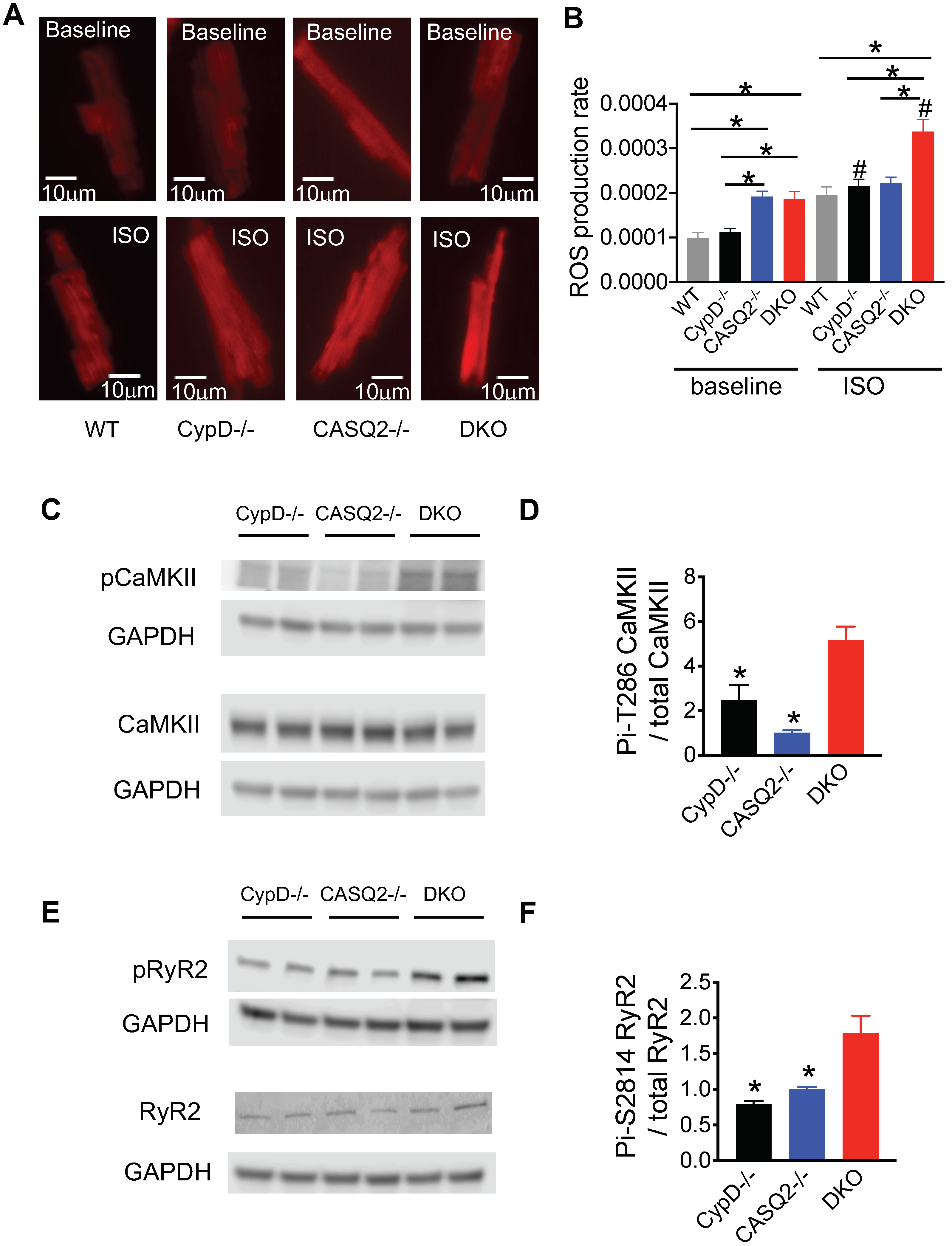
3.3. Genetic Inhibition of mPTP in the CPVT Model Led to CaMKII-Mediated Hyperphosphorylation of RyR2
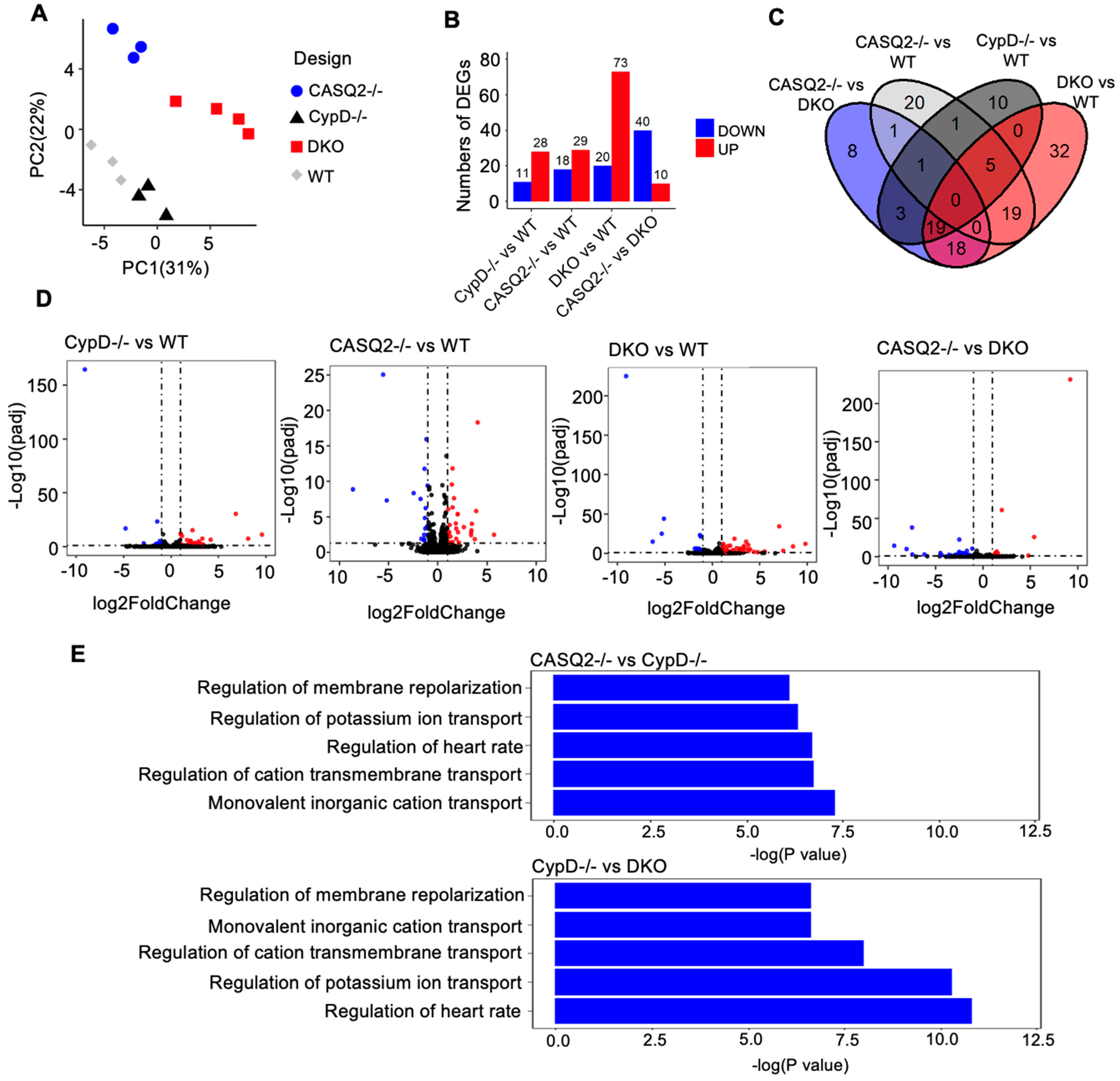
3.4. Transcriptome Analysis Identified Altered Gene Expression Patterns Associated with Electrical Instability in DKO
4. Discussion
4.1. Frequency-Dependent Exacerbation of Ca2+ Handling
4.2. Role of CaMKII in CPVT
4.3. The Detrimental Effect of mPTP Inhibition in Cardiac Dysfunctions Marked by Leaky RyR2
4.4. Electrical Remodeling Identified with Transcriptome Analysis
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACR | Aberrant calcium release |
| BSA | Bovine Serum Albumin |
| Ca2+ | Calcium |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| DEGs | Differentially expressed genes |
| DKO | Double Knockout |
| E/A-wave ratio | E-wave and A-wave that represent the left ventricular early Filling and atrial contraction filling, respectively |
| EF | Ejection fraction |
| FC | Fold Change |
| GO | Gene Ontology |
| H&E | Hematoxylin and Eosin |
| HW/BW | Heart weight/body weight |
| ISO | Isoproterenol |
| IVS;d | Interventricular septal end diastole |
| IVS;s | Interventricular septal end systole |
| KO | Knockout |
| LVID;d | Left ventricular internal diameter end diastole |
| LVID;s | Left ventricular internal diameter end systole |
| LVPW;d | Left ventricular posterior wall end diastole |
| LVPW;s | Left ventricular posterior wall end systole |
| mCa2+ | Mitochondrial Calcium |
| MCU | Mitochondrial calcium uniporter |
| MitoWinks | Transient opening of mPTP |
| mNCX | Mitochondrial Na/Ca (sodium/calcium) exchanger |
| mPTP | Mitochondrial permeability transition pore |
| PCA | Principal component analysis |
| PCR | Polymerase chain reactions |
| RyR2 | Ryanodine receptor 2 |
| SCWs | Spontaneous Ca waves |
| SEM | Standard error of the mean |
| SR | Sarcoplasmic reticulum |
| SRA | Sequence Read Archive |
| TLR | Toll-like receptor |
| WGA | Wheat germ agglutinin |
| WT | Wild type |
References
- Belevych, A.E.; Radwanski, P.B.; Carnes, C.A.; Gyorke, S. ‘Ryanopathy’: Causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc. Res. 2013, 98, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Chen, S.R. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaras, N.; Ugur, M.; Ozdemir, S.; Gurdal, H.; Purali, N.; Lacampagne, A.; Vassort, G.; Turan, B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes 2005, 54, 3082–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorke, S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2009, 6, 123–129. [Google Scholar] [CrossRef]
- De la Fuente, S.; Sheu, S.S. SR-mitochondria communication in adult cardiomyocytes: A close relationship where the Ca2+ has a lot to say. Arch. Biochem. Biophys. 2019, 663, 259–268. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Boyman, L.; Williams, G.S.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.; Hoek, J.; Sheu, S.S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Lim, S.Y.; Davidson, S.M.; Hausenloy, D.J.; Yellon, D.M. Preconditioning and postconditioning: The essential role of the mitochondrial permeability transition pore. Cardiovasc. Res. 2007, 75, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Tow, B.D.; Deb, A.; Neupane, S.; Patel, S.M.; Reed, M.; Loper, A.B.; Eliseev, R.A.; Knollmann, B.C.; Gyorke, S.; Liu, B. SR-Mitochondria Crosstalk Shapes Ca Signalling to Impact Pathophenotype in Disease Models Marked by Dysregulated Intracellular Ca Release. Cardiovasc. Res. 2022, 118, 2819–2832. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef]
- Gyorke, S.; Terentyev, D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc. Res. 2008, 77, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ho, H.T.; Brunello, L.; Unudurthi, S.D.; Lou, Q.; Belevych, A.E.; Qian, L.; Kim, D.H.; Cho, C.; Janssen, P.M.; et al. Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovasc. Res. 2015, 108, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidlmayer, L.K.; Mages, C.; Berbner, A.; Eder-Negrin, P.; Arias-Loza, P.A.; Kaspar, M.; Song, M.; Dorn, G.W., II; Kohlhaas, M.; Frantz, S.; et al. Mitofusin 2 Is Essential for IP(3)-Mediated SR/Mitochondria Metabolic Feedback in Ventricular Myocytes. Front. Physiol. 2019, 10, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Joung, J.G.; Zheng, Y.; Chen, Y.R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 2011, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wullschleger, M.; Blanch, J.; Egger, M. Functional local crosstalk of inositol 1,4,5-trisphosphate receptor- and ryanodine receptor-dependent Ca2+ release in atrial cardiomyocytes. Cardiovasc. Res. 2017, 113, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Park, K.H.; Yamazaki, D.; Lin, P.H.; Nishi, M.; Ma, Z.; Qiu, L.; Murayama, T.; Zou, X.; Takeshima, H.; et al. TRIC-A Channel Maintains Store Calcium Handling by Interacting with Type 2 Ryanodine Receptor in Cardiac Muscle. Circ. Res. 2020, 126, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lin, P.; Yamazaki, D.; Park, K.H.; Komazaki, S.; Chen, S.R.; Takeshima, H.; Ma, J. Trimeric intracellular cation channels and sarcoplasmic/endoplasmic reticulum calcium homeostasis. Circ. Res. 2014, 114, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, I.M.; Belevych, A.E.; Baine, S.; Stepanov, A.; Mezache, L.; Bodnar, T.; Liu, B.; Volpe, P.; Priori, S.; Weisleder, N.; et al. Enhancement of Cardiac Store Operated Calcium Entry (SOCE) within Novel Intercalated Disk Microdomains in Arrhythmic Disease. Sci. Rep. 2019, 9, 10179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Min, L.; Qiu, X.; Wu, X.; Liu, C.; Ma, J.; Zhang, D.; Zhu, L. Biological Function of Long Non-coding RNA (LncRNA) Xist. Front. Cell Dev. Biol. 2021, 9, 645647. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Chen, L.; Chen, W.; Zhang, Y.; Chen, J.; Wu, X.; Zhao, Y.; Wu, X.; Sun, G. The long noncoding RNA XIST protects cardiomyocyte hypertrophy by targeting miR-330-3p. Biochem. Biophys. Res. Commun. 2018, 505, 807–815. [Google Scholar] [CrossRef]
- Brunello, L.; Slabaugh, J.L.; Radwanski, P.B.; Ho, H.T.; Belevych, A.E.; Lou, Q.; Chen, H.; Napolitano, C.; Lodola, F.; Priori, S.G.; et al. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc. Natl. Acad. Sci. USA 2013, 110, 10312–10317. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.R. Mechanisms of CaMKII Activation in the Heart. Front. Pharmacol. 2014, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Wu, Y.; Gao, Z.; Chen, B.; Koval, O.M.; Singh, M.V.; Guan, X.; Hund, T.J.; Kutschke, W.; Sarma, S.; Grumbach, I.M.; et al. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. USA 2009, 106, 5972–5977. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Metrich, M.; Fernandez-Velasco, M.; Lucas, A.; Leroy, J.; Perrier, R.; Morel, E.; Fischmeister, R.; Richard, S.; Benitah, J.P.; et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 2007, 583, 685–694. [Google Scholar] [CrossRef]
- Shan, J.; Betzenhauser, M.J.; Kushnir, A.; Reiken, S.; Meli, A.C.; Wronska, A.; Dura, M.; Chen, B.X.; Marks, A.R. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Investig. 2010, 120, 4375–4387. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Makarewich, C.A.; Kubo, H.; Wang, W.; Duran, J.M.; Li, Y.; Berretta, R.M.; Koch, W.J.; Chen, X.; Gao, E.; et al. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ. Res. 2012, 110, 831–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonnell, S.M.; Garcia-Rivas, G.; Scherman, J.A.; Kubo, H.; Chen, X.; Valdivia, H.; Houser, S.R. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ. Res. 2008, 102, e65–e72. [Google Scholar] [CrossRef]
- Liu, B.; Ho, H.T.; Velez-Cortes, F.; Lou, Q.; Valdivia, C.R.; Knollmann, B.C.; Valdivia, H.H.; Gyorke, S. Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca2+-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J. Physiol. 2014, 592, 1957–1973. [Google Scholar] [CrossRef] [Green Version]
- Respress, J.L.; van Oort, R.J.; Li, N.; Rolim, N.; Dixit, S.S.; deAlmeida, A.; Voigt, N.; Lawrence, W.S.; Skapura, D.G.; Skardal, K.; et al. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 2012, 110, 1474–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.T.; Liu, B.; Snyder, J.S.; Lou, Q.; Brundage, E.A.; Velez-Cortes, F.; Wang, H.; Ziolo, M.T.; Anderson, M.E.; Sen, C.K.; et al. Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc. Res. 2014, 101, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wang, T.; Wang, W.; Cutler, M.J.; Wang, Q.; Voigt, N.; Rosenbaum, D.S.; Dobrev, D.; Wehrens, X.H. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ. Res. 2012, 110, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Zhang, D.; Qi, Y.; Li, Y.; Lee, K.Y.; Bezzerides, V.J.; Yang, P.; Xia, S.; Kim, S.L.; Liu, X.; et al. Insights Into the Pathogenesis of Catecholaminergic Polymorphic Ventricular Tachycardia From Engineered Human Heart Tissue. Circulation 2019, 140, 390–404. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca2+/Calmodulin-Dependent Kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Erickson, J.R.; He, B.J.; Grumbach, I.M.; Anderson, M.E. CaMKII in the cardiovascular system: Sensing redox states. Physiol. Rev. 2011, 91, 889–915. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Noujaim, S.F.; Tolkacheva, E.G.; Talkachou, A.; O’Connell, R.; Berenfeld, O.; Anumonwo, J.; Pandit, S.V.; Vikstrom, K.; Napolitano, C.; et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2007, 101, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Loaiza, R.; Benkusky, N.A.; Powers, P.P.; Hacker, T.; Noujaim, S.; Ackerman, M.J.; Jalife, J.; Valdivia, H.H. Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2013, 112, 298–308. [Google Scholar] [CrossRef]
- Rizzi, N.; Liu, N.; Napolitano, C.; Nori, A.; Turcato, F.; Colombi, B.; Bicciato, S.; Arcelli, D.; Spedito, A.; Scelsi, M.; et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: A complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 2008, 103, 298–306. [Google Scholar] [CrossRef]
- Houser, S.R. Can novel therapies for arrhythmias caused by spontaneous sarcoplasmic reticulum Ca2+ release be developed using mouse models? Circ. Res. 2005, 96, 1031–1032. [Google Scholar] [CrossRef] [Green Version]
- Denegri, M.; Bongianino, R.; Lodola, F.; Boncompagni, S.; De Giusti, V.C.; Avelino-Cruz, J.E.; Liu, N.; Persampieri, S.; Curcio, A.; Esposito, F.; et al. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation 2014, 129, 2673–2681. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Walton, S.D.; Ho, H.T.; Belevych, A.E.; Tikunova, S.B.; Bonilla, I.; Shettigar, V.; Knollmann, B.C.; Priori, S.G.; Volpe, P.; et al. Gene Transfer of Engineered Calmodulin Alleviates Ventricular Arrhythmias in a Calsequestrin-Associated Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2018, 7, e008155. [Google Scholar] [CrossRef] [Green Version]
- Kashimura, T.; Briston, S.J.; Trafford, A.W.; Napolitano, C.; Priori, S.G.; Eisner, D.A.; Venetucci, L.A. In the RyR2(R4496C) mouse model of CPVT, beta-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ. Res. 2010, 107, 1483–1489. [Google Scholar] [CrossRef] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Gordan, R.; Fefelova, N.; Gwathmey, J.K.; Xie, L.H. Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: Evidence from cyclophilin D knockout mice. Cell Calcium 2016, 60, 363–372. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deb, A.; Tow, B.D.; Qing, Y.; Walker, M.; Hodges, E.R.; Stewart, J.A., Jr.; Knollmann, B.C.; Zheng, Y.; Wang, Y.; Liu, B. Genetic Inhibition of Mitochondrial Permeability Transition Pore Exacerbates Ryanodine Receptor 2 Dysfunction in Arrhythmic Disease. Cells 2023, 12, 204. https://doi.org/10.3390/cells12020204
Deb A, Tow BD, Qing Y, Walker M, Hodges ER, Stewart JA Jr., Knollmann BC, Zheng Y, Wang Y, Liu B. Genetic Inhibition of Mitochondrial Permeability Transition Pore Exacerbates Ryanodine Receptor 2 Dysfunction in Arrhythmic Disease. Cells. 2023; 12(2):204. https://doi.org/10.3390/cells12020204
Chicago/Turabian StyleDeb, Arpita, Brian D. Tow, You Qing, Madelyn Walker, Emmanuel R. Hodges, James A. Stewart, Jr., Björn C. Knollmann, Yi Zheng, Ying Wang, and Bin Liu. 2023. "Genetic Inhibition of Mitochondrial Permeability Transition Pore Exacerbates Ryanodine Receptor 2 Dysfunction in Arrhythmic Disease" Cells 12, no. 2: 204. https://doi.org/10.3390/cells12020204